
Obesity Arrhythmias: Role of IL-6 Trans-Signaling

{kind=link}
{kind=link}
{kind=link}
Abstract
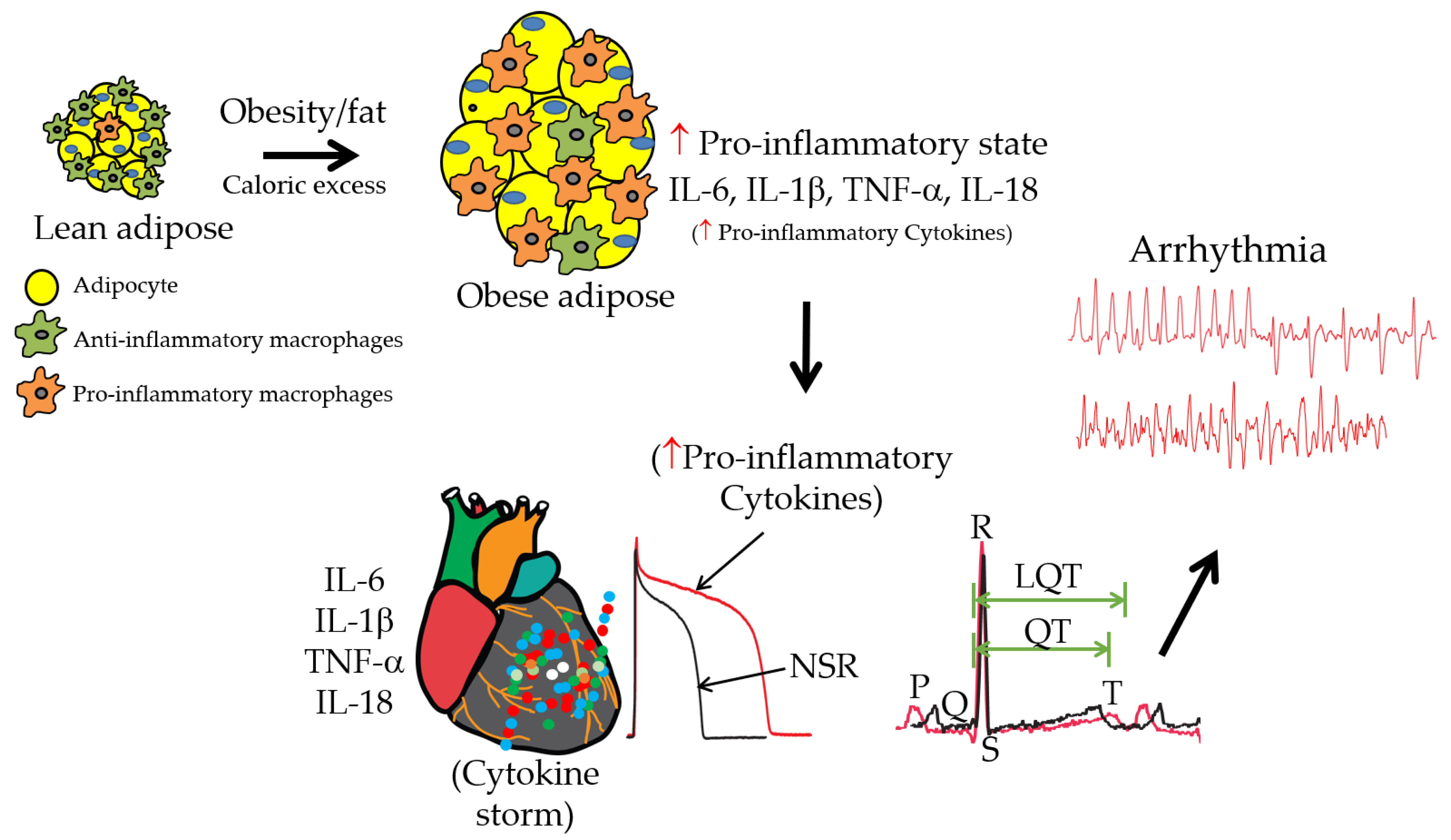
:1. Introduction
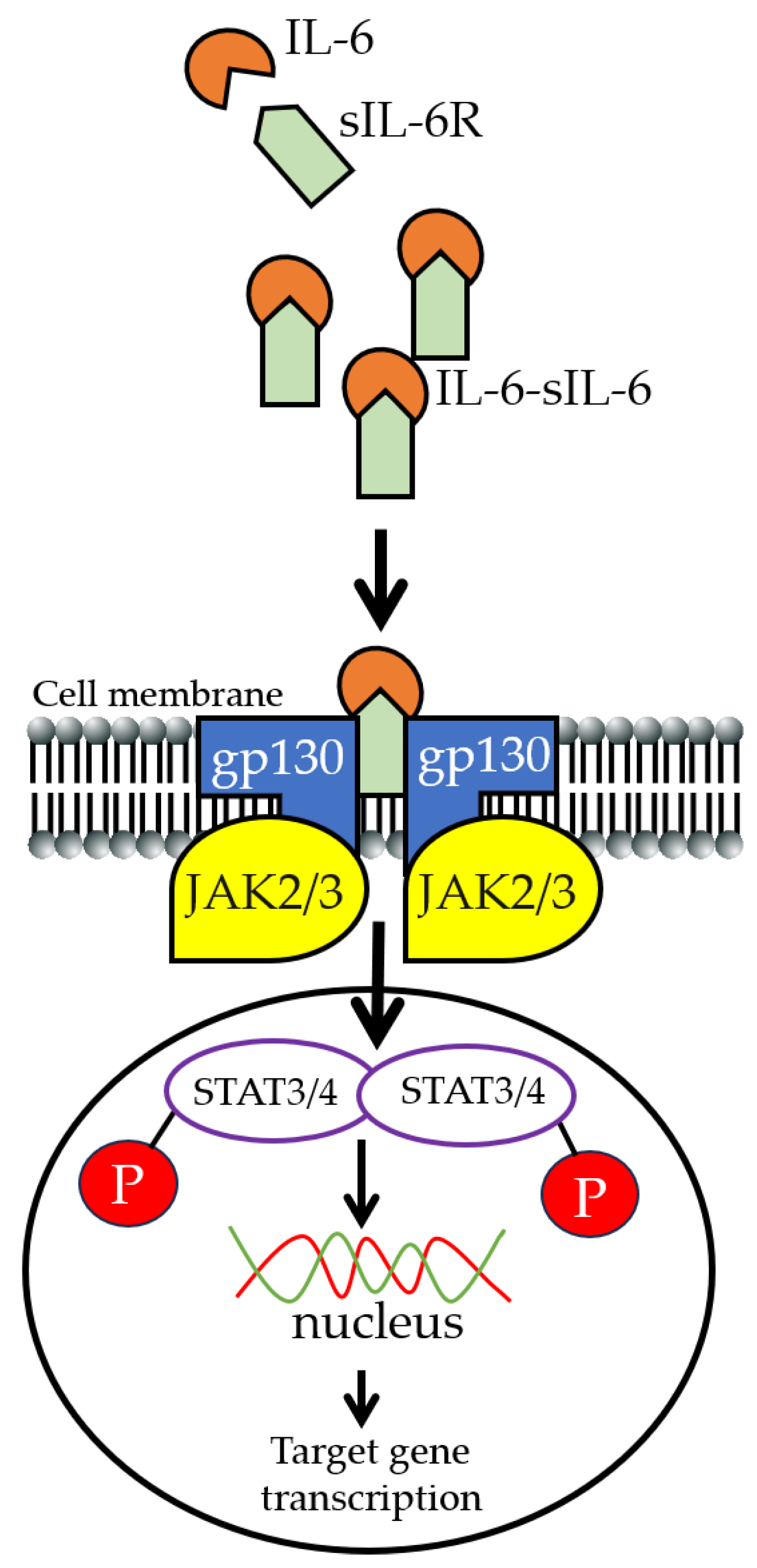
2. IL-6 Signaling
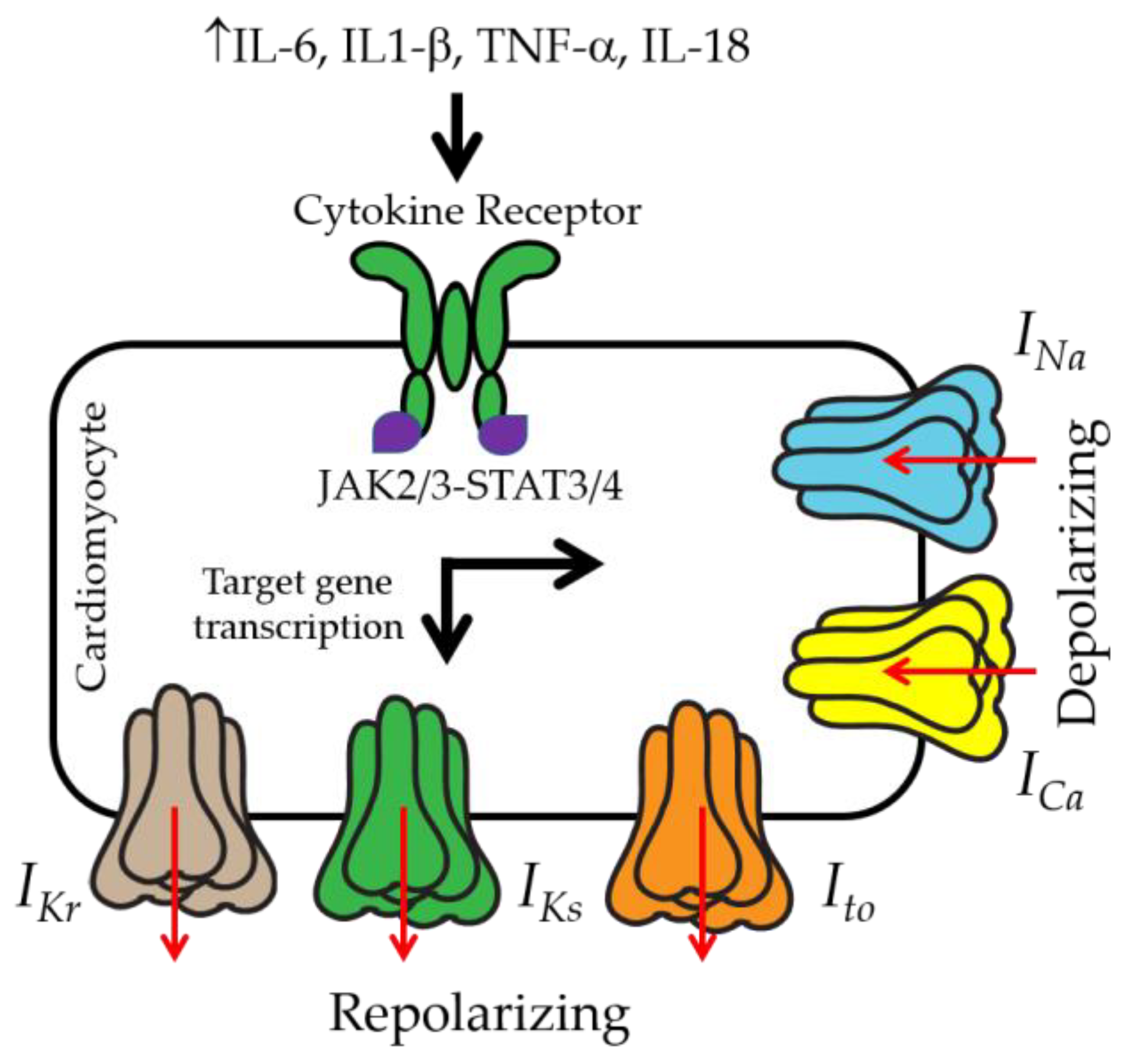
3. IL-6 and Ventricular Electrical Activity
4. Anti-IL-6 Inhibitors
5. Importance of Preclinical Models for Studying IL-6 Trans-Signaling
6. Insights into Significant Knowledge Gaps and Future Directions in Obesity, Inflammation, and Ventricular Arrhythmias
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Nonstandard Abbreviations and Acronyms | |
| TCZ | Tocilizumab |
| IKr | Rapidly activating delayed rectifier K current |
| IKs | Slowly activating delayed rectifier K current |
| INa | Sodium current |
| ICa,L | L-type Ca current |
| IK1 | Inwardly rectifying K current |
| LQT | Long QT |
| QTc | QT interval corrected for heart rate |
| FFAs | Free fatty acids |
| AF | Atrial fibrillation |
| hERG | Human ether-á-go-go-related gene |
| HEK | Human embryonic kidney |
| ERG | Ether-á-go-go-related gene |
| APD | Action potential duration |
| EAT | Epicardial adipose tissue |
| SCD | Sudden cardiac death |
| VF | Ventricular fibrillation |
| VT | Ventricular tachycardia |
| PC | Purkinje cells |
References
- Srinivasan, N.T.; Schilling, R.J. Sudden Cardiac Death and Arrhythmias. Arrhythm. Electrophysiol. Rev. 2018, 7, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Reinier, K.; Teodorescu, C.; Evanado, A.; Kehr, E.; Al Samara, M.; Mariani, R.; Gunson, K.; Jui, J. Epidemiology of Sudden Cardiac Death: Clinical and Research Implications. Prog. Cardiovasc. Dis. 2008, 51, 213–228. [Google Scholar] [CrossRef]
- Huikuri, H.V.; Castellanos, A.; Myerburg, R.J. Sudden death due to cardiac arrhythmias. N. Engl. J. Med. 2001, 345, 1473–1482. [Google Scholar] [CrossRef]
- Podrid, P.J.; Myerburg, R.J. Epidemiology and stratification of risk for sudden cardiac death. Clin. Cardiol. 2005, 28 (Suppl S1), I3–I11. [Google Scholar] [CrossRef] [PubMed]
- Kallergis, E.M.; Goudis, C.A.; Simantirakis, E.N.; Kochiadakis, G.E.; Vardas, P.E. Mechanisms, Risk Factors, and Management of Acquired Long QT Syndrome: A Comprehensive Review. Sci. World J. 2012, 2012, 212178. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.J.; Ackerman, M.J.; Funk, M.; Gibler, W.B.; Kligfield, P.; Menon, V.; Philippides, G.J.; Roden, D.M.; Zareba, W.; on behalf of the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology; et al. Prevention of torsade de pointes in hospital settings: A scientific statement from the American Heart Association and the American College of Cardiology Foundation. J. Am. Coll. Cardiol. 2010, 55, 934–947. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F.; Boutjdir, M. Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat. Rev. Cardiol. 2017, 14, 521–535. [Google Scholar] [CrossRef]
- Pietrasik, G.; Goldenberg, I.; McNitt, S.; Moss, A.J.; Zareba, W. Obesity as a risk factor for sustained ventricular tachyarrhythmias in MADIT II patients. J. Cardiovasc. Electrophysiol. 2007, 18, 181–184. [Google Scholar] [CrossRef]
- Sabbag, A.; Goldenberg, I.; Moss, A.J.; McNitt, S.; Glikson, M.; Biton, Y.; Jackson, L.; Polonsky, B.; Zareba, W.; Kutyifa, V. Predictors and Risk of Ventricular Tachyarrhythmias or Death in Black and White Cardiac Patients: A MADIT-CRT Trial Substudy. JACC Clin. Electrophysiol. 2016, 2, 448–455. [Google Scholar] [CrossRef]
