Abstract
The study investigates the antibiotic resistance (AR) profiles and genetic determinants in three strains of guaiacol-producing Alicyclobacillus spp. isolated from orchard soil and pears. Their phenotypic characteristics, such as spore formation; resistance to different factors, including drugs or disinfectants; or production of off-flavor compounds, can affect the taste and aroma of spoiled products. Food and beverages are potential vectors for the transfer of antibiotic resistance genes, which is a growing health concern; thus, microorganisms in food and beverages should not be a potential source of drug resistance to consumers. Whole-genome sequencing (WGS) was utilized to identify antibiotic resistance genes, metabolic pathways, and elements associated with guaiacol and halophenol production. Minimum inhibitory concentration (MIC) testing revealed that all strains were susceptible to eight out of nine tested antibiotics (ampicillin, gentamycin, kanamycin, streptomycin, clindamycin, tetracycline, chloramphenicol, and vancomycin) but exhibited high resistance to erythromycin. Analysis indicated that the erythromycin resistance gene, ribosomal RNA small subunit methyltransferase A (RsmA), was intrinsic and likely acquired through horizontal gene transfer (HGT). The comprehensive genomic analysis provides insights into the molecular mechanisms of antibiotic resistance in Alicyclobacillus spp., highlighting the potential risk of these bacteria as vectors for antibiotic resistance genes in the food chain. This study expands the understanding of the genetic makeup of these spoilage bacteria and their role in antimicrobial resistance dissemination.
1. Introduction
Alicyclobacillus spp. are common, spoiling, nonpathogenic, Gram (+), spore-forming, thermo-acidophilic bacteria. They are found in various natural and anthropogenic environments, such as soils, hot springs, mine waters, different fruits, juices, and beverages, as they have the ability to survive in acidic, high temperature, and high-pressure conditions [1,2,3,4,5,6,7,8,9,10] This genus includes species that produce off-flavors and antiseptic-, smoky-, and medicinal-tasting compounds, such as guaiacol and halophenols, 2,6-dibromophenol, and 2,6-dichlorophenol [1,3,11]. Currently, 29 species of Alicyclobacillus are known [4,6,11], with 9 of them reported to produce guaiacol: A. acidophilus, A. acidoterrestris, A. daucy, A. herbarius, A. cycloheptanicus, A. pomorum, A. contaminans, some strains of A. hesperidum, and (as recently reported) A. fastidiosus [11].
Alicyclobacillus spp. pose a significant concern for the food and beverage industry due to their potential to cause substantial economic losses. Consequently, most studies have focused on identifying and characterizing their growth, germination, behavior, adaptation, metabolite production, and control measures [1,2,3,11,12,13,14,15,16]. Agricultural fields and fertilized orchards may serve as hotspots for the duplication of genes that confer drug resistance, facilitating their transfer to soil bacteria and, subsequently, the food chain. This pathway could allow resistance determinants to reach the human body, making it crucial to uncover potential antibiotic resistance (AR) transmission pathways and the critical mechanisms involved in antibiotic resistance. Limited information is available regarding antibiotic susceptibility among Alicyclobacillus spp. Aulitto et al. [2] identified the brcC gene involved in bacitracin resistance in two A. mali genomes. They also reported that A. mali FL18 exhibited resistance to ampicillin, bacitracin, kanamycin, streptomycin, erythromycin, ciprofloxacin, and vancomycin. Based on genomic analysis, they hypothesized that such a broad spectrum of antibiotic resistance is associated with the expression of multiple multidrug efflux transporters. The potential for food and beverages to act as vectors for the transfer of antibiotic resistance genes is a growing health concern [17,18]. The removal of antibiotics from the market in accordance with Regulation (EC) N 1831/2003 has underscored the need to reduce antimicrobial resistance in the food chain [18]. Microorganisms in food and beverages should not be a source of drug resistance to consumers. Resistance to antimicrobial agents can be intrinsic, as natural phenotypic traits typical of all strains of all species, or acquired through additional genes carried on mobile genetic elements (plasmids or transposons) or mutations of indigenous genes [17,19].
One of the key mechanisms for the formation of new genes, including antibiotic resistance genes, is amplification and subsequent mutations, as shown for trimethoprim [20], sulfonamides [21], and β-lactams [22]. Antibiotic resistance predates the 20th-century “antibiotic era”. Prokaryotic antibiotic resistance genes have been found in environments minimally affected by human activity. For instance, the Sul2–strA–strB gene cluster was identified in a 1200- to 1400-year-old Antarctic ice core sample [23]; genes conferring resistance to β-lactams, tetracycline, and glycopeptides were found in 30,000-year-old Beringian permafrost sediments [24]. Additionally, aminoglycosides, β-lactams, fluoroquinolones, and macrolide resistance factors were isolated from Tibetan glacial lake sediments and soil [25]. Genes encoding resistance to standard antibiotics in these samples were associated with efflux pumps, distinct from the modern antibiotic resistome. The natural source of AR genes is environmental bacteria, especially Actinomycetota [26,27,28]. Since metabolic pathways for antibiotic synthesis are estimated to be 200 to 500 million years old, antibiotic resistance must be of similar age to counterbalance the antibiotic effects [29,30,31].
Intrinsic resistance presents minimal potential for horizontal spread among microorganisms. In contrast, acquired resistance poses a greater risk for horizontal dissemination, enabling rapid inactivation of selected antibiotics through degradation, export from the cell via efflux systems, or alteration of the antibiotic’s target site [19]. However, the distinction between intrinsic and acquired resistance may be blurred; due to the action of transposases or integrases, resistance factors may transfer between chromosomes to plasmids, and the ability to transmit genes horizontally can be gained or lost. An additional important factor in the evolution and spread of antibiotic resistance is that if the genes associated with antibiotic resistance are located on high-copy-number plasmids, this type of amplification enables faster evolution and increased resistance to the agent. Bacteria in heterogeneous environments can develop antibiotic resistance faster than bacteria living in homogeneous environments, especially when the potential relationship between AR and flagella and chemotaxis can be found through efflux mechanisms, biofilm formation, or mobile genetic elements [32]. Piskovsky et al., 2023, suggest that bacterial motility can facilitate dispersal on the surface, which may lead to greater nutrient availability and potentially higher antibiotic resistance. Bacterial swarming is the best-understood relationship between cell motility and the ability of bacteria to cope with antibiotics. Swarming is a form of group motility on surfaces where cells are particularly resistant to antibiotic stress. Reduced exposure of individual cells to antibiotics, leads to tolerance of antibiotics. In addition to swarming, other connections between bacterial motility and the antibiotic landscape have been reported. In particular, antibiotics have been shown to induce motor responses. However, it is not known what impact they have on the evolution of antibiotic resistance.
The FEEDAP Panel (The Panel on Additives and Products or Substances used in Animal Feed) defines the microbiological breakpoints to distinguish resistant from susceptible strains [17]. Microbiological cut-off values are established by examining the distribution of MIC values of selected antimicrobials in bacterial populations of a single species or genus. These cut-off values are used to evaluate microbial products for the presence of antimicrobial resistance. Genetic investigations are required to further determine the nature of resistance, especially when limited or no information on the MIC distribution within the species is known, as is true of Alicyclobacillus spp.
This work aims to deepen the understanding of the genetic determinants associated with antibiotic resistance in three guaiacol-producing Alicyclobacillus spp. strains. To achieve this, the three strains were sequenced to annotate relevant regions. Additionally, loci involved in functional characteristics were identified using comprehensive bioinformatic analyses, focusing on the presence of prophage regions, mobile elements, and plasmids. Finally, clusters for the production of metabolites involved in the spoilage phenotype were also recognized, which may be significant for comparative and functional genome analysis within this group of food spoilage bacteria.
2. Results and Discussion
2.1. Assessment of Antibiotic Resistance
The MIC for 9 antibiotics was performed for three strains, KKP 3000, KKP 3001, and KKP 3002, and the reference strain Alicyclobacillus fastidiosus DSM 17978. The antibiotics chosen were based on their diverse mechanisms of action belonging to different classes, such as aminoglycosides (kanamycin, streptomycin, gentamycin), macrolides (erythromycin), chloramphenicol, tetracycline, and clindamycin, which inhibit protein synthesis, as well as β-lactams (ampicillin) and glycopeptides (vancomycin), which inhibit the synthesis of peptidoglycan in Gram (+) bacteria. The assumption was made that the strains should be considered resistant if MICs were >256 μg/mL and sensitive if MICs were <8 μg/mL.
The profiles of antibiotic resistance for examined strains of Alicyclobacillus spp. were very similar to each other. All four strains exhibited significant susceptibility to ampicillin, gentamycin, kanamycin, streptomycin, clindamycin, tetracycline, chloramphenicol, and vancomycin (all values < 1 μg/mL). However, all strains displayed high resistance (256 μg/mL) to erythromycin; even at the maximum applied concentration, no inhibition of the growth of the examined Alicyclobacillus strains was observed. The MIC values with the lowest concentration (μg/mL) of an antibiotic that inhibits the growth of a given strain of bacteria are presented in Table 1. These results suggest that Alicyclobacillus spp. strains are generally susceptible to a broad spectrum of antibiotics commonly used in clinical and laboratory settings.

Table 1.
Minimum inhibitory concentration (MIC) of selected antibiotics in studied Alicyclobacillus strains.
A highly important aspect of AR in the environment is that the ecosystem is a complex structure, and the usage of antibiotics in agriculture or horticulture may lead to the dissemination of AR genes. Using animal fertilizers or manure, with possible antibiotic-resistant intestinal bacteria, may result in the introduction of antibiotic resistance genes into the soil, which has the potential for horizontal gene transfer (HGT). Similarly, using recirculated water for irrigation may lead to exposure of soil microorganisms to pharmaceuticals. Exposure of soil to macrolide antibiotics increases the relative abundance of numerous gene targets associated with resistance to macrolides and other antibiotics, as well as mobile genetic elements. This occurred at an exposure dose that was unrealistically high but did not occur at the lower, more realistic exposure dose [33]. Georgakakos et al., 2023 [34] suggested that erythromycin is more mobile in the environment when introduced with manure, which is likely the largest source of agriculturally sourced environmental antibiotics. And since it is particularly resistant to natural environmental degradation, it can increase the influence on the microbial communities within it.
