
The Inferential Binding Sites of GCGR for Small Molecules Using Protein Dynamic Conformations and Crystal Structures

Abstract
:1. Introduction
2. Results
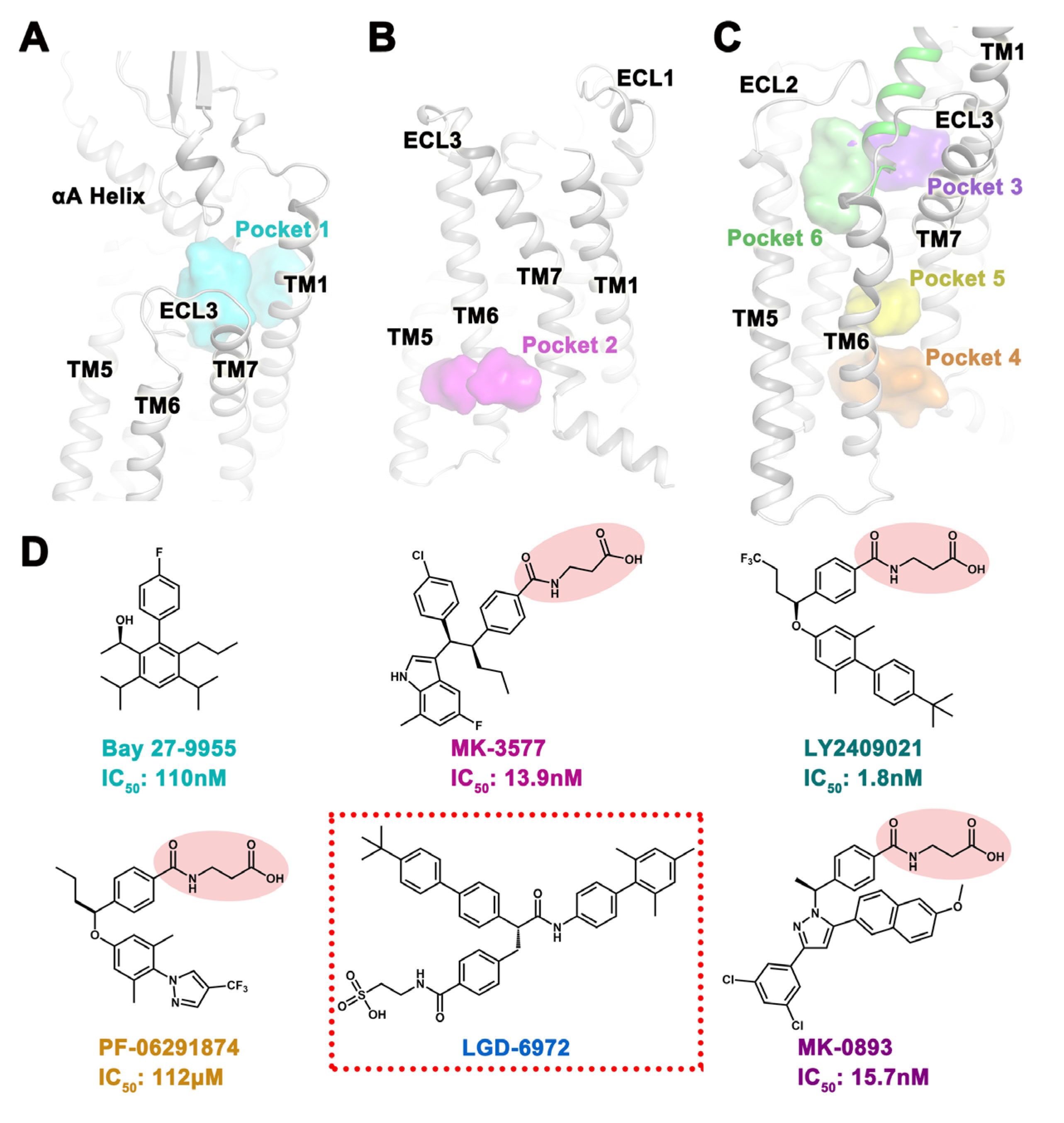
2.1. Potential Binding Sites for Small Molecules
2.2. Verify the Accuracy of the Experimental Method Using MK-0893
2.3. Docking Situation of Five Small Molecules in Six Pockets
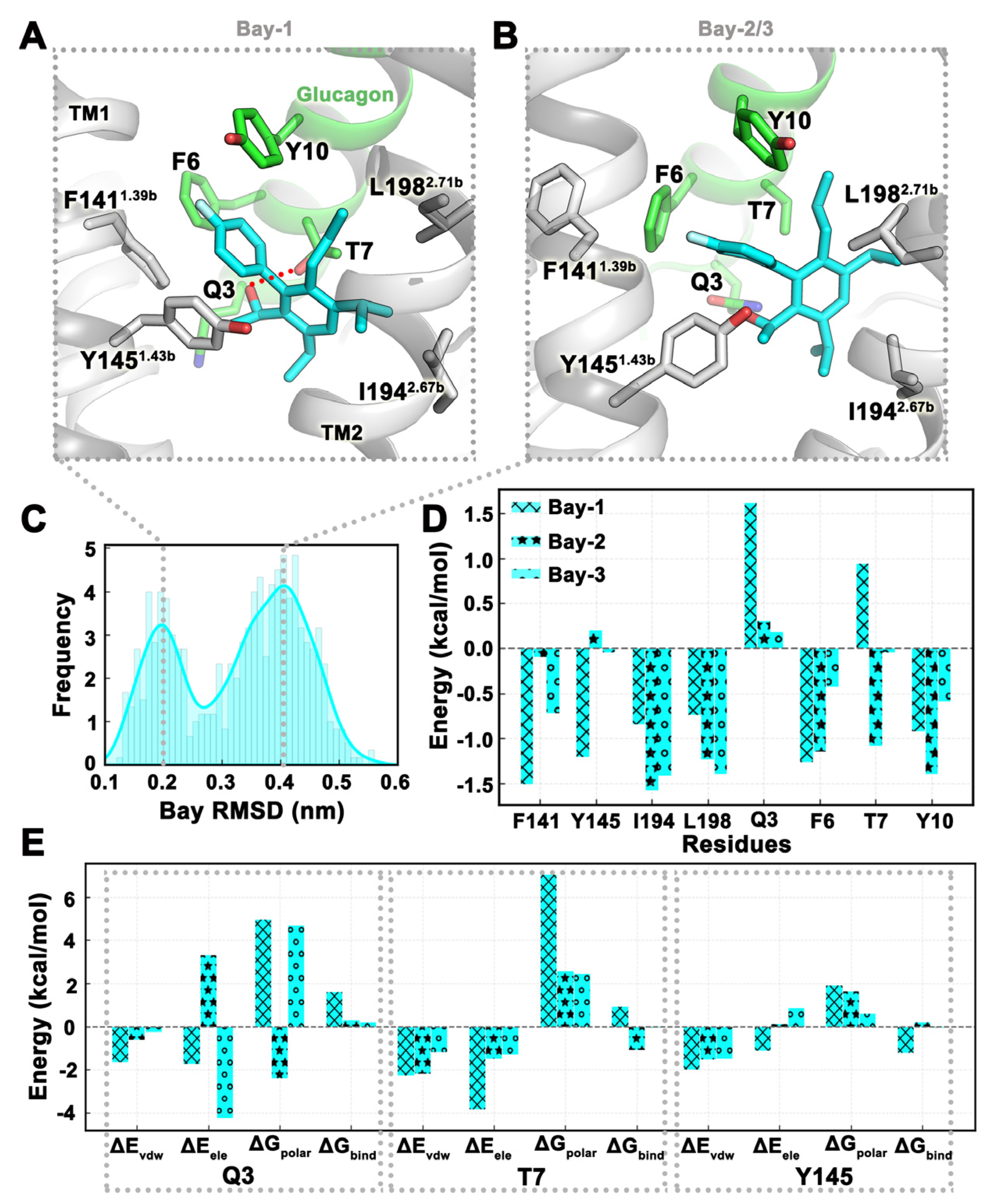
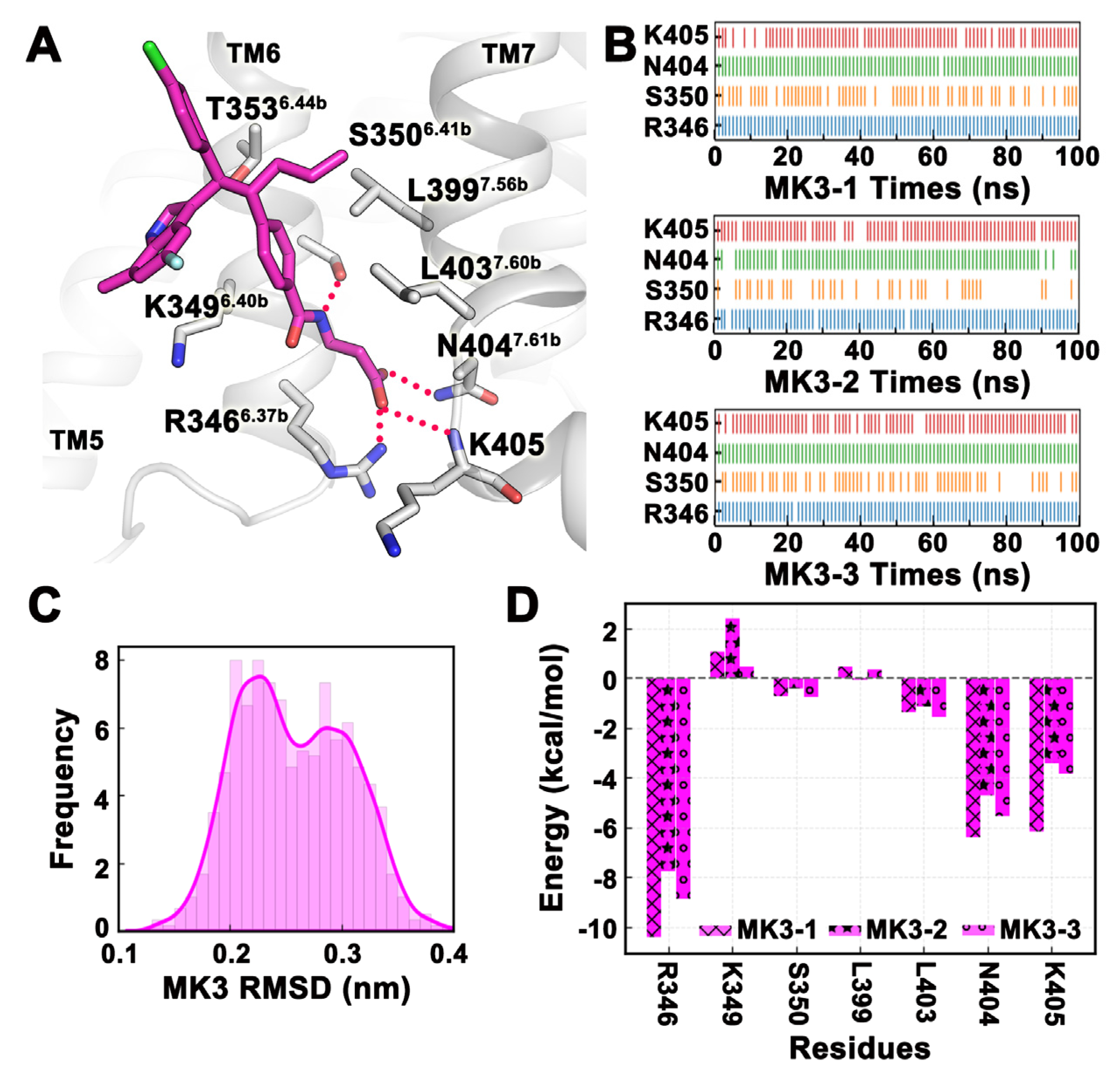
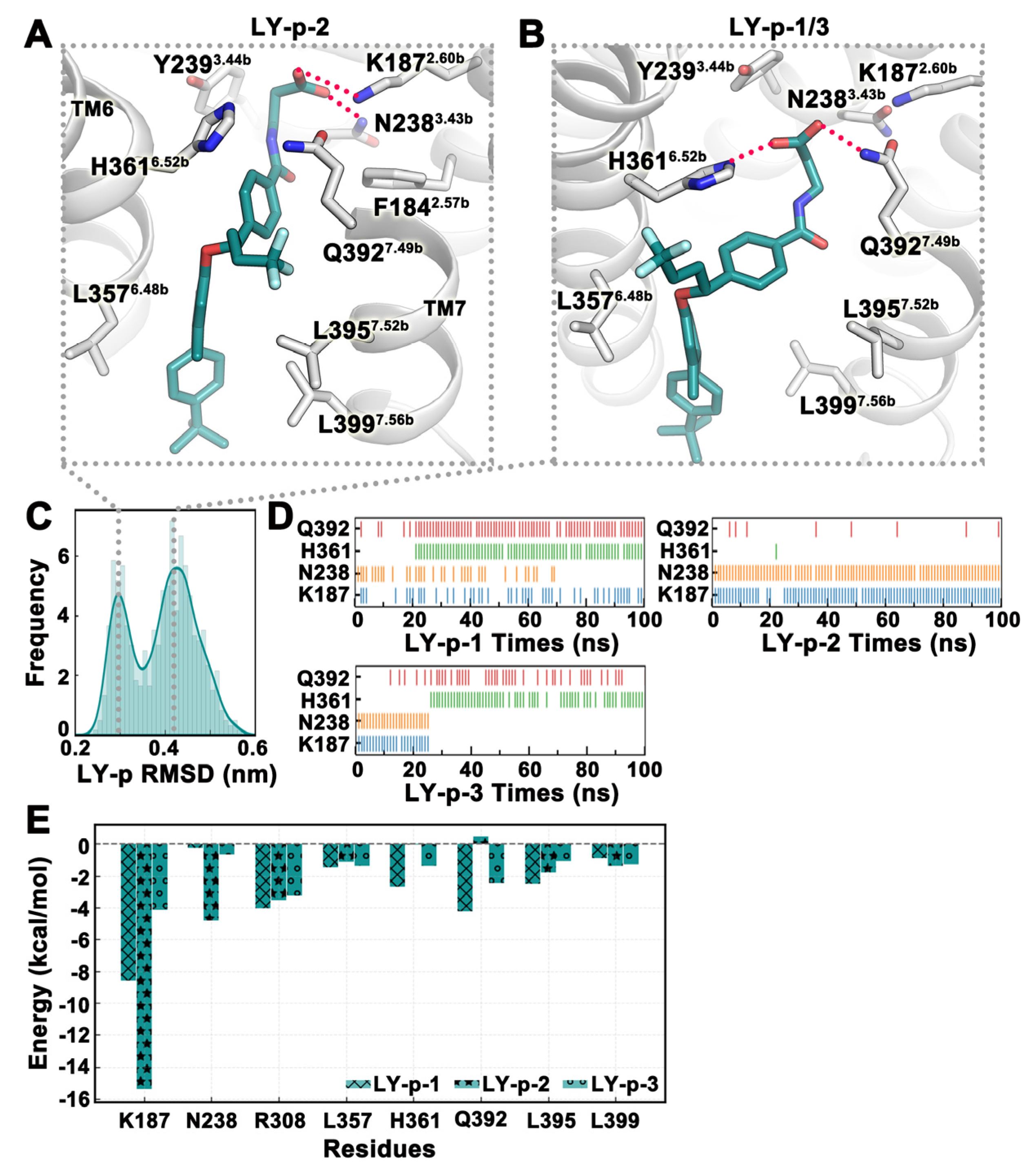
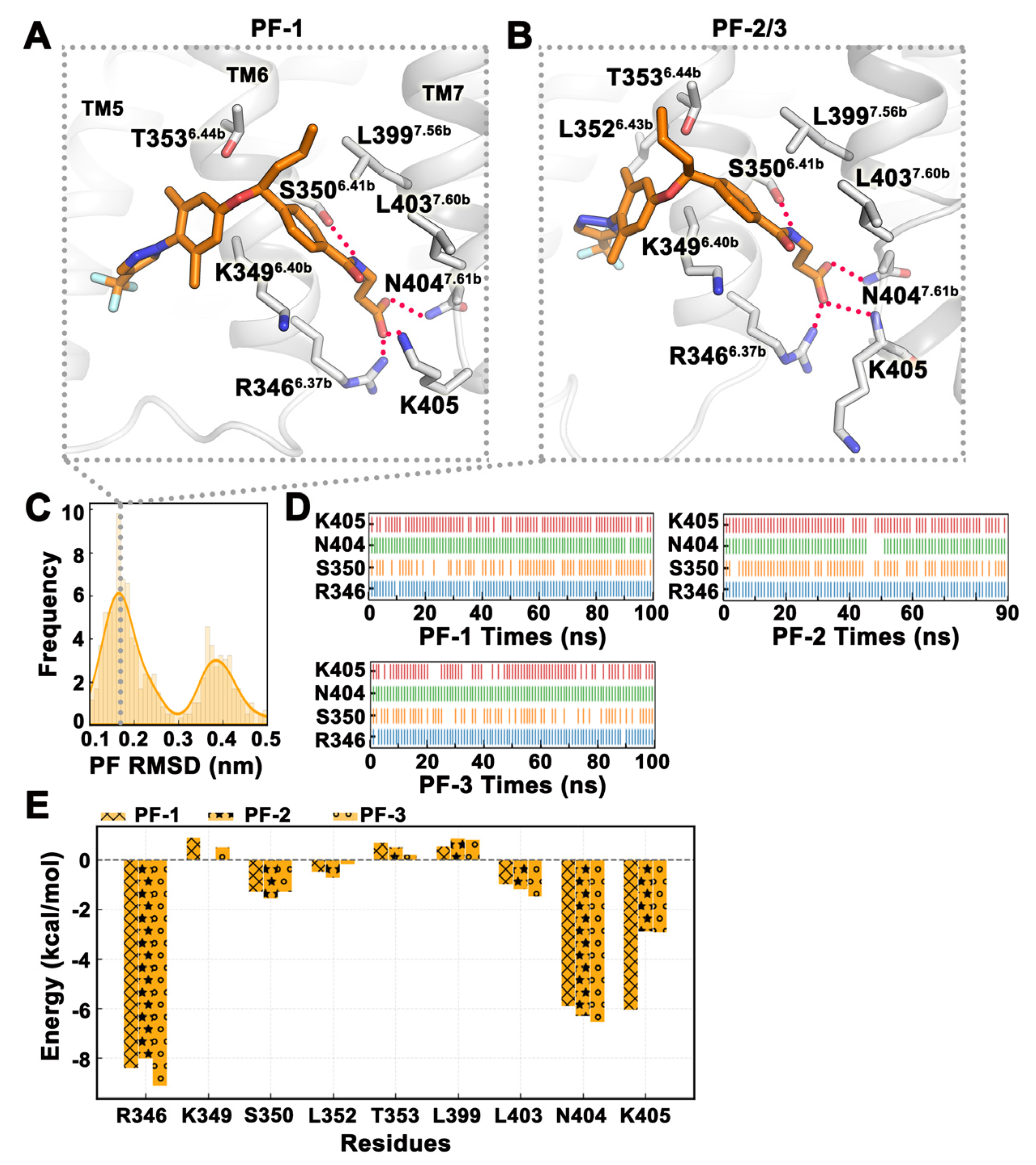
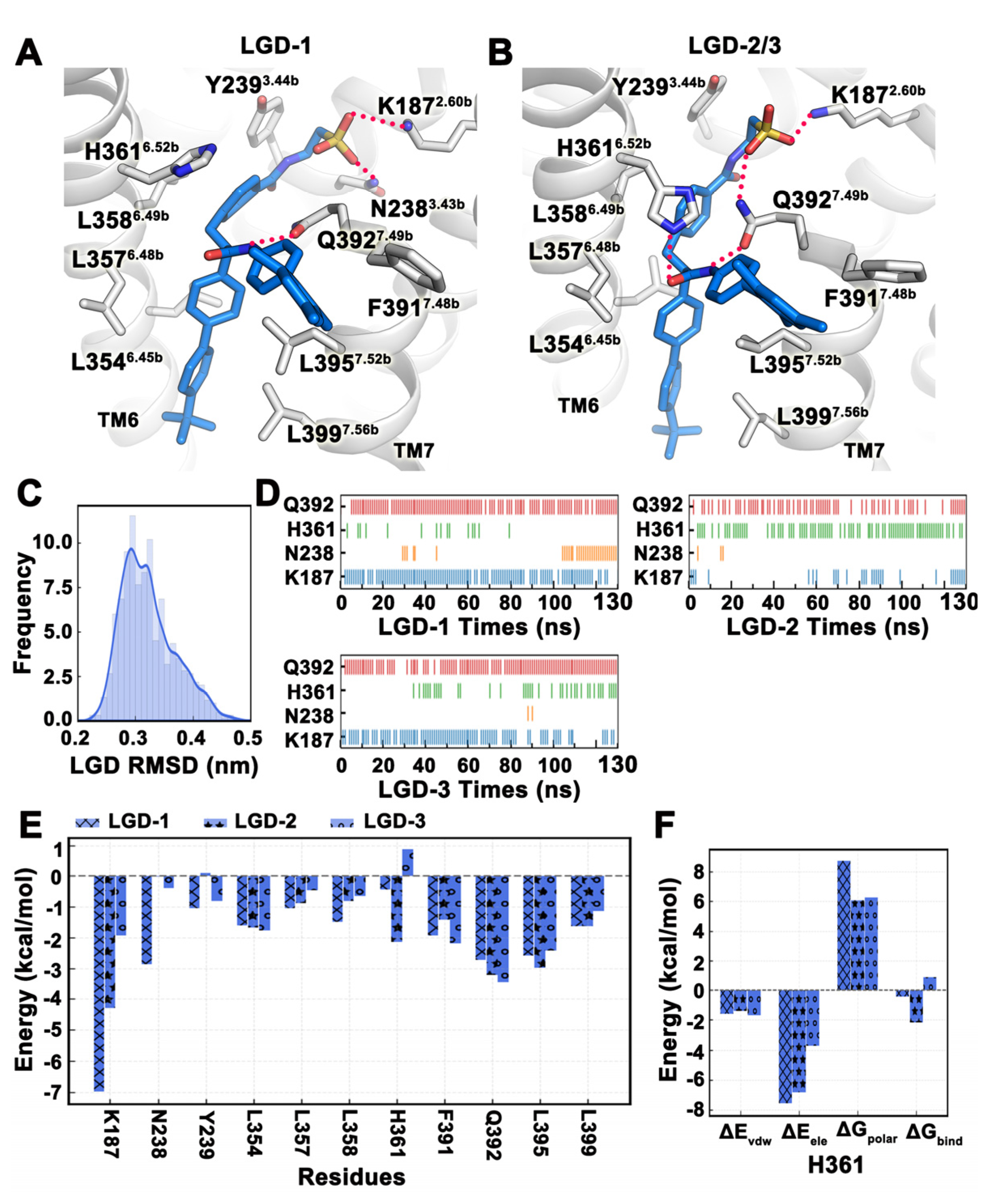
2.4. MD Simulation Was Used to Confirm Binding Modes of Small Molecules
2.5. Structural Modification Based on MD Results
3. Discussion
4. Materials and Methods
4.1. Homology Modeling
4.2. Molecular Dynamics Simulation
4.3. MDpocket Detects Pockets in MD Trajectories
4.4. Molecular Docking
4.5. Calculation and Decomposition the Binding Free Energy
4.6. Prediction of Pharmacokinetic Properties
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. S1), S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, A.; Sabbour, H.; Echtay, A.; Rahmah, A.M.; Alhozali, A.M.; Al Sabaan, F.S.; Haddad, F.H.; Iraqi, H.; Elebrashy, I.; Assaad, S.N.; et al. Current gaps in management and timely referral of cardiorenal complications among people with type 2 diabetes mellitus in the middle east and african countries: Expert recommendations. J. Diabetes 2022, 14, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; de Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Zhang, H.; Han, G.W.; et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 