- Sabbag, A.; Sidi, Y.; Kivity, S.; Beinart, R.; Glikson, M.; Segev, S.; Goldenberg, I.; Maor, E. Obesity and exercise-induced ectopic ventricular arrhythmias in apparently healthy middle aged adults. Eur. J. Prev. Cardiol. 2016, 23, 511–517. [Google Scholar] [CrossRef]
- Remme, C.A. Sudden Cardiac Death in Diabetes and Obesity: Mechanisms and Therapeutic Strategies. Can. J. Cardiol. 2022, 38, 418–426. [Google Scholar] [CrossRef]
- Mukerji, R.; Terry, B.E.; Fresen, J.L.; Petruc, M.; Govindarajan, G.; Alpert, M.A. Relation of left ventricular mass to QTc in normotensive severely obese patients. Obesity 2012, 20, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef] [PubMed]
- Hookana, E.; Junttila, M.J.; Puurunen, V.P.; Tikkanen, J.T.; Kaikkonen, K.S.; Kortelainen, M.L.; Myerburg, R.J.; Huikuri, H.V. Causes of nonischemic sudden cardiac death in the current era. Heart Rhythm. 2011, 8, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.A.; Gottdiener, J.S.; Bartz, T.M.; Sotoodehnia, N.; DeFilippi, C.; See, V.; Deo, R.; Siscovick, D.; Stein, P.K.; Lloyd-Jones, D. Inflammation and sudden cardiac death in a community-based population of older adults: The Cardiovascular Health Study. Heart Rhythm. 2013, 10, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity among Adults: United States, 2017–2018. NCHS Data Brief 2020, 360, 1–8. [Google Scholar]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.W.; D’Agostino, R.B.; Sullivan, L.; Parise, H.; Kannel, W.B. Overweight and obesity as determinants of cardiovascular risk: The Framingham experience. Arch. Intern. Med. 2002, 162, 1867–1872. [Google Scholar] [CrossRef]
- Ades, P.A.; Savage, P.D. Obesity in coronary heart disease: An unaddressed behavioral risk factor. Prev. Med. 2017, 104, 117–119. [Google Scholar] [CrossRef]
- Choi, S.; Kim, K.; Kim, S.M.; Lee, G.; Jeong, S.-M.; Park, S.Y.; Kim, Y.-Y.; Son, J.S.; Yun, J.-M.; Park, S.M. Association of Obesity or Weight Change with Coronary Heart Disease among Young Adults in South Korea. JAMA Intern. Med. 2018, 178, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Ebong, I.A.; Goff, D.C., Jr.; Rodriguez, C.J.; Chen, H.; Bertoni, A.G. Mechanisms of heart failure in obesity. Obes. Res. Clin. Pr. 2014, 8, e540–e548. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J. Molecular autopsy in victims of inherited arrhythmias. J. Arrhythm. 2016, 32, 359–365. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018, 138, e272–e391. [Google Scholar]
- Kien, C.L.; Bunn, J.Y.; Ugrasbul, F. Increasing dietary palmitic acid decreases fat oxidation and daily energy expenditure. Am. J. Clin. Nutr. 2005, 82, 320–326. [Google Scholar] [CrossRef]
- Park, T.S.; Goldberg, I.J. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail. Clin. 2012, 8, 633–641. [Google Scholar] [CrossRef]
- Haim, T.E.; Wang, W.; Flagg, T.P.; Tones, M.A.; Bahinski, A.; Numann, R.E.; Nichols, C.G.; Nerbonne, J.M. Palmitate attenuates myocardial contractility through augmentation of repolarizing Kv currents. J. Mol. Cell. Cardiol. 2010, 48, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Redfors, B.; Stahlman, M.; Tang, M.S.; Miljanovic, A.; Mollmann, H.; Troidl, C.; Szardien, S.; Hamm, C.; Nef, H.; et al. A mouse model reveals an important role for catecholamine-induced lipotoxicity in the pathogenesis of stress-induced cardiomyopathy. Eur. J. Heart Fail. 2013, 15, 9–22. [Google Scholar] [CrossRef]
- O’Connell, R.P.; Musa, H.; Gomez, M.S.; Avula, U.M.; Herron, T.J.; Kalifa, J.; Anumonwo, J.M. Free Fatty Acid Effects on the Atrial Myocardium: Membrane Ionic Currents Are Remodeled by the Disruption of T-Tubular Architecture. PLoS ONE 2015, 10, e0133052. [Google Scholar] [CrossRef]