2.2. Genome Characteristics
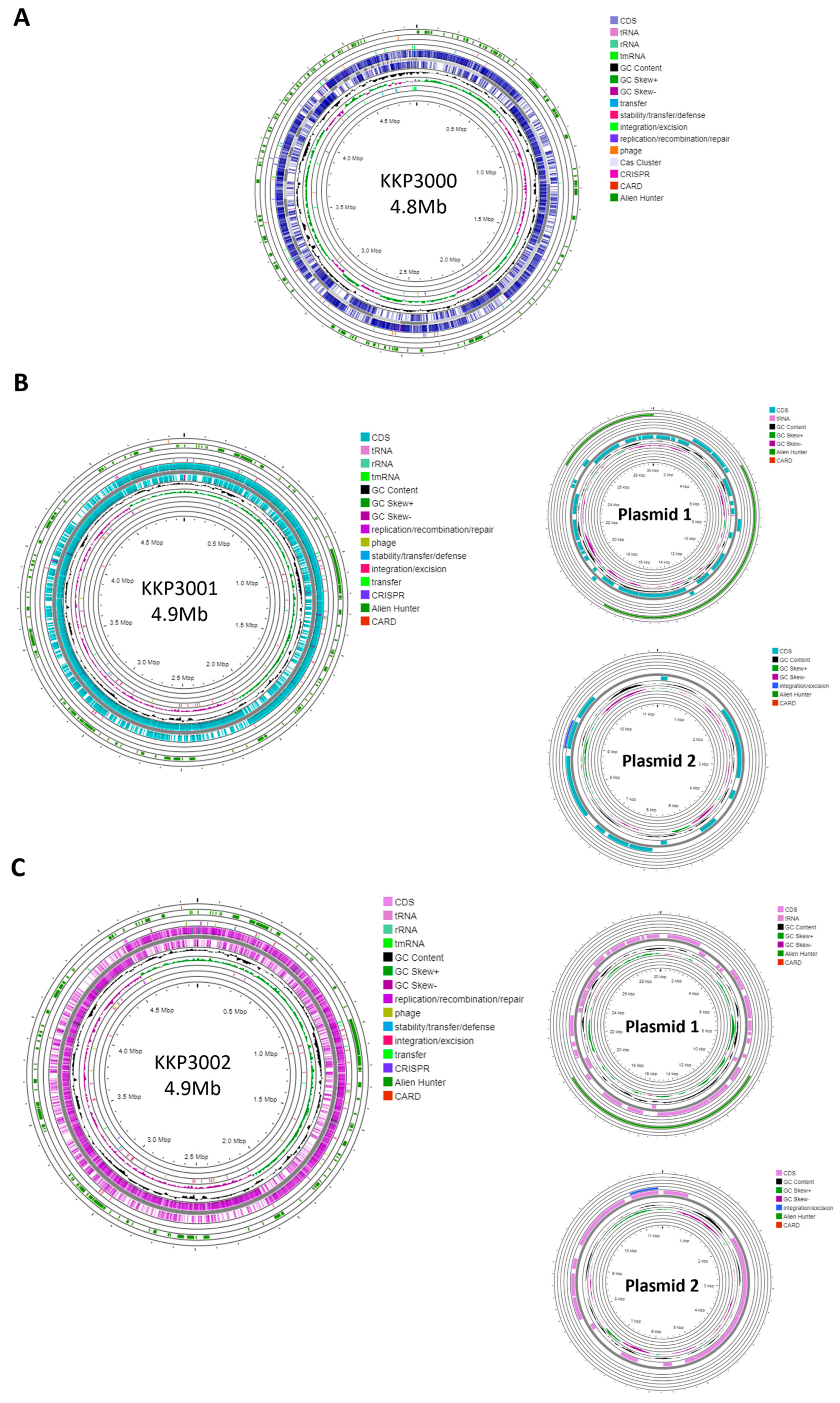
Strain KKP 3000 consists of a single chromosome with a length of 4,859,599 bp and a GC content of 52.95% (Table 2). The WGS of strains KKP 3001 and KKP 3002 consists of a chromosome and two plasmids. In greater detail, the chromosome length of KKP 3001 is 4,972,367 bp, and that of KKP 3002 is 4,977,437 bp. The GC content of the chromosomes is 53 and 52.92%, respectively. Strains KKP 3001 and KKP 3002 each carry two plasmids with a size of 30,081 and 11,548 bp, respectively. The chromosome and plasmid maps of the strains are presented in Figure 1.
Table 2.
Genome characteristics of the three Alicyclobacillus spp. strains.
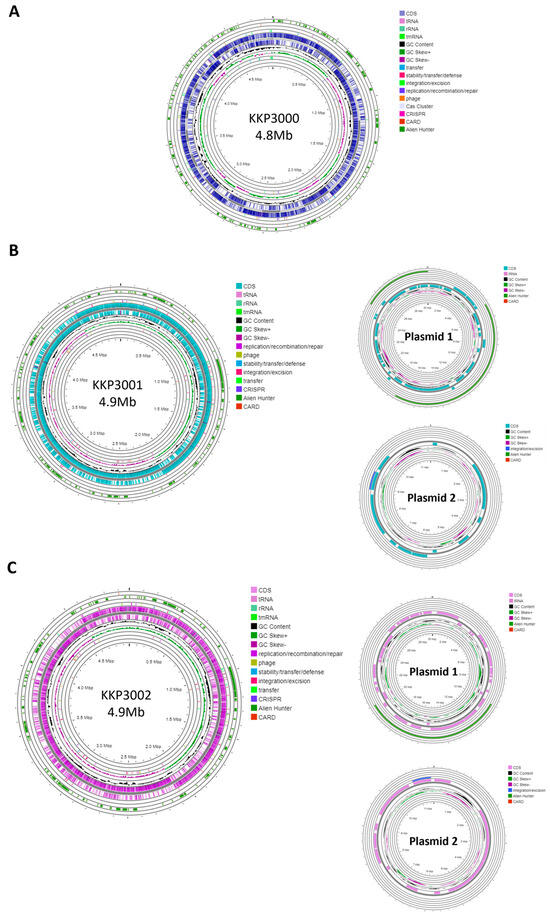
Figure 1.
Genomic maps of the chromosome and plasmids of (A) KKP 3001, (B) KKP 3001, and (C) KKP 3002 constructed with Proksee.
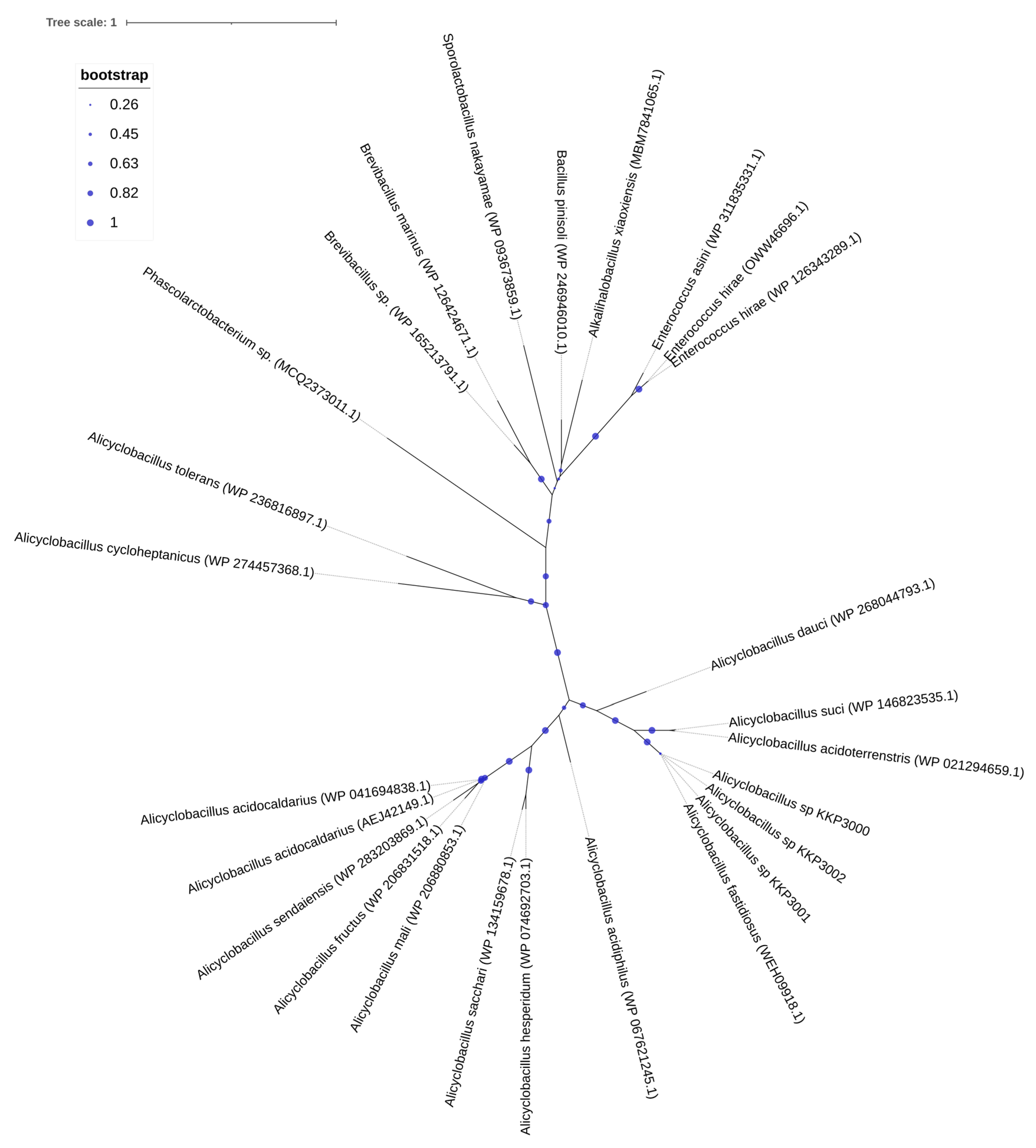
One CRISPR array was identified in each of the strains: KKP 3000, KKP 3001, and KKP 3002 (Table 2, Figure 1). Concerning transposable elements, 52 IS were identified in the genome of KKP 3000 and 14 in the genome of the two other strains (Table S1). Furthermore, 5 incomplete prophages were annotated for KKP 3000 and 7 for KKP 3001 and KKP 3002 (Table S2). AlienHunter was used to pinpoint regions containing mobile elements. To this end, 121, 97, and 98 regions were identified in the genome of KKP 3000, KKP 3001, and KKP 3002, respectively. Accordingly, 76 elements related to mobile element transfer, replication, and integration were identified by mobileOG in the genome of KKP 3000 and 92 in the chromosome of KKP 3001 and of KKP 3002 (Tables S3–S5). Five strict hits (<70% identity) for transferable antibiotic resistance genes were found in the genome of the strains, as shown in Table S6. Specifically, all three strains code for qacG, FosBx1, VanT, and VanW, which may confer resistance to disinfectant agents and antiseptics, as well as glycopeptides and Fosfomycin (Table S6). QacG protein acts as a proton-dependent efflux, and FosBx1 is an Mn+ dependent fosfomycin thiol transferase. The VanT is a membrane-bound serine racemase that converts L-Ser to D-Ser, resulting in D-Ser being used for peptidoglycan synthesis. The VanW protein is possibly involved in antibiotic target alteration, but its mode of action is unknown. In all three analyzed strains downstream of the vanT gene, are three genes encoding alanine dehydrogenase, dipeptidase, and permease, which strongly resemble parts of van resistance operons found in other bacteria [35]. The CARD database confirmed the alanine dehydrogenase gene as a distant analog of VanH in the vanF cluster; the dipeptidase gene found downstream vanT gene in analyzed strains does not show any resemblance to known vanX genes that encode dipeptidase in vancomycin resistance operons. Additionally, the permease gene mentioned above does not show any resemblance to any known van genes. The region encoding part of the putative van operon is limited by IS21 upstream and by IS110 downstream and is not functional. Downstream IS110, there are two genes, a sensor histidine kinase and an OmpR superfamily response regulator transcription factor, which functionally resemble vanS and vanR genes, respectively. At the protein sequence level, the response regulator shows significant similarity to VanR and the kinase to VanS in various clusters. The same two genes were identified in direct proximity to the putative vanW gene. It is possible the described genes were parts of the functional van operon, which was transferred and disrupted by transposition events. Evidence for the acquisition of these genes through HGT were provided by AlienHunter. Of note, the same HGT region contains the gene fosBx1. According to CARD, this gene is chromosomally encoded in Bacillus spp. and Staphylococcus spp., while mobile fosfomycin resistance genes have been identified in the genomes of clinically relevant Enterobacteriaceae, including members of the Escherichia and Salmonella species [36]. Furthermore, genes coding for ribosomal RNA small subunit methyltransferase A (RsmA) that could confer resistance to erythromycin were identified in the genome of the three strains (KFAPOJEI_01910, PKEAFELD_04479, PLKKBLCC_04475), that were not correlated with mobile or transposable elements. To further investigate the origins of these genes, an unrooted tree of homologous proteins was constructed. As shown in Figure 2, proteins encoded by Alicyclobacillus spp. are clustered together possibly indicating that genes are intrinsic and not acquired through HGT, or that they were acquired through a common ancestor in the past. Gene rsmA is homologous to ermC encoded by Bacillus spp. The Erm family of adenine-N(6) methyltransferases confer resistance to macrolide–lincosamide–streptogramin B antibiotics via alteration of their target, as they are responsible for the demethylation of the adenine residue at position 2085 in 23S rRNA interfering with the antibiotic binding [37].