2017, 546, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J. Family-B G-protein-coupled receptors. Genome Biol. 2001, 2, reviews3013.1. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A.; Faloona, G.R.; Unger, R.H. The effect of experimental insulin deficiency on glucagon secretion. J. Clin. Investig. 1971, 50, 1992–1999. [Google Scholar] [CrossRef] [PubMed]
- Raskin, P.; Unger, R.H. Hyperglucagonemia and its suppression. Importance in the metabolic control of diabetes. N. Engl. J. Med. 1978, 299, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Ling, Z.; Pipeleers, D.; Schuit, F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 2000, 43, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Ambery, P.D.; Klammt, S.; Posch, M.G.; Petrone, M.; Pu, W.; Rondinone, C.; Jermutus, L.; Hirshberg, B. MEDI0382, a GLP-1/glucagon receptor dual agonist, meets safety and tolerability endpoints in a single-dose, healthy-subject, randomized, phase 1 study. Br. J. Clin. Pharmacol. 2018, 84, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Tillner, J.; Posch, M.G.; Wagner, F.; Teichert, L.; Hijazi, Y.; Einig, C.; Keil, S.; Haack, T.; Wagner, M.; Bossart, M.; et al. Novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: Results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes. Metab. 2019, 21, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Yan, H.; Shi, Z.; Evans, M.R.; Yu, X.; Lee, Y.; Chen, S.; Williams, A.; Philippe, J.; Roth, M.G.; et al. Glucagon receptor antibody completely suppresses type 1 diabetes phenotype without insulin by disrupting a novel diabetogenic pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 2503–2508. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Kim, J.; Aglione, J.; Lee, J.; Cavino, K.; Na, E.; Rafique, A.; Kim, J.H.; Harp, J.; Valenzuela, D.M.; et al. Glucagon receptor blockade with a human antibody normalizes blood glucose in diabetic mice and monkeys. Endocrinology 2015, 156, 2781–2794. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.; Esteves, B.; Dell, V.; Joh, T.; Garzone, P.D.; Forgie, A.; Udata, C. Single and multiple ascending-dose study of glucagon-receptor antagonist RN909 in type 2 diabetes: A phase 1, randomized, double-blind, placebo-controlled trial. Endocrine 2018, 62, 371–380. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, M.G.; Geerts, B.F.; Morgan, E.S.; Brandt, T.A.; de Kam, M.L.; Romijn, J.A.; Cohen, A.F.; Bhanot, S.; Burggraaf, J. First proof of pharmacology in humans of a novel glucagon receptor antisense drug. J. Clin. Pharmacol. 2015, 55, 298–306. [Google Scholar] [CrossRef]
- Scheen, A.J.; Paquot, N.; Lefebvre, P.J. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert. Opin. Investig. Drugs 2017, 26, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Roth, B.L. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 117–144. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Sullivan, J.T. Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia 2001, 44, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Dore, A.S.; Lamb, D.; Krishnamurthy, H.; Southall, S.M.; Baig, A.H.; Bortolato, A.; Koglin, M.; Robertson, N.J.; Errey, J.C.; et al. Extra-helical binding site of a glucagon receptor antagonist. Nature 2016, 533, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Guo, J.; Candelore, M.R.; Liang, R.; Miller, C.; Dallas-Yang, Q.; Jiang, G.; McCann, P.E.; Qureshi, S.A.; Tong, X.; et al. Discovery of a novel glucagon receptor antagonist n-[(4-(1s)-1-[3-(3, 5-dichlorophenyl)-5-(6-methoxynaphthalen-2-yl)-1h-pyrazol-1-yl]ethylphenyl)carbonyl]-beta-alanine (mk-0893) for the treatment of type II diabetes. J. Med. Chem. 2012, 55, 6137–6148. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.Z.; Denney, W.S.; Musser, B.J.; Liu, R.; Tsai, K.; Fang, L.; Reitman, M.L.; Troyer, M.D.; Engel, S.S.; Xu, L.; et al. A semi-mechanistic model for the effects of a novel glucagon receptor antagonist on glucagon and the interaction between glucose, glucagon, and insulin applied to adaptive phase II design. AAPS J. 2014, 16, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Kazda, C.M.; Ding, Y.; Kelly, R.P.; Garhyan, P.; Shi, C.; Lim, C.N.; Fu, H.; Watson, D.E.; Lewin, A.J.; Landschulz, W.H.; et al. Evaluation of efficacy and safety of the glucagon receptor antagonist LY2409021 in patients with type 2 diabetes: 12- and 24-week phase 2 studies. Diabetes Care 2016, 39, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.P.; Garhyan, P.; Raddad, E.; Fu, H.; Lim, C.N.; Prince, M.J.; Pinaire, J.A.; Loh, M.T.; Deeg, M.A. Short-term