- Aromolaran, A.S.; Colecraft, H.M.; Boutjdir, M. High-fat diet-dependent modulation of the delayed rectifier K+ current in adult guinea pig atrial myocytes. Biochem. Biophys. Res. Commun. 2016, 474, 554–559. [Google Scholar] [CrossRef]
- Anumonwo, J.M.B.; Herron, T. Fatty Infiltration of the Myocardium and Arrhythmogenesis: Potential Cellular and Molecular Mechanisms. Front. Physiol. 2018, 9, 2. [Google Scholar] [CrossRef]
- Feng, Y.; Ye, D.; Wang, Z.; Pan, H.; Lu, X.; Wang, M.; Xu, Y.; Yu, J.; Zhang, J.; Zhao, M.; et al. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 818890. [Google Scholar] [CrossRef]
- Aromolaran, A.S.; Srivastava, U.; Ali, A.; Chahine, M.; Lazaro, D.; El-Sherif, N.; Capecchi, P.L.; Laghi-Pasini, F.; Lazzerini, P.E.; Boutjdir, M. Interleukin-6 inhibition of hERG underlies risk for acquired long QT in cardiac and systemic inflammation. PLoS ONE 2018, 13, e0208321. [Google Scholar] [CrossRef]
- Chowdhury, M.K.H.; Martinez-Mateu, L.; Do, J.; Aromolaran, K.A.; Saiz, J.; Aromolaran, A.S. Macrophage-Dependent Interleukin-6-Production and Inhibition of I (K) Contributes to Acquired QT Prolongation in Lipotoxic Guinea Pig Heart. Int. J. Mol. Sci. 2021, 22, 11249. [Google Scholar] [CrossRef]
- Teng, K.T.; Chang, C.Y.; Chang, L.F.; Nesaretnam, K. Modulation of obesity-induced inflammation by dietary fats: Mechanisms and clinical evidence. Nutr. J. 2014, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Ancey, C.; Corbi, P.; Froger, J.; Delwail, A.; Wijdenes, J.; Gascan, H.; Potreau, D.; Lecron, J.C. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine 2002, 18, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zheng, R.; Hu, S.; Ma, Y.; Choudhry, M.A.; Messina, J.L.; Rue, L.W., 3rd; Bland, K.I.; Chaudry, I.H. Mechanism of cardiac depression after trauma-hemorrhage: Increased cardiomyocyte IL-6 and effect of sex steroids on IL-6 regulation and cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2183–H2191. [Google Scholar] [CrossRef]
- Lust, J.A.; Donovan, K.A.; Kline, M.P.; Greipp, P.R.; Kyle, R.A.; Maihle, N.J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96–100. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef]
- Taga, T.; Kishimoto, T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.A.; Rose, N.R.; Cihakova, D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [PubMed]
- IL6R Genetics Consortium Emerging Risk Factors Collaboration; Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar]
- Akira, S.; Isshiki, H.; Sugita, T.; Tanabe, O.; Kinoshita, S.; Nishio, Y.; Nakajima, T.; Hirano, T.; Kishimoto, T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. Embo J. 1990, 9, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef]
- Werner-Klein, M.; Grujovic, A.; Irlbeck, C.; Obradović, M.; Hoffmann, M.; Koerkel-Qu, H.; Lu, X.; Treitschke, S.; Köstler, C.; Botteron, C.; et al. Interleukin-6 trans-signaling is a candidate mechanism to drive progression of human DCCs during clinical latency. Nat. Commun. 2020, 11, 4977. [Google Scholar] [CrossRef]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M.; Darnell, J.E. The state of the STATs: Recent developments in the study of signal transduction to the nucleus. Curr. Opin. Cell Biol. 1997, 9, 233–239. [Google Scholar] [CrossRef]