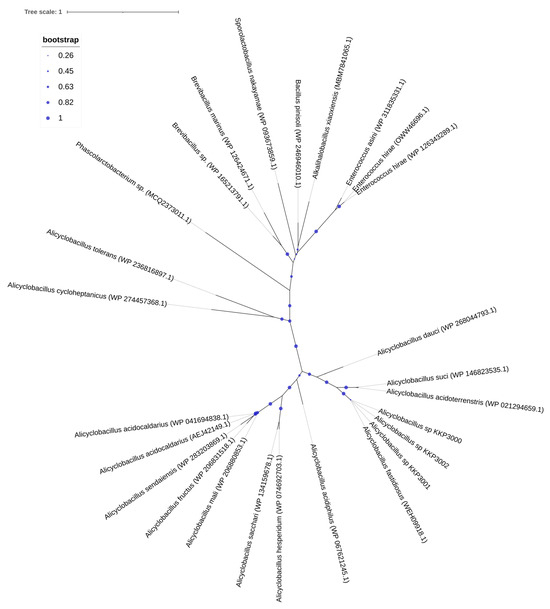
Figure 2.
An unrooted tree of homologous RsmA proteins identified and constructed using RAxML and visualized on the iTOL server.
2.3. Functional Annotation
The annotated proteins of the three strains were categorized into 19 COGs. As shown in Table 3, the most represented COG in all three strains is amino acid metabolism and transport (class E), followed by transcription (class K) and carbohydrate metabolism and transport (class G).
Table 3.
Categorization of genes contained in the genomes of the three strains into clusters of orthologous groups.
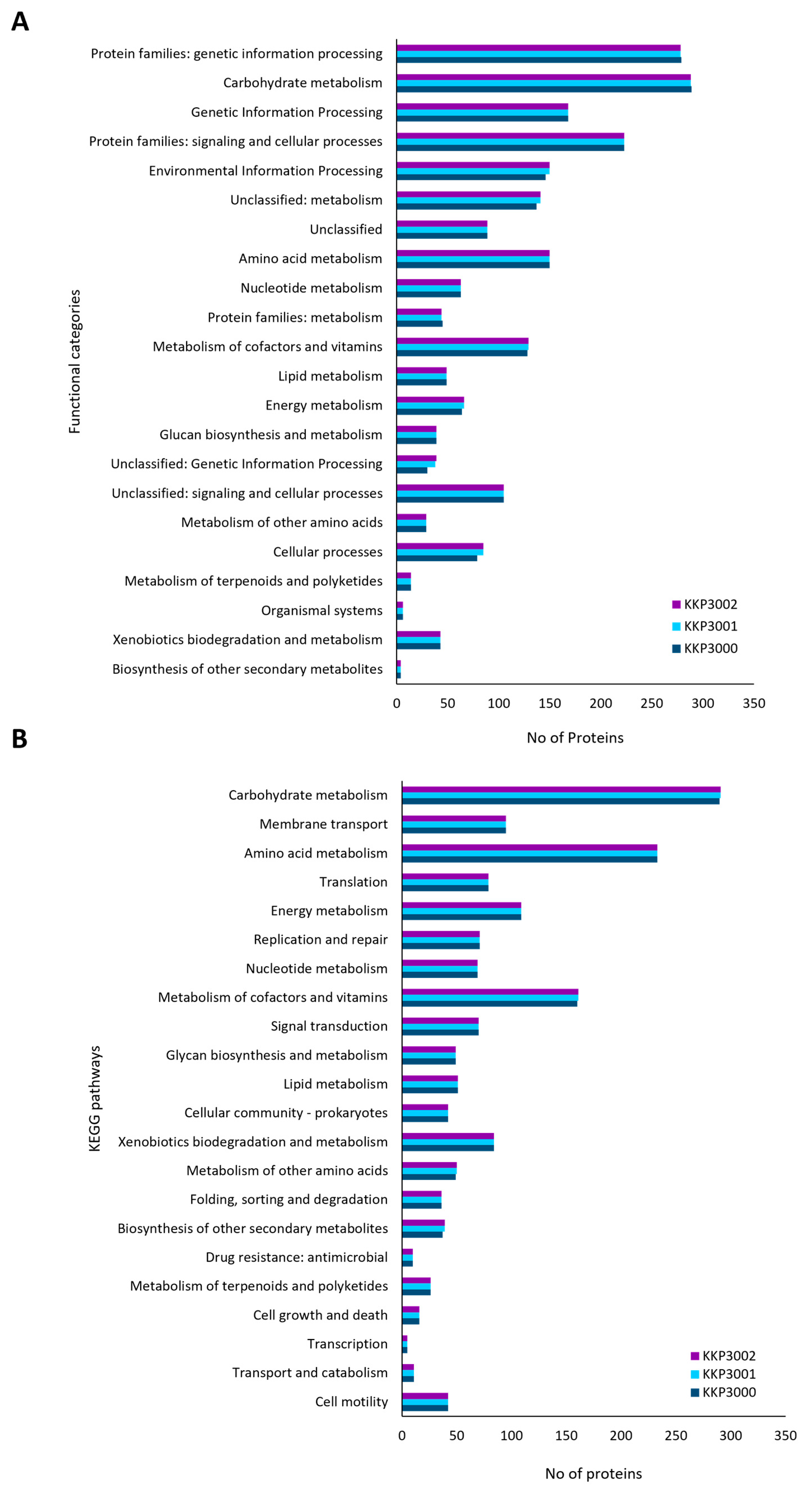
Accordingly, annotated proteins were categorized into KEGG functional categories and pathways (Figure 3). The most represented category in all three strains is carbohydrate metabolism, followed by amino acid metabolism and genetic information processing. No significant differences in the distribution of proteins into these categories were noted for the three novel strains.
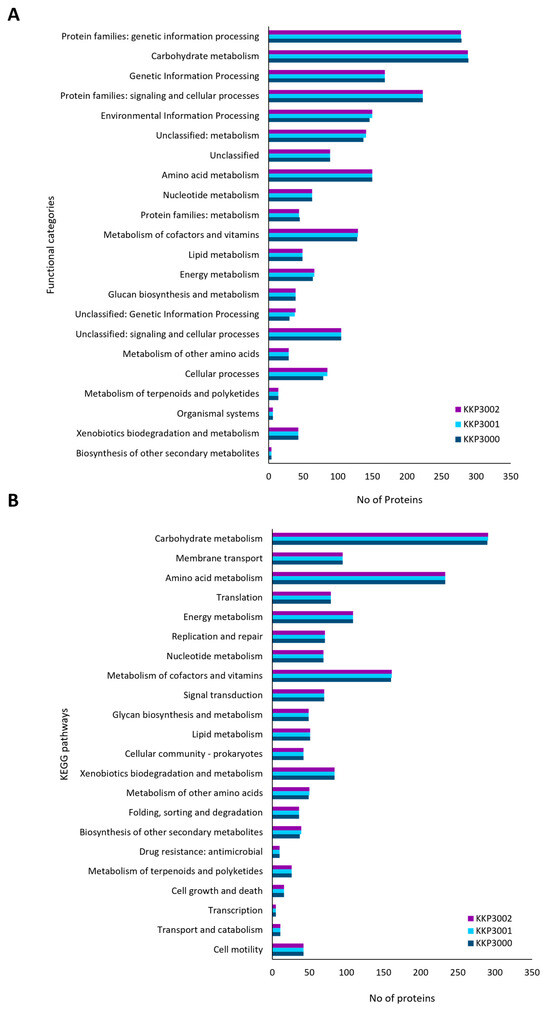
Figure 3.
Classification of proteins encoded by KKP 3000, KKP 3001, and KKP 3002 into (A) KEGG functional categories and (B) KEGG pathways.