administration of the glucagon receptor antagonist LY2409021 lowers blood glucose in healthy people and in those with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 414–422. [Google Scholar] [CrossRef]
- Guzman-Perez, A.; Pfefferkorn, J.A.; Lee, E.C.; Stevens, B.D.; Aspnes, G.E.; Bian, J.; Didiuk, M.T.; Filipski, K.J.; Moore, D.; Perreault, C.; et al. The design and synthesis of a potent glucagon receptor antagonist with favorable physicochemical and pharmacokinetic properties as a candidate for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. Lett. 2013, 23, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Vajda, E.G.; Logan, D.; Lasseter, K.; Armas, D.; Plotkin, D.J.; Pipkin, J.D.; Li, Y.X.; Zhou, R.; Klein, D.; Wei, X.; et al. Pharmacokinetics and pharmacodynamics of single and multiple doses of the glucagon receptor antagonist LGD-6972 in healthy subjects and subjects with type 2 diabetes mellitus. Diabetes Obes. Metab. 2017, 19, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Pettus, J.H.; D’Alessio, D.; Frias, J.P.; Vajda, E.G.; Pipkin, J.D.; Rosenstock, J.; Williamson, G.; Zangmeister, M.A.; Zhi, L.; Marschke, K.B. Efficacy and safety of the glucagon receptor antagonist RVT-1502 in type 2 diabetes uncontrolled on metformin monotherapy: A 12-week dose-ranging study. Diabetes Care 2020, 43, 161–168. [Google Scholar] [CrossRef]