- Imada, K.; Leonard, W.J. The Jak-STAT pathway. Mol. Immunol. 2000, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, O.P.; Mandrup-Poulsen, T. Interleukin-6 and diabetes: The good, the bad, or the indifferent? Diabetes 2005, 54 (Suppl. 2), S114–S124. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, C.M.; Hövelmeyer, N.; Wunderlich, F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAK-STAT 2013, 2, e23878. [Google Scholar] [CrossRef]
- Galic, S.; Sachithanandan, N.; Kay, T.W.; Steinberg, G.R. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem. J. 2014, 461, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, P.A.; Singh, A.K.; Nguyen, N.; Dvorak, N.M.; Tapia, C.M.; Russell, W.K.; Stephan, C.; Laezza, F. JAK2 regulates Nav1.6 channel function via FGF14 (Y158) phosphorylation. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118786. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, H. Identification of WP1066, an inhibitor of JAK2 and STAT3, as a K (V) 1.3 potassium channel blocker. Br. J. Pharmacol. 2021, 178, 2617–2631. [Google Scholar] [CrossRef]
- Hosseinzadeh, Z.; Almilaji, A.; Honisch, S.; Pakladok, T.; Liu, G.; Bhavsar, S.K.; Ruth, P.; Shumilina, E.; Lang, F. Upregulation of the large conductance voltage- and Ca2+-activated K+ channels by Janus kinase 2. Am. J. Physiol. Cell Physiol. 2014, 306, C1041–C1049. [Google Scholar] [CrossRef]
- Hosseinzadeh, Z.; Bhavsar, S.K.; Lang, F. Downregulation of ClC-2 by JAK2. Cell. Physiol. Biochem. 2012, 29, 737–742. [Google Scholar] [CrossRef]
- Hosseinzadeh, Z.; Luo, D.; Sopjani, M.; Bhavsar, S.K.; Lang, F. Down-regulation of the epithelial Na+ channel ENaC by Janus kinase 2. J. Membr. Biol. 2014, 247, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hosseinzadeh, Z.; Zhang, B.; Froeschl, M.; Schulze-Osthoff, K.; Stournaras, C.; Lang, F. Decrease of Store-Operated Ca2+ Entry and Increase of Na+/Ca2+ Exchange by Pharmacological JAK2 Inhibition. Cell. Physiol. Biochem. 2016, 38, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Zhang, W.; Karle, C.A.; Kathofer, S.; Schols, W.; Kubler, W.; Kiehn, J. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J. Biol. Chem. 1999, 274, 27457–27462. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Melman, Y.; Palma, E.; Fishman, G.I.; McDonald, T.V. Cyclic AMP regulates the HERG K+ channel by dual pathways. Curr. Biol. 2000, 10, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Cockerill, S.L.; Tobin, A.B.; Torrecilla, I.; Willars, G.B.; Standen, N.B.; Mitcheson, J.S. Modulation of hERG potassium currents in HEK-293 cells by protein kinase C. Evidence for direct phosphorylation of pore forming subunits. J. Physiol. 2007, 581 Pt 2, 479–493. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Zhang, H.; Shen, L.; Xu, Y. Different protein kinase C isoenzymes mediate inhibition of cardiac rapidly activating delayed rectifier K+ current by different G-protein coupled receptors. Br. J. Pharmacol. 2017, 174, 4464–4477. [Google Scholar] [CrossRef] [PubMed]
- WU, L.-M.; Ueda, K.; Hirano, Y.; Furukawa, T.; Hiraoka, M. Herg Potassium Channel Is Regulated by Protein Tyrosine Kinase (Ptk) in Human Embryonic Kidney Cells. In Advances in Electrocardiology 2004; World Scientific: Singapore, 2004; pp. 54–56. [Google Scholar]
- Zhang, F.; Wang, Y.; Liu, Y.; Han, H.; Zhang, D.; Fan, X.; Du, X.; Gamper, N.; Zhang, H. Transcriptional Regulation of Voltage-Gated Sodium Channels Contributes to GM-CSF-Induced Pain. J. Neurosci. 2019, 39, 5222–5233. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Zhao, F.; Wang, J.; Zhao, Y.; Luo, Z.; Gao, Y.; Shi, J. Regulation of TRPM7 Function by IL-6 through the JAK2-STAT3 Signaling Pathway. PLoS ONE 2016, 11, e0152120. [Google Scholar] [CrossRef]