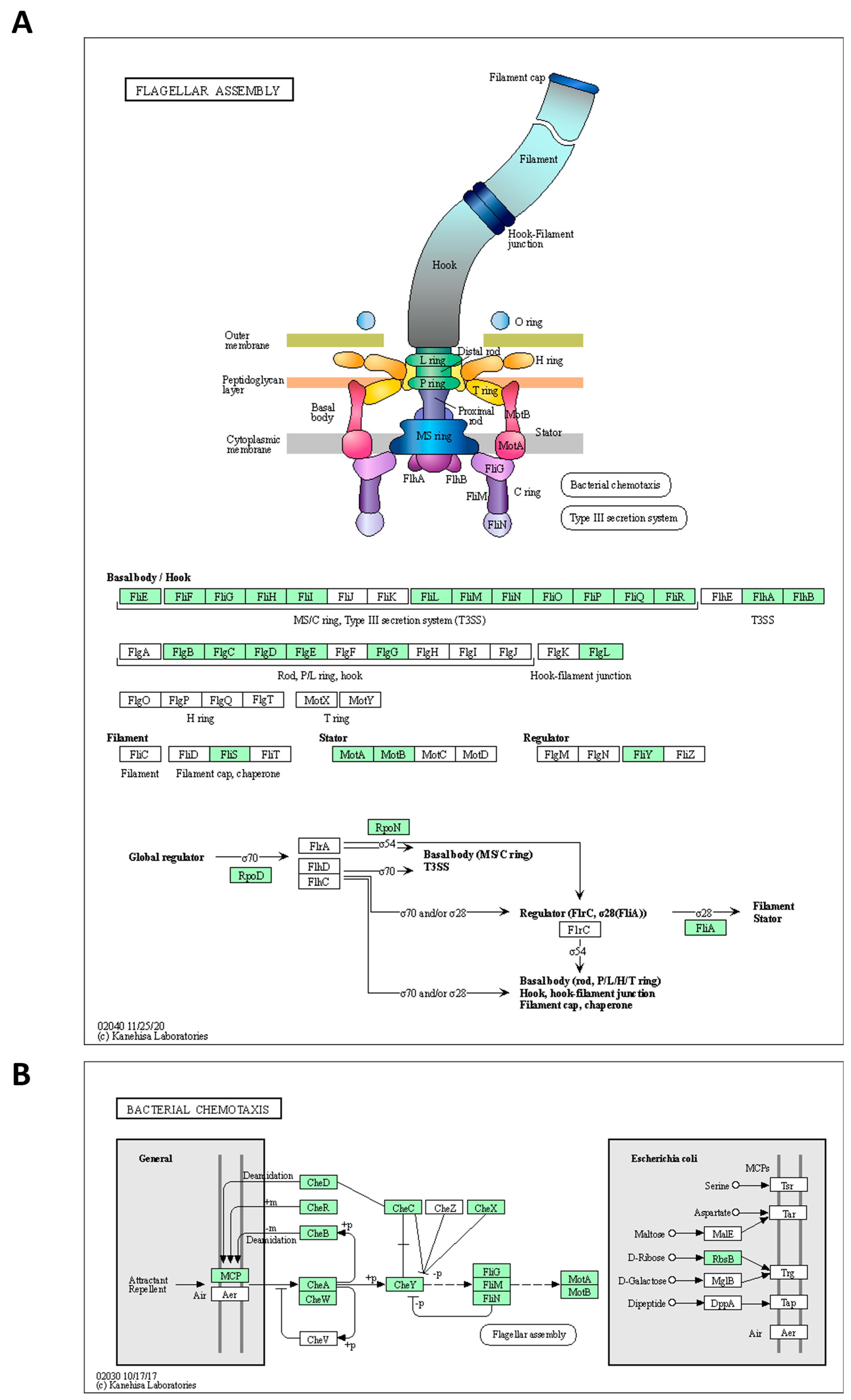
The genome of the strains was searched for genes involved in spore formation, germination, flagellar assembly, and chemotaxis using annotation algorithms and KEGG modules. The annotated genes are comprehensibly presented in Table 4. In greater detail, 27 genes involved in various processes of sporulation and germination were annotated in the genome of the Alicyclobacillus KKP 3000 and KKP 3001 strains. Of note, KKP 3002 does not code for germination protease gpr (Table 4). Furthermore, 41 genes related to flagellar assembly were identified. However, as illustrated by KEGG Mapper (Figure 4), the strains do not code for proteins involved in the formation of the H and T ring, while also missing some important regulatory proteins. On the other hand, the strains code for a full module for bacterial chemotaxis. In this context, flagellar production and swarming behavior were previously recorded for A. acidoterrestris and was dependent on pH conditions. Of note, these phenotypes were abolished in low pH, and cells were organized into biofilms to maximize survival [38]. For bacteria, an important adaptive element is the ability to be motile. According to Shemesh et al. [37] bacteria in heterogeneous environments can develop antibiotic resistance faster than bacteria living in homogeneous environments. Since the Alicyclobacillus spp. is a species highly correlated with food, it is presumed that motility can facilitate spreading onto surfaces to make nutrients more accessible. Thus, future studies will focus on the investigation of flagellar production and biofilm formation capacity to decipher the ability of the three strains to withstand unfavorable environmental conditions.
Table 4.
Annotation of genes in the genomes of KPP 3000, KKP 3001, and KKP 3002 involved in sporulation, germination, flagellar assembly, and chemotaxis.
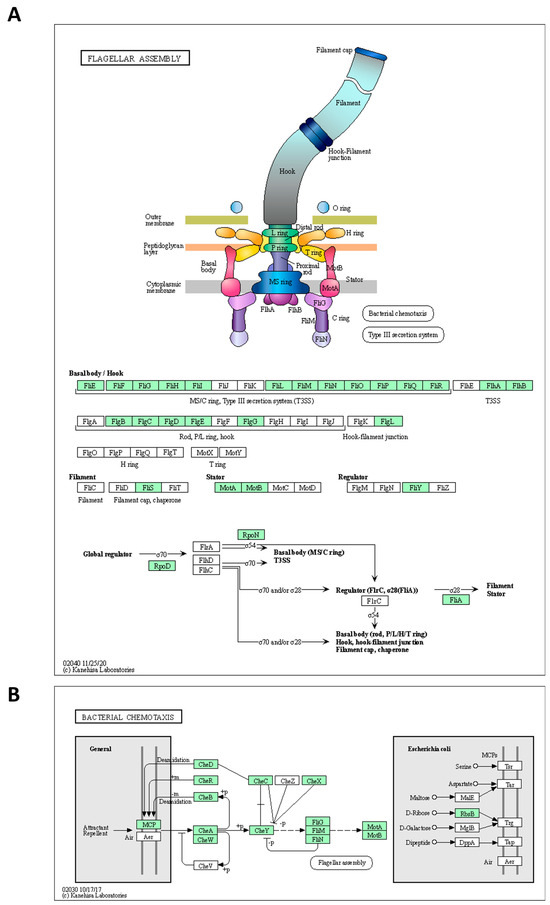
Figure 4.
Alicyclobacillus strains KKP 3000, KKP 3001, and KKP 3002 code for proteins involved in (A) flagellar assembly and (B) chemotaxis. The maps were produced using KEGG Mapper. The proteins encoded by the strains are indicated in green.
2.4. Vdc Operons
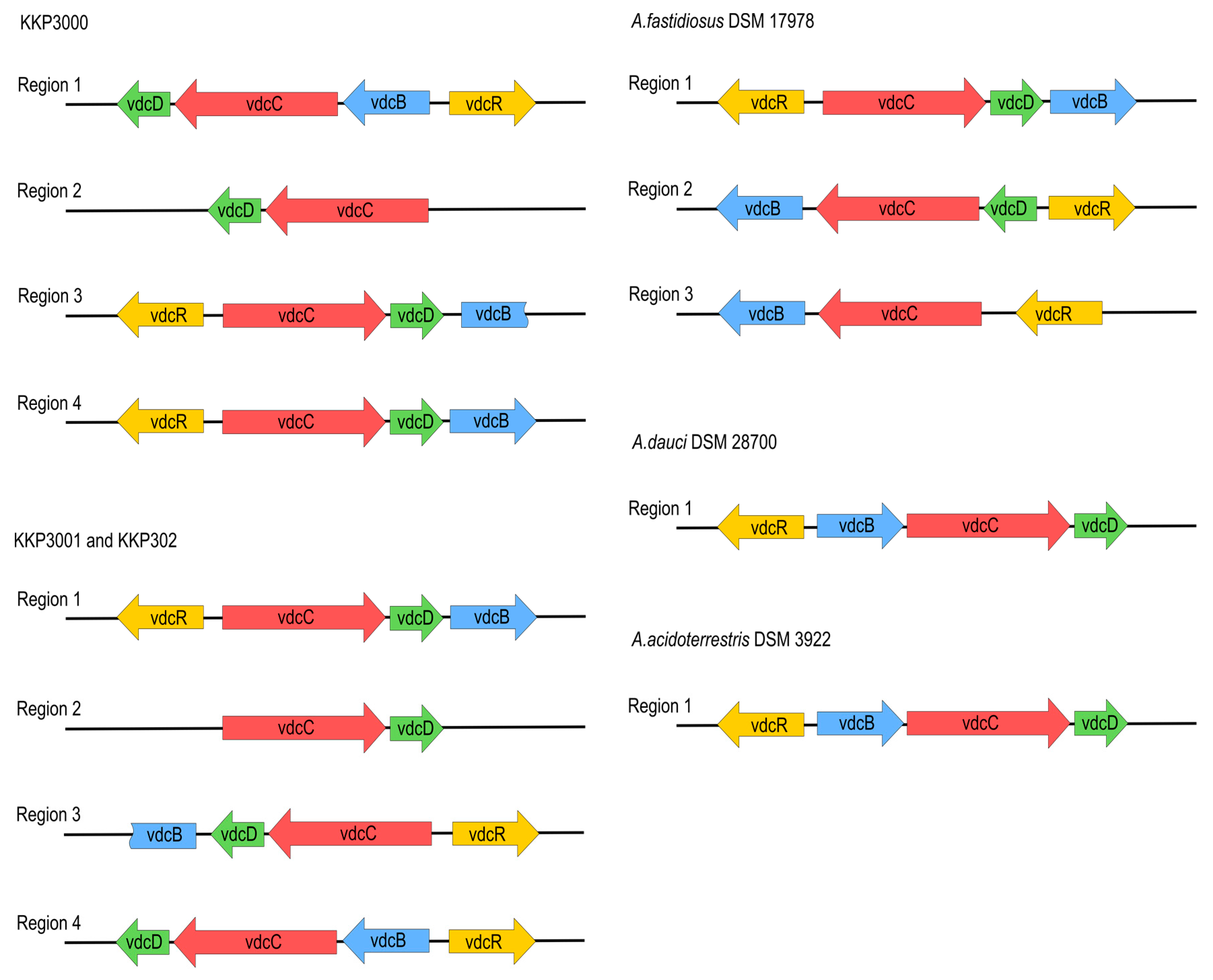
All analyzed Alicyclobacillus strains, KKP 3000, KKP 3001, and KKP 3002 have the ability to produce guaiacol [11]. To this end, proteins associated with the guaiacol production from vanillin were annotated in the chromosomes of the three strains (Table S7). The metabolic pathway for the production of guaiacol from vanillin involves two consecutive enzymatic reactions; the transformation of vanillin to vanillic acid performed by vanillin dehydrogenase, and of vanillic acid to guaiacol by vanillic acid decarboxylase [39]. Local blastp was performed to determine the presence of the genes involved in these enzymatic reactions. All three strains possess the gene coding for benzaldehyde dehydrogenase, the enzyme involved in the first step of the transformation (44% sequence identity and 66% positive substitutions). Accordingly, the strains code for phenolic acid decarboxylase, which is involved in the last step of guaiacol production, with higher sequence identity (78%), and 91% positive substitutions (Table S7). The nucleotide sequences of known A. acidoterrestris vdc clusters [11] were compared to genomic sequences of strains, KKP 3000, KKP 3001, and KKP 3002. A. acidoterrestris is linked to off-odor production in fruit juices, mainly attributed to guaiacol production in the beverage matrix [39]. The complete vdc operon consists of three structural genes, vdcB, vdcC, and vdcD, encoding subunits of non-oxidative vanillic acid decarboxylase Vdc, and a regulator gene vdcR, which is located upstream of the structural genes and orientated in the opposite direction [40]. The four regions in each of their genomes related to guaiacol production were found. However, three of them are incomplete or rearranged, probably due to transpositional events. The location and composition of vdc regions in all novel strains are similar, especially between KKP 3001 and KKP 3002. To obtain a more complete picture of the vdc regions of Alicyclobacillus, we also searched for similar regions in complete genome sequences known as guaiacol producers, A. acidoterrestris DSM 3922 and A. dauci DSM 28700, as well as non-guaiacol producing A. fastidiosus DSM 17978 and A. acidocaldarius DSM 446. Both guaiacol-producing species carry one complete vdc operon in their genomes. We have not found sequences of significant similarity to the vdc region in the A. acidocaldarius DSM446 genome. A. fastidiosus DSM 17978 carries three vdc-related regions, although one of them is incomplete, and two others are arranged in a different order than in guaiacol-producing species, which probably is the cause for this strain of A. fastidiosus not producing guaiacol. The arrangement of vdc-related regions in all analyzed species is shown in Figure 5.