- Cheng, C.; Jabri, S.; Taoka, B.M.; Sinz, C.J. Small molecule glucagon receptor antagonists: An updated patent review (2015–2019). Expert. Opin. Ther. Pat. 2020, 30, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hubner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar]
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; Pillai, S.G.; Kazda, C.; Chalasani, N.; Hardy, T.A. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528. [Google Scholar] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Wootten, D.; Simms, J.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. USA 2013, 110, 5211–5216. [Google Scholar] [CrossRef] [PubMed]
- Mattedi, G.; Acosta-Gutierrez, S.; Clark, T.; Gervasio, F.L. A combined activation mechanism for the glucagon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 15414–15422. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiao, A.; Yang, L.; Van Eps, N.; Frederiksen, K.S.; Yang, D.; Dai, A.; Cai, X.; Zhang, H.; Yi, C.; et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. Charmm36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Tresanco, M.S.; Valdes-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Bo, Z.; Xu, T.; Xu, B.; Wang, D.; Zheng, H. Uni-GBSA: An open-source and web-based automatic workflow to perform MM/GB(PB)SA calculations for virtual screening. Brief. Bioinform. 2023, 24, bbad218. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Pocket 1 | Pocket 2 | Pocket 3 | Pocket 4 | Pocket 5 | Pocket 6 |
|---|---|---|---|---|---|---|
| Bay 27-9955 | −9.54 | −5.12 | −6.85 | −8.54 | ||