- Koglin, J.; Glysing-Jensen, T.; Gadiraju, S.; Russell, M.E. Attenuated Cardiac Allograft Vasculopathy in Mice with Targeted Deletion of the Transcription Factor STAT4. Circulation 2000, 101, 1034–1039. [Google Scholar] [CrossRef]
- Svenungsson, E.; Gustafsson, J.; Leonard, D.; Sandling, J.; Gunnarsson, I.; Nordmark, G.; Jönsen, A.; Bengtsson, A.A.; Sturfelt, G.; Rantapää-Dahlqvist, S.; et al. A STAT4 risk allele is associated with ischaemic cerebrovascular events and anti-phospholipid antibodies in systemic lupus erythematosus. Ann. Rheum. Dis. 2010, 69, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Meinert, C.; Gembardt, F.; Böhme, I.; Tetzner, A.; Wieland, T.; Greenberg, B.; Walther, T. Identification of intracellular proteins and signaling pathways in human endothelial cells regulated by angiotensin-(1–7). J. Proteom. 2016, 130, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.J.; Shakespear, M.R.; Kamal, N.A.; Fairlie, D.P. HDAC inhibitors: Modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol. Cell Biol. 2012, 90, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.Z.; Liu, W.; Xia, Y.; Yin, H.M.; Zhang, C.Y.; Su, D.; Yan, L.F.; Gu, A.H.; Zhou, Y. The pro-inflammatory signalling regulator Stat4 promotes vasculogenesis of great vessels derived from endothelial precursors. Nat. Commun. 2017, 8, 14640. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Singh, N.; Mullican, S.E.; Everett, L.J.; Li, L.; Yuan, L.; Liu, X.; Epstein, J.A.; Lazar, M.A. Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J. Biol. Chem. 2011, 286, 33301–33309. [Google Scholar] [CrossRef]
- Meraviglia, V.; Alcalde, M.; Campuzano, O.; Bellin, M. Inflammation in the Pathogenesis of Arrhythmogenic Cardiomyopathy: Secondary Event or Active Driver? Front. Cardiovasc. Med. 2021, 8, 784715. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef]
- Bers, D.M.; Despa, S. Na+ transport in cardiac myocytes; Implications for excitation-contraction coupling. IUBMB Life 2009, 61, 215–221. [Google Scholar] [CrossRef]
- Varro, A.; Nanasi, P.P.; Lathrop, D.A. Potassium currents in isolated human atrial and ventricular cardiocytes. Acta Physiol. Scand. 1993, 149, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, A.S.; Subramanyam, P.; Chang, D.D.; Kobertz, W.R.; Colecraft, H.M. LQT1 mutations in KCNQ1 C-terminus assembly domain suppress IKs using different mechanisms. Cardiovasc. Res. 2014, 104, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Puckerin, A.; Aromolaran, K.A.; Chang, D.D.; Zukin, R.S.; Colecraft, H.M.; Boutjdir, M.; Aromolaran, A.S. hERG 1a LQT2 C-terminus truncation mutants display hERG 1b-dependent dominant negative mechanisms. Heart Rhythm. 2016, 13, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.P.; Yuan, C.; Navedo, M.F.; Dixon, R.E.; Nieves-Cintron, M.; Scott, J.D.; Santana, L.F. Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ. Res. 2011, 109, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. 2018, 41, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.D.; Kumar, S.; Kalman, J.M.; Sanders, P.; Sacher, F.; Hocini, M.; Jais, P.; Haisaguerre, M.; Lee, G. Sudden Cardiac Death and Ventricular Arrhythmias: State of the Art in 2018–2019: Chapter 13: Catheter Ablation of Ventricular Fibrillation. Heart Lung Circ. 2018. [Google Scholar]