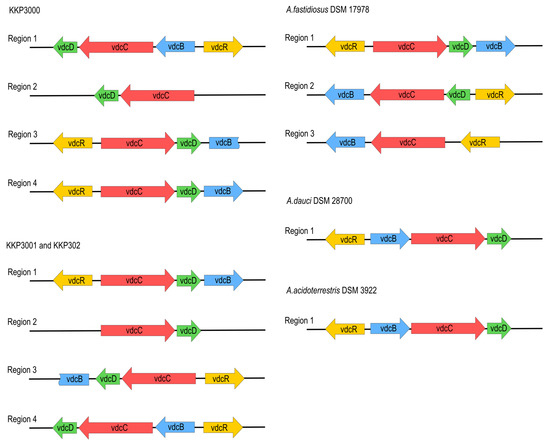
Figure 5.
Arrangement of vdc operons in novel strains, compared to A. fastidiosus DSM17978, A. acidoterrestris DSM3922, and A.dauci DSM28700.
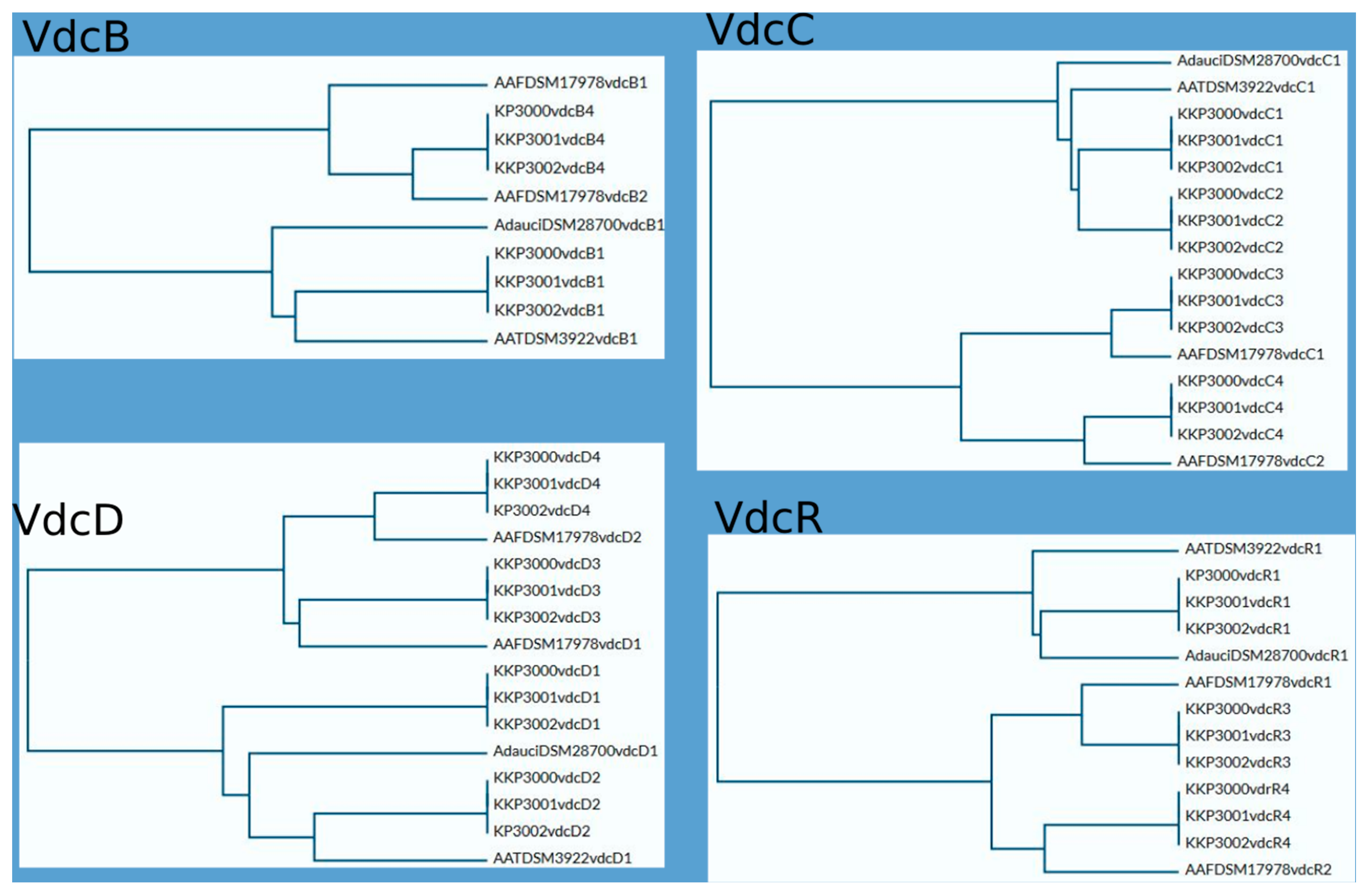
As mentioned above, all novel species share significant similarities regarding locations and composition of vdc regions in their genomes. This similarity is particularly evident between KKP 3001 and KKP 3002. Sequence comparison of VdcB, VdcC, VdcD, and VdcR proteins show a higher level of similarity between respective Vdc proteins of particular regions of KKP 3000, KKP 3001, and KKP 3002 than intragenomically, which suggests that divergence of those strains may have occurred later than the duplication of the vdc genes. Phylograms based on Vdc proteins sequence alignment are shown in Figure 6.
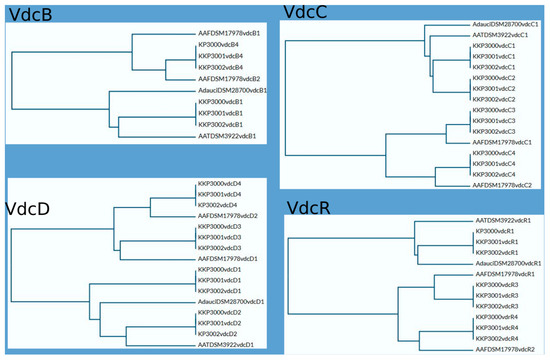
Figure 6.
Comparison of Vdc proteins. Phylograms were prepared using UniProt software https://www.uniprot.org/ (accessed on 24 April 2024).