| MK-3577 | −10.65 | −8.65 | −9.53 | −12.69 | −12.11 | −11.42 |
| LY2409021 | −11.22 | −12.00 | −10.34 | −12.55 | −16.01 | −13.13 |
| PF-06291874 | −11.06 | −12.05 | −9.26 | −8.18 | −10.75 | −11.54 |
| LGD-6972 | −13.06 | −9.41 | −10.86 | −10.70 | −18.09 | −14.29 |
| Molecule | Pocket 1 | Pocket 2 | Pocket 3 | Pocket 4 | Pocket 5 | Pocket 6 |
|---|---|---|---|---|---|---|
| Bay 27-9955 | −88.59 | −47.35 | −63.94 | −74.75 | ||
| MK-3577 | −96.70 | −68.75 | −76.62 | −105.20 | −85.88 | −77.54 |
| LY2409021 | −95.56 | −105.84 | −87.94 | −111.16 | −111.19 | −100.29 |
| PF-06291874 | −85.33 | −85.62 | −73.21 | −57.33 | −81.46 | −72.30 |
| LGD-6972 | −119.53 | −78.44 | −99.56 | −116.25 | −157.91 | −142.12 |
| System | ΔGbind | ΔEvdw | ΔEele | ΔGpolar |
|---|---|---|---|---|
| Bay-1 | −19.43 | −44.65 | 29.89 | −4.67 |
| Bay-2 | −29.21 | −40.75 | 16.07 | −4.53 |
| Bay-3 | −28.12 | −42.34 | 18.79 | −4.57 |
| Mean ± SD | −25.59 ± 3.09 | −42.58 ± 1.13 | 21.58 ± 4.23 | −4.59 ± 0.04 |
| MK-1 | −31.97 | −25.69 | −2.69 | −3.60 |
| MK-2 | −26.12 | −26.66 | 4.23 | −3.69 |
| MK-3 | −31.06 | −27.62 | 0.17 | −3.62 |
| Mean ± SD | −29.72 ± 1.82 | −26.66 ± 0.56 | 0.57 ± 2.01 | −3.64 ± 0.03 |
| LY-1 | −43.87 | −39.36 | 0.32 | −4.83 |
| LY-2 | −56.02 | −32.74 | −18.92 | −4.36 |
| LY-3 | −43.66 | −40.11 | 1.24 | −4.79 |
| Mean ± SD | −47.85 ± 4.09 | −37.40 ± 2.34 | −5.79 ± 6.57 | −4.66 ± 0.15 |
| LY-p-1 | −29.15 | −43.42 | 19.68 | −5.41 |
| LY-p-2 | −49.05 | −45.75 | 2.58 | −5.88 |
| LY-p-3 | −26.13 | −38.82 | 18.01 | −5.31 |