- Liu, X.; Shi, J.; Xiao, P. Associations between common ion channel single nucleotide polymorphisms and sudden cardiac death in adults: A MOOSE-compliant meta-analysis. Medicine 2018, 97, e12428. [Google Scholar] [CrossRef] [PubMed]
- Cubeddu, L.X. Drug-induced Inhibition and Trafficking Disruption of ion Channels: Pathogenesis of QT Abnormalities and Drug-Induced Fatal Arrhythmias. Curr. Cardiol. Rev. 2016, 12, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Medenwald, D.; Kors, J.A.; Loppnow, H.; Thiery, J.; Kluttig, A.; Nuding, S.; Tiller, D.; Greiser, K.H.; Werdan, K.; Haerting, J. Inflammation and prolonged QT time: Results from the Cardiovascular Disease, Living and Ageing in Halle (CARLA) study. PLoS ONE 2014, 9, e95994. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Acampa, M.; Capecchi, P.L.; Fineschi, I.; Selvi, E.; Moscadelli, V.; Zimbone, S.; Gentile, D.; Galeazzi, M.; Laghi-Pasini, F. Antiarrhythmic potential of anticytokine therapy in rheumatoid arthritis: Tocilizumab reduces corrected QT interval by controlling systemic inflammation. Arthritis Care Res. 2015, 67, 332–339. [Google Scholar] [CrossRef]
- Adlan, A.M.; Panoulas, V.F.; Smith, J.P.; Fisher, J.P.; Kitas, G.D. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J. Rheumatol. 2015, 42, 421–428. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Laghi-Pasini, F.; Bertolozzi, I.; Morozzi, G.; Lorenzini, S.; Simpatico, A.; Selvi, E.; Bacarelli, M.R.; Finizola, F.; Vanni, F.; et al. Systemic inflammation as a novel QT-prolonging risk factor in patients with torsades de pointes. Heart 2017, 103, 1821–1829. [Google Scholar] [CrossRef]
- Carella, M.J.; Mantz, S.L.; Rovner, D.R.; Willis, P.W., 3rd; Gossain, V.V.; Bouknight, R.R.; Ferenchick, G.S. Obesity, adiposity, and lengthening of the QT interval: Improvement after weight loss. Int. J. Obes. Relat. Metab. Disord. 1996, 20, 938–942. [Google Scholar]
- Milovančev, A.; Stokic, E. In Corrected QT Interval and Corrected QT Dispersion in Obesity. CJournal Clin. Cardiol. Diagn. 2019, 2, 1–4. [Google Scholar]
- Hagiwara, Y.; Miyoshi, S.; Fukuda, K.; Nishiyama, N.; Ikegami, Y.; Tanimoto, K.; Murata, M.; Takahashi, E.; Shimoda, K.; Hirano, T.; et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+] i transient, and APD increase in cardiomyocytes. J. Mol. Cell. Cardiol. 2007, 43, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, Y.; Xiao, Y.; Gao, Q.; Gao, L.; Zhang, W.; Xin, X.; Chen, K.; Srivastava, U.; Ginjupalli, V.K.M.; et al. Arrhythmogenic mechanisms of interleukin-6 combination with hydroxychloroquine and azithromycin in inflammatory diseases. Sci. Rep. 2022, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Rozanski, G.J. Effects of human recombinant interleukin-1 on electrical properties of guinea pig ventricular cells. Cardiovasc. Res. 1993, 27, 525–530. [Google Scholar] [CrossRef]
- Bänsch, D.; Oyang, F.; Antz, M.; Arentz, T.; Weber, R.; Val-Mejias, J.E.; Ernst, S.; Kuck, K.-H. Successful Catheter Ablation of Electrical Storm after Myocardial Infarction. Circulation 2003, 108, 3011–3016. [Google Scholar] [CrossRef]
- Ideker, R.E.; Kong, W.; Pogwizd, S. Purkinje Fibers and Arrhythmias. Pacing Clin. Electrophysiol. 2009, 32, 283–285. [Google Scholar] [CrossRef]
- Sinha, A.-M.; Schmidt, M.; Marschang, H.; Gutleben, K.; Ritscher, G.; Brachmann, J.; Marrouche, N.F. Role of Left Ventricular Scar and Purkinje-Like Potentials During Mapping and Ablation of Ventricular Fibrillation in Dilated Cardiomyopathy. Pacing Clin. Electrophysiol. 2009, 32, 286–290. [Google Scholar] [CrossRef]