2.5. ANI, and Phylogenomic Analysis
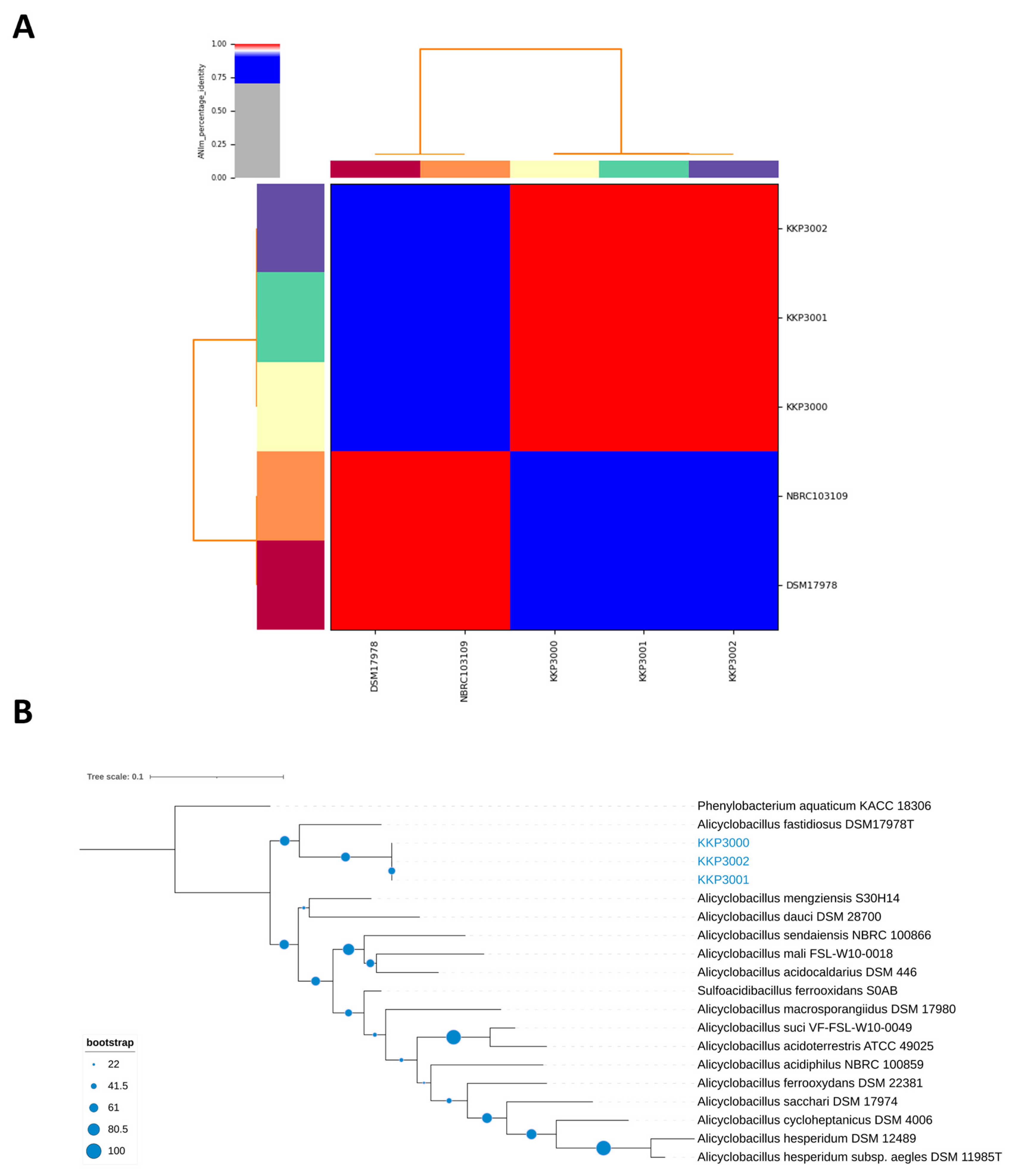
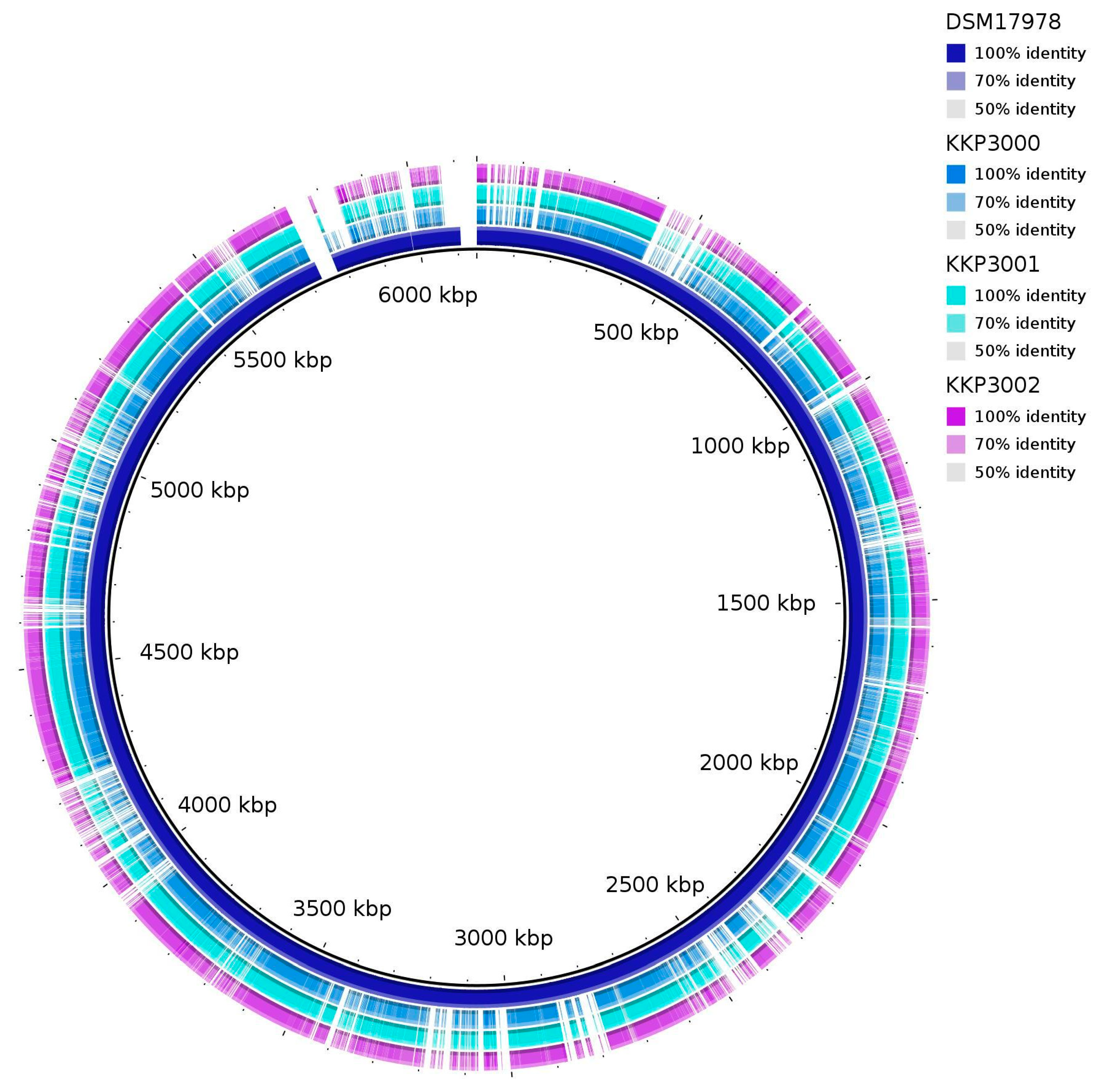
ANI (Average Nucleotide Identity) analysis between members of the Alicyclobacillus spp. and the three novel strains showed that strains KKP 3000, KKP 3001, and KKP 3002 share very high genome similarity (>99%). However, the three strains do not present high ANI with other known Alicyclobacillus species. Moreover, the phylogenomic analysis exhibited that the three strains cluster together in a distinct phylogenetic clade, that is closely related to A. fastidiosus (Figure 7). The ANI between the three novel strains and members of the A. fastiodiosus species was recorded to be 88%, lower than the species cut-off (>96%) (Figure 7). Furthermore, a comparative genetic analysis between the three strains and the type strain A. fastidiosus DSM 17978 with BRIG showed distinct genomic differences (Figure 8). Indeed, several regions in the genome of strain DSM 17978 show very low sequence identity (<50%) to the genomic regions of the three novel strains. Based on these findings, further phylogenomic analysis will be pursued to elucidate the taxonomy of the three novel strains.
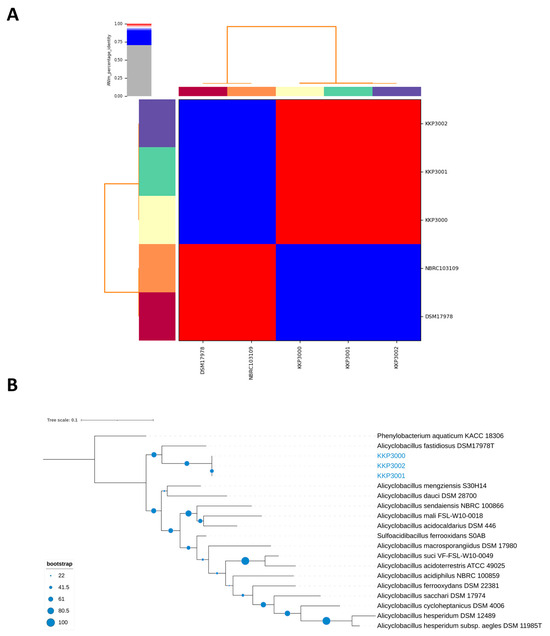
Figure 7.
(A) ANI of KKP 3000, KKP 3001, and KKP 3002 with members of the A. fastidiosus species (i.e., DSM 17978 and NBRC103109). (B) The phylogenomic tree containing the three novel strains and members of the Alicyclobacillus genus. Phenylobacterium aquaticum KACC 18306 was used as an outgroup. The tree was calculated on the TYGS server and visualized using iTOL.
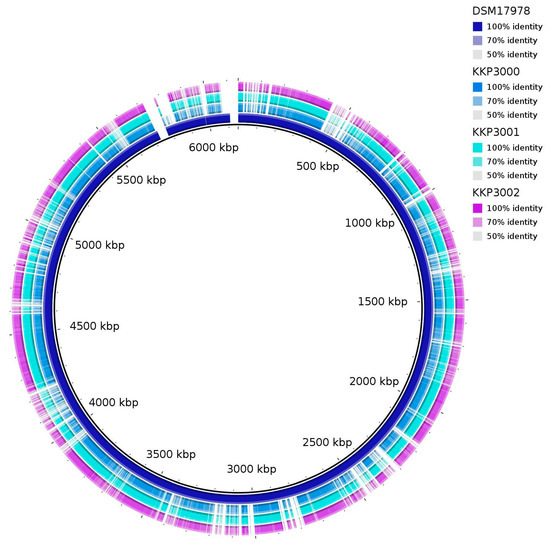
Figure 8.
BRIG diagram showing homologous chromosome segments of the three novel strains (KKP 3000, KKP 3001, KKP 3002) to the A. fastidiosus type strain DSM 17978.
3. Materials and Methods
3.1. Bacterial Strains and Growth Conditions
The Alicyclobacillus strains used in this study were isolated from Polish pear orchards and pear fruits, as described by Połaska et al. [11] Briefly, the samples were processed according to The International Fruit and Vegetable Juice Association (IFU) Method No. 12, including initial heat shock (80 °C, 10′), enrichment in BAT broth (Merck, Darmstadt, Germany) for 5 days (45 °C), and transferring 10 μL of the enriched culture on the BAT agar (Merck, Darmstadt, Germany). After 3 days of incubation, the obtained colonies were subjected to additional tests, to confirm their affiliation to taint-producing Alicyclobacillus group: incubation at 65 °C, guaiacol detection test [41], and erythritol utilization test [42].
All strains, based on the 16S rDNA identification, were classified as Alicyclobacillus fastidiosus and deposited in the Culture Collection of Industrial Microorganisms—Microbiological Resource Center (IAFB, Warsaw, Poland), under collection number KKP 3000, KKP 3001, and KKP 3002. The 16S rDNA GenBank accession numbers are available for all three strains, KY088044, KY088045, and KY088046.
3.2. Genomic DNA Isolation and Sequencing
The total genomic DNA extraction for strain KKP 3000 was performed following the manufacturer protocol with the DNeasy PowerFood Microbial Kit (Qiagen, GmbH, Hilden, Germany), as described by Bucka-Kolendo et al., 2023, and Kiousi et al., 2023 [43,44]. Briefly, the purity of DNA was verified with a Nanodrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Watertown, MA, USA), and the concentration was measured with a Qubit 4.0 Fluorometer (Qubit dsDNA BR Assay Kit, Invitrogen, Carlsbad, CA, USA). Library preparation and sequencing were performed with the Illumina MiSeq NGS platform (Illumina, San Diego, CA, USA), with an Illumina DNA Prep kit (number #1000000025416v09), the 2 × 151 bp paired-end MiSeq protocol, the 343 reagent v3 (600-cycle) kit.