| Mean ± SD | −34.78 ± 7.19 | −42.66 ± 2.04 | 13.42 ± 5.44 | −5.53 ± 0.18 |
| PF-1 | −33.10 | −35.68 | 6.86 | −4.28 |
| PF-2 | −37.01 | −34.49 | 1.79 | −4.31 |
| PF-3 | −25.17 | −24.52 | 2.65 | −3.29 |
| Mean ± SD | −31.76 ± 3.48 | −31.56 ± 3.54 | 3.77 ± 1.57 | −3.96 ± 0.34 |
| LGD-1 | −63.51 | −67.33 | 11.09 | −7.26 |
| LGD-2 | −53.78 | −66.80 | 20.29 | −7.27 |
| LGD-3 | −36.81 | −67.83 | 38.32 | −7.29 |
| Mean ± SD | −51.37 ± 7.80 | −67.32 ± 0.30 | 23.23 ± 8.00 | −7.27 ± 0.01 |
| Molecule | HBA (≤10) | HBD (≤5) | TPSA (Å2) | Synthetic Accessibility (0–10) |
|---|---|---|---|---|
| LGD-6972 | 5 | 3 | 120.95 | 5.19 |
| Compound1 | 7 | 4 | 158.25 | 5.43 |
| Compound2 | 6 | 4 | 141.18 | 5.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Fu, X.; Du, L.; Shi, F.; Huang, Z.; Yang, L. The Inferential Binding Sites of GCGR for Small Molecules Using Protein Dynamic Conformations and Crystal Structures. Int. J. Mol. Sci. 2024, 25, 8389. https://doi.org/10.3390/ijms25158389
Wang M, Fu X, Du L, Shi F, Huang Z, Yang L. The Inferential Binding Sites of GCGR for Small Molecules Using Protein Dynamic Conformations and Crystal Structures. International Journal of Molecular Sciences. 2024; 25(15):8389. https://doi.org/10.3390/ijms25158389
Chicago/Turabian StyleWang, Mengru, Xulei Fu, Limin Du, Fan Shi, Zichong Huang, and Linlin Yang. 2024. "The Inferential Binding Sites of GCGR for Small Molecules Using Protein Dynamic Conformations and Crystal Structures" International Journal of Molecular Sciences 25, no. 15: 8389. https://doi.org/10.3390/ijms25158389