- Gintant, G. An evaluation of hERG current assay performance: Translating preclinical safety studies to clinical QT prolongation. Pharmacol. Ther. 2011, 129, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.J.M.D.; Roden, D.M.M.D. DRUG THERAPY: Drug-Induced Prolongation of the QT Interval. New Engl. J. Med. 2004, 350, 1013–1022. [Google Scholar]
- Szabo, B.; Sweidan, R.; Rajagopalan, C.V.; Lazzara, R. Role of Na+:Ca2+ exchange current in Cs+-induced early afterdepolarizations in Purkinje fibers. J. Cardiovasc. Electrophysiol. 1994, 5, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Surawicz, B. Role of potassium channels in cycle length dependent regulation of action potential duration in mammalian cardiac Purkinje and ventricular muscle fibres. Cardiovasc. Res. 1992, 26, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Dun, W.; Boyden, P.A. The Purkinje cell; 2008 style. J. Mol. Cell. Cardiol. 2008, 45, 617–624. [Google Scholar] [CrossRef] [PubMed]
- András, V.; Tomek, J.; Nagy, N.; Virág, L.; Passini, E.; Rodriguez, B.; Baczkó, I. Cardiac transmembrane ion channels and action potentials: Cellular physiology and arrhythmogenic behavior. Physiol. Rev. 2021, 101, 1083–1176. [Google Scholar]
- Damiano, B.P.; Rosen, M.R. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation 1984, 69, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Monserrat, M.; Saiz, J.; Ferrero, J.M.; Torres, V.; Ferrero, J.M.; Thakor, N.V. Influence of Purkinje-Muscle Coupling on EAD Development: A Simulation Study. In Proceedings of the Computers in Cardiology 1997, Lund, Sweden, 7–10 September 1997; pp. 513–516. [Google Scholar]
- Boyden, P.A.; Hirose, M.; Dun, W. Cardiac Purkinje cells. Heart Rhythm. 2010, 7, 127–135. [Google Scholar] [CrossRef]
- Iyer, V.; Sampson, K.J.; Kass, R.S. Modeling tissue- and mutation- specific electrophysiological effects in the long QT syndrome: Role of the Purkinje fiber. PLoS ONE 2014, 9, e97720. [Google Scholar] [CrossRef] [PubMed]
- Boyden, P.A. Purkinje physiology and pathophysiology. J. Interv. Card. Electrophysiol. 2018, 52, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Zhang, Y.; Gao, H.; Nattel, S.; Wang, Z. Impairment of HERG K+ channel function by tumor necrosis factor-alpha: Role of reactive oxygen species as a mediator. J. Biol. Chem. 2004, 279, 13289–13292. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, G.; Alarcon, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepulveda, M.; et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Fei, Y.D.; Kim, T.Y.; Xie, A.; Batai, K.; Greener, I.; Tang, H.; Ciftci-Yilmaz, S.; Juneman, E.; Indik, J.H.; et al. IL-18 mediates sickle cell cardiomyopathy and ventricular arrhythmias. Blood 2021, 137, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Heymes, C.; Corbaz, A.; Logeart, D.; Alouani, S.; Cohen-Solal, A.; Seidler, T.; Hasenfuss, G.; Chvatchko, Y.; Shah, A.M.; et al. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. Faseb J. 2004, 18, 1752–1754. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.C.; Mezzaroma, E.; Van Tassell, B.W.; Marchetti, C.; Carbone, S.; Abbate, A.; Toldo, S. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol. Med. 2014, 20, 221–229. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; O’Brien, L.; Marchetti, C.; Seropian, I.M.; Voelkel, N.F.; Van Tassell, B.W.; Dinarello, C.A.; Abbate, A. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1025–H1031. [Google Scholar] [CrossRef]