For KKP 3001 and KKP 3002, the whole-genome sequencing was commissioned to sequence by GenXone SA (Złotniki, Poland). Libraries were prepared with Rapid Barcoding kit reagents (Oxford Nanopore Technologies, Oxford, UK), with assured 50X genome coverage. WGS was performed in the nanopore technology on the GridlON X5 sequencing device (Oxford Nanopore Technologies, Oxford, UK), with MinKnow v22.10.5 control, as described by Wójcicki et al., 2023 [45] base calling and barcode demultiplexing a Guppy v6.3.8 and Gruppy Barcoder v6.3.8 (Oxford Nanopore Technologies, Oxford, UK) were used. De novo assembly of genomes and annotations were made with Flye v2.8.1. software and Prokka server, respectively.
3.3. Antibiotic Resistance
Nine antibiotics were utilized for Minimum Inhibitory Concentration (MIC) evaluations. The antibiotic resistance profiles of Alicyclobacillus spp. strains were assessed through the utilization of the E-test (bioMérieux, Craponne, France) on BAT agar (Merck, Darmstadt, Germany), following the manufacturer’s instructions. E-test strips encompassing a gradient of antibiotics (with a concentration range of 0.016–256 µg/mL) were used and included the following antibiotics: chloramphenicol (catalog no. 412309), clindamycin (catalog no. 412315), erythromycin (catalog no. 412334), gentamicin (catalog no. 412368), kanamycin (catalog no. 412382), streptomycin (catalog no. 526800), and tetracycline (catalog no. 412471), serving as inhibitors of protein synthesis. Additionally, vancomycin (catalog no. 412488) and ampicillin (catalog no. 412253) were used as cell wall synthesis inhibitors.
It is noteworthy that all selected antibiotics are included in the list recommended by the European Food Safety Authority (EFSA) Panel on Additives and Products or Substances used in Animal Feed (FEEDAP) [46]. This list specifies antibiotics against where microbial strains are intended for human use as live culture or probiotics and which animal feed additives should be tested.
The resistance profile of the examined Alicyclobacillus strains was established in strict accordance with the manufacturer’s guidelines. After cultivating the strains on BAT agar for 24 h, they underwent centrifugation, followed by two washes with phosphate-buffered saline (PBS). The resulting bacterial pellet was resuspended in PBS buffer with a pH of 7.2, aiming for an optical density of 0.5 McFarland units. Using a swab, the bacterial cultures were evenly applied to BAT agar, covering the entire petri dish. Subsequently, E-tests containing the appropriate antibiotics were placed on the agar dishes inoculated with bacteria. The plates were subsequently incubated under aerobic conditions at 45 °C for 3 days. The MIC results were compared with the reference strain A. fastidiosus DSM 17978.
The experiment was carried out in three replicates.
3.4. Genome Annotation and Analysis
Genome annotation of the three strains (KKP 3000, KKP 3001, and KKP 3002) was performed with Prokka (version 1.14.5) [47]. EggNOG-mapper (version 2.1.9) and BlastKOALA (version 3) were used for functional annotation of the genomes. Plasmid sequences in the WGS of KKP3000 were investigated with PlasmidFinder [48]. Insertion elements, prophages, and CRISPR arrays were annotated with ISFinder (e-value cut-off: 0.01) [49], PHAge Search Tool Enhanced Release (PHASTER) for prophage regions [50], and CRISPRDetect (version 2.4), and PILER-CR [51], respectively. Acquired antimicrobial resistance genes were identified using Resistance Gene Identifier (RGI; version 5.2.0) and ResFider [51]. Genes coding for virulence factors were annotated with VirulenceFinder 2.0 and PathogenFinder 2.0 [52]. Genome maps were constructed with Proksee, and chromosomes were annotated for antimicrobial resistance genes, mobile elements, and horizontal gene transfer regions with CARD-RGI (version 1.2.0), mobileOG-db (version 1.1.2) and Alien Hunter (version 1.1.0) [53]. The annotated genome of the three strains was manually searched for genes involved in guaiacol production and resistance to erythromycin. To this end, Blastp+ was utilized for the detection of genes encoded by the three novel strains that are homologous to ribosomal RNA small subunit methyltransferase A (RsmA) (WP_021294659.1), benzaldehyde dehydrogenase YfmT (WP_003244104.1), and phenolic acid decarboxylase (WP_003246683.1). Homologous proteins to RsmA were identified using Blastp. The top 24 entries and proteins encoded by the three strains were aligned with ClustalW [54] and produce an unrooted phylogenetic tree was constructed with RAxML. The tree was visualized on the iTol server [55].
3.5. Phylogenomic Analysis
The genomic sequences of A. fastidiosus DSM 17978 were obtained from the NCBI Assembly database. The Average Nucleotide Identity (ANI) between the three novel strains and the two deposited genomes was calculated with Pyani (version 0.2.10). BLAST Ring Image Generator (BRIG) was used to determine the homology of chromosomal regions of the three novel strains with that of A. fastidiosus DSM 17978 (type strain). Phylogenomic trees were constructed based on the whole-genome sequences of the three strains on the TYGS server [56]. Trees were visualized with iTOL [55]. Phenylobacterium aquaticum KACC 18306 was used as an outgroup.
4. Conclusions
This study determined the antibiotic susceptibility of three novel Alicyclobacillus spp. strains isolated from orchard soil and pear both in vitro and in silico. The strains Alicyclobacillus spp. KKP 3000, KKP 3001, and KKP 3002 were susceptible to most of the studied antibiotics, excluding erythromycin, to which high resistance was observed. The erythromycin resistance was attributed to the presence of the rsmA gene in all three genomes; however, no correlation between antibiotic resistance genes and mobile or transposable elements was found. Furthermore, we identified and characterized the arrangement of the vdc operon, which may be involved in the guaiacol production. By analyzing the structure of the operon in the three novel strains and other guaiacol producers, we arrived at the hypothesis that the divergence among the strains may be a result of duplication events that occurred later in time. However, this does not affect their guaiacol production capability. Notably, operons for flagellar assembly and chemotaxis were located, which may be part of the adaptational mechanisms of these strains that will be further explored. Finally, the strains present low Average Nucleotide Identity (ANI) with other members of the Alicyclobacillus genus, exhibiting the highest identity to A. fastidiosus strains, which was still lower than the species cut-off. Future studies will aim to decipher these novel strains’ evolutionary history and taxonomy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25158144/s1. References [57,58] are cited in Supplementary Materials.
Author Contributions
Conceptualization, J.B.-K. and B.S.; methodology, J.B.-K.; software, D.E.K., D.M.K., P.M. and A.D.; validation, J.B.-K. and D.E.K.; formal analysis, J.B.-K., D.E.K., A.D. and A.M.-S.; investigation, J.B.-K.; resources, J.B.-K. and A.D; data curation, J.B.-K., D.E.K., D.M.K., P.M. and A.D.; writing—original draft preparation, J.B.-K., D.E.K., A.D. and A.M.-S.; writing—review and editing, J.B.-K., D.E.K., A.G. and A.D.; visualization, J.B.-K., D.E.K., D.M.K., P.M. and A.D.; supervision, A.G and B.S.; project administration, B.S.; funding acquisition, B.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The Alicyclobacillus strain KKP 3000, KKP 3001, and KKP 3002 genome sequences have been deposited at DDBJ/ENA/GenBank under the accessions SAMN41503782, SAMN41503783, and SAMN41503784, respectively.
Acknowledgments
The authors wish to acknowledge Marzena Połaska for her support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dekowska, A.; Niezgoda, J.; Sokołowska, B.S. Genetic Heterogeneity of Alicyclobacillus Strains Revealed by RFLP Analysis of VDC Region and RpoB Gene. Biomed. Res. Int. 2018, 2018, 9608756. [Google Scholar] [CrossRef]
- Aulitto, M.; Gallo, G.; Puopolo, R.; Mormone, A.; Limauro, D.; Contursi, P.; Piochi, M.; Bartolucci, S.; Fiorentino, G. Genomic Insight of Alicyclobacillus Mali FL18 Isolated From an Arsenic-Rich Hot Spring. Front. Microbiol. 2021, 12, 639697. [Google Scholar] [CrossRef]
- Leonardo, I.C.; Crespo, M.T.B.; Gaspar, F.B. Unveiling the Complete Genome Sequence of Alicyclobacillus Acidoterrestris DSM 3922T, a Taint-Producing Strain. G3 Genes Genomes Genet. 2022, 12, jkac225. [Google Scholar] [CrossRef]
- Sourri, P.; Tassou, C.C.; Nychas, G.J.E.; Panagou, E.Z. Fruit Juice Spoilage by Alicyclobacillus: Detection and Control Methods—A Comprehensive Review. Foods 2022, 11, 747. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Y.F.; Song, J.L.; Huang, Z.S.; Wang, B.J.; Liu, S.J.; Jiang, C.Y. Alicyclobacillus Fodiniaquatilis Sp. Nov, Isolated from Acid Mine Water. Int. J. Syst. Evol. Microbiol. 2015, 65, 4915–4920. [Google Scholar] [CrossRef]
- Parte, A.C.; Carbasse, J.S.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic Names with Standing in Nomenclature (LPSN) Moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Shymialevich, D.; Wójcicki, M.; Świder, O.; Średnicka, P.; Sokołowska, B. Characterization and Genome Study of a Newly Isolated Temperate Phage Belonging to a New Genus Targeting Alicyclobacillus Acidoterrestris. Genes 2023, 14, 1303. [Google Scholar] [CrossRef]
- Smit, Y.; Cameron, M.; Venter, P.; Witthuhn, R.C. Alicyclobacillus Spoilage and Isolation–A Review. Food Microbiol. 2011, 28, 331–349. [Google Scholar]
- Porębska, I.; Sokołowska, B.; Skąpska, S.; Rzoska, S.J. Treatment with High Hydrostatic Pressure and Supercritical Carbon Dioxide to Control Alicyclobacillus Acidoterrestris Spores in Apple Juice. Food Control 2017, 73, 24–30. [Google Scholar] [CrossRef]
- Sourri, P.; Dekowska, A.; Joanna Bucka-Kolendo, J.; Nychas, G.-J.; Tassou, C.; Doulgeraki, A.I. Identification of Guaiacol Producing Alicyclobacillus Recovered from Commercial Orange Juices Distributed in Greek Markets. Ital. J. Food Sci. 2024, 36, 205–215. [Google Scholar] [CrossRef]
- Połaska, M.; Dekowska, A.; Sokołowska, B. Isolation and Identification of Guaiacol-Producing Alicyclobacillus Fastidiosus Strains from Orchards in Poland. Acta Biochim. Pol. 2021, 68, 301–307. [Google Scholar] [CrossRef]
- Van Luong, T.S.; Moir, C.; Bowman, J.P.; Chandry, P.S. Heat Resistance and Genomics of Spoilage Alicyclobacillus spp. Isolated from Fruit Juice and Fruit-Based Beverages. Food Microbiol. 2021, 94, 103662. [Google Scholar] [CrossRef]
- Dekowska, A. Zastosowanie Metod Biologii Molekularnej W Diagnostyce Bakterii Z Rodzaju Alicyclobacillus. 2011, Volume 66. Available online: https://www.ibprs.pl/wp-content/uploads/2018/08/PNiTPRS-2011-2-rozdzia-iii.pdf (accessed on 21 March 2024).
- Goto, K.; Mochida, K.; Kato, Y.; Asahara, M.; Fujita, R.; An, S.Y.; Kasai, H.; Yokota, A. Proposal of Six Species of Moderately Thermophilic, Acidophilic, Endospore-Forming Bacteria: Alicyclobacillus contaminans sp. Nov., Alicyclobacillus fastidiosus sp. Nov., Alicyclobacillus kakegawensis sp. Nov., Alicyclobacillus macrosporangiidus sp. Nov., Alicyclobacillus sacchari sp. Nov. and Alicyclobacillus shizuokensis sp. Nov. Int. J. Syst. Evol. Microbiol. 2007, 57, 1276–1285. [Google Scholar] [CrossRef]
- Tianli, Y.; Jiangbo, Z.; Yahong, Y. Spoilage by Alicyclobacillus Bacteria in Juice and Beverage Products: Chemical, Physical, and Combined Control Methods. Compr. Rev. Food Sci. Food Saf. 2014, 13, 771–797. [Google Scholar] [CrossRef]
- Osopale, B.A.; Witthuhn, C.R.; Albertyn, J.; Oguntoyinbo, F.A. Inhibitory Spectrum of Diverse Guaiacol-Producing Alicyclobacillus Acidoterrestris by Poly Dimethyl Ammonium Chloride Disinfectant. LWT 2017, 84, 241–247. [Google Scholar] [CrossRef]
- EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP). Guidance on the Assessment of Bacterial Susceptibility to Antimicrobials of Human and Veterinary Importance. EFSA J. 2012, 10, 2740. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Opinion of the Scientific Panel on Additives and Products or Substances Used in Animal Feed (FEEDAP) on the Updating of the Criteria Used in the Assessment of Bacteria for Resistance to Antibiotics of Human or Veterinary Importance. EFSA J. 2005, 3, 223. [Google Scholar] [CrossRef]
- Adimpong, D.B.; Sørensen, K.I.; Thorsen, L.; Stuer-Lauridsen, B.; Abdelgadir, W.S.; Nielsen, D.S.; Derkx, P.M.F.; Jespersen, L. Antimicrobial Susceptibility of Bacillus Strains Isolated from Primary Starters for African Traditional Bread Production and Characterization of the Bacitracin Operon and Bacitracin Biosynthesis. Appl. Env. Microbiol. 2012, 78, 7903–7914. [Google Scholar] [CrossRef]
- Kashmir, S.V.S.; Hotchkiss, R.D. Evidence of Tandem Duplication of Genes in a Merodiploid Region of Pneumococcal Mutants Resistant to Sulfonamide. Genetics 1975, 81, 21–31. [Google Scholar] [CrossRef]
- Brochet, M.; Couvé, E.; Zouine, M.; Poyart, C.; Glaser, P. A Naturally Occurring Gene Amplification Leading to Sulfonamide and Trimethoprim Resistance in Streptococcus Agalactiae. J. Bacteriol. 2008, 190, 672–680. [Google Scholar] [CrossRef]
- Bertini, A.; Poirel, L.; Bernabeu, S.; Fortini, D.; Villa, L.; Nordmann, P.; Carattoli, A. Multicopy BlaOXA-58 Gene as a Source of High-Level Resistance to Carbapenems in Acinetobacter Baumannii. Antimicrob. Agents Chemother. 2007, 51, 2324–2328. [Google Scholar] [CrossRef]
- Okubo, T.; Ae, R.; Noda, J.; Iizuka, Y.; Usui, M.; Tamura, Y. Detection of the Sul2–StrA–StrB Gene Cluster in an Ice Core from Dome Fuji Station, East Antarctica. J. Glob. Antimicrob. Resist. 2019, 17, 72–78. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Chen, B.; Yuan, K.; Chen, X.; Yang, Y.; Zhang, T.; Wang, Y.; Luan, T.; Zou, S.; Li, X. Metagenomic Analysis Revealing Antibiotic Resistance Genes (ARGs) and Their Genetic Compartments in the Tibetan Environment. Env. Sci. Technol. 2016, 50, 6670–6679. [Google Scholar] [CrossRef]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the Antibiotic Resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef]
- Aminov, R.I.; Mackie, R.I. Evolution and Ecology of Antibiotic Resistance Genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef]
- Davies, J. Origins and Evolution of Antibiotic Resistance. Microbiologia 1996, 12, 9–16. [Google Scholar] [CrossRef]
- Hopwood, D.A. How Do Antibiotic-Producing Bacteria Ensure Their Self-Resistance before Antibiotic Biosynthesis Incapacitates Them? Mol. Microbiol. 2007, 63, 937–940. [Google Scholar] [CrossRef]
- Wright, G.D. The Antibiotic Resistome: The Nexus of Chemical and Genetic Diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Li, L.; Ashbolt, N.; Wang, X.; Cui, Y.; Zhu, X.; Xu, Y.; Yang, Y.; Mao, D.; Luo, Y. Arctic Antibiotic Resistance Gene Contamination, a Result of Anthropogenic Activities and Natural Origin. Sci. Total Environ. 2018, 621, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Piskovsky, V.; Oliveira, N.M. Bacterial Motility Can Govern the Dynamics of Antibiotic Resistance Evolution. Nat. Commun. 2023, 14, 5584. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.F.; Tien, Y.C.; Stedtfeld, R.D.; Topp, E. Impacts of Multi-Year Field Exposure of Agricultural Soil to Macrolide Antibiotics on the Abundance of Antibiotic Resistance Genes and Selected Mobile Genetic Elements. Sci. Total Environ. 2020, 727, 138520. [Google Scholar] [CrossRef] [PubMed]
- Georgakakos, C.B.; Martínez, C.E.; Helbling, D.E.; Walter, M.T. More Movement with Manure: Increased Mobility of Erythromycin through Agricultural Soil in the Presence of Manure. J. Water Health 2023, 21, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Lessardl, I.A.; Pratt, S.D.; Mccaffertyl, D.G.; Bussiere, D.E.; Hutchins, C.; Wanner, B.L.; Katz, L.; Walsh’, C.T. Homologs of the Vancomycin Resistance D-Ala-D-Ala Dipeptidase VanX in Streptomyces Toyocaensis, Escherichia Co/i and Synechocystis: Attributes of Catalytic Efficiency, Stereoselectivity and Regulation with Implications for Function. Chem. Biol. 1998, 5, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Zurfluh, K.; Treier, A.; Schmitt, K.; Stephan, R. Mobile Fosfomycin Resistance Genes in Enterobacteriaceae—An Increasing Threat. Microbiologyopen 2020, 9, e1135. [Google Scholar] [CrossRef] [PubMed]
- Maravić, G.; Bujnicki, J.M.; Feder, M.; Pongor, S.; Flögel, M. Alanine-Scanning Mutagenesis of the Predicted RRNA-Binding Domain of ErmC’ Redefines the Substrate-Binding Site and Suggests a Model for Protein-RNA Interactions. Nucleic Acids Res. 2003, 31, 4941–4949. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, M.; Pasvolsky, R.; Zakin, V. External PH Is a Cue for the Behavioral Switch That Determines Surface Motility and Biofilm Formation of Alicyclobacillus Acidoterrestris. J. Food Prot. 2014, 77, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Yuan, Y.; Wang, Z.; Guo, C.; Liu, B.; Liu, L.; Wang, Y.; Yue, T. Precursors and Metabolic Pathway for Guaiacol Production by Alicyclobacillus Acidoterrestris. Int. J. Food Microbiol. 2015, 214, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, Y.; Wang, Q.; Jia, H.; Yue, T.; Yuan, Y.; Gao, Z.; Cai, R. Integrated Analysis of Transcriptome and Proteome for Exploring the Mechanism of Guaiacol Production by Alicyclobacillus Acidoterrestris. Food Res. Int. 2021, 148, 110621. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Kuriyama, A.A. Acidoterrestris Rapid Detection Kit. Fruit Process. 2003, 13, 328–331. [Google Scholar]
- Baumgart, J. Handbook of Culture Media for Food Microbiology; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Bucka-Kolendo, J.; Kiousi, D.E.; Wojtczak, A.; Doulgeraki, A.I.; Galanis, A.; Sokołowska, B. Depiction of the In Vitro and Genomic Basis of Resistance to Hop and High Hydrostatic Pressure of Lactiplantibacillus Plantarum Isolated from Spoiled Beer. Genes 2023, 14, 1710. [Google Scholar] [CrossRef] [PubMed]
- Kiousi, D.E.; Bucka-Kolendo, J.; Wojtczak, A.; Sokołowska, B.; Doulgeraki, A.I.; Galanis, A. Genomic Analysis and In Vitro Investigation of the Hop Resistance Phenotype of Two Novel Loigolactobacillus Backii Strains, Isolated from Spoiled Beer. Microorganisms 2023, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Wójcicki, M.; Świder, O.; Średnicka, P.; Shymialevich, D.; Ilczuk, T.; Koperski, Ł.; Cieślak, H.; Sokołowska, B.; Juszczuk-Kubiak, E. Newly Isolated Virulent Salmophages for Biocontrol of Multidrug-Resistant Salmonella in Ready-to-Eat Plant-Based Food. Int. J. Mol. Sci. 2023, 24, 10134. [Google Scholar] [CrossRef] [PubMed]
- Rychen, G.; Aquilina, G.; Azimonti, G.; Bampidis, V.; de Lourdes Bastos, M.; Bories, G.; Chesson, A.; Cocconcelli, P.S.; Flachowsky, G.; Gropp, J.; et al. Guidance on the Characterisation of Microorganisms Used as Feed Additives or as Production Organisms. EFSA J. 2018, 16, 5206. [Google Scholar] [CrossRef]
- Seemann, T. Genome Analysis Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The Reference Centre for Bacterial Insertion Sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder--Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-Depth Characterization and Visualization of Bacterial Genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Schaffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).