- Vm, M.; Al, S.; Aa, A.; As, Z.; Av, K.; Rs, O.; Im, M.; Ga, K. Circulating interleukin-18: Association with IL-8, IL-10 and VEGF serum levels in patients with and without heart rhythm disorders. Int. J. Cardiol. 2016, 215, 105–109. [Google Scholar] [CrossRef]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef]
- Narazaki, M.; Kishimoto, T. Current status and prospects of IL-6–targeting therapy. Expert. Rev. Clin. Pharmacol. 2022, 15, 575–592. [Google Scholar] [CrossRef]
- Alvi, R.M.; Frigault, M.J.; Fradley, M.G.; Jain, M.D.; Mahmood, S.S.; Awadalla, M.; Lee, D.H.; Zlotoff, D.A.; Zhang, L.; Drobni, Z.D.; et al. Cardiovascular Events among Adults Treated with Chimeric Antigen Receptor T-Cells (CAR-T). J. Am. Coll. Cardiol. 2019, 74, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.F.; Ettich, J.; Weitz, H.T.; Krusche, M.; Floss, D.M.; Scheller, J.; Moll, J.M. Exclusive inhibition of IL-6 trans-signaling by soluble gp130 (FlyR) Fc. Cytokine X 2021, 3, 100058. [Google Scholar] [CrossRef] [PubMed]
- George, M.J.; Jasmin, N.H.; Cummings, V.T.; Richard-Loendt, A.; Launchbury, F.; Woollard, K.; Turner-Stokes, T.; Garcia Diaz, A.I.; Lythgoe, M.; Stuckey, D.J.; et al. Selective Interleukin-6 Trans-Signaling Blockade Is More Effective than Panantagonism in Reperfused Myocardial Infarction. JACC Basic. Transl. Sci. 2021, 6, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Conceicao, M.; Forcina, L.; Wiklander, O.P.B.; Gupta, D.; Nordin, J.Z.; Vrellaku, B.; McClorey, G.; Mager, I.; Grgens, A.; Lundin, P.; et al. Engineered extracellular vesicle decoy receptor-mediated modulation of the IL6 trans-signalling pathway in muscle. Biomaterials 2021, 266, 120435. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.J.; Zhang, Z.; Jung, D.Y.; Jun, J.Y.; Ma, Z.; Jones, K.E.; Chan, S.Y.; Kim, J.K. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 2009, 58, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Zhang, S.; Yang, S.; Lu, Y.; Lu, K.; Wu, Y.; Wu, Q.; Zhao, N.; Dong, Q.; Chen, L.; et al. Interleukin-6-Mediated-Ca2+ Handling Abnormalities Contributes to Atrial Fibrillation in Sterile Pericarditis Rats. Front. Immunol. 2021, 12, 758157. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar]
- Kotake, S.; Sato, K.; Kim, K.J.; Takahashi, N.; Udagawa, N.; Nakamura, I.; Yamaguchi, A.; Kishimoto, T.; Suda, T.; Kashiwazaki, S. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J. Bone Miner. Res. 1996, 11, 88–95. [Google Scholar] [CrossRef]
- Campbell, L.; Chen, C.; Bhagat, S.S.; Parker, R.A.; Ostor, A.J. Risk of adverse events including serious infections in rheumatoid arthritis patients treated with tocilizumab: A systematic literature review and meta-analysis of randomized controlled trials. Rheumatology 2011, 50, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Beaulieu, A.; Rubbert-Roth, A.; Ramos-Remus, C.; Rovensky, J.; Alecock, E.; Woodworth, T.; Alten, R.; Investigators, O. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): A double-blind, placebo-controlled, randomised trial. Lancet 2008, 371, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Jones, G. Anti-interleukin-6 receptor antibody treatment in inflammatory autoimmune diseases. Rev. Recent Clin. Trials 2006, 1, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, S.Y.; Kawakami, A.; Yamasaki, S.; Imazato, T.; Iwamoto, N.; Fujikawa, K.; Aramaki, T.; Tamai, M.; Nakamura, H.; Ida, H.; et al. Effects of the anti-interleukin-6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis. Rheumatol. Int. 2011, 31, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, S.; Wang, B.; Chen, H.; Li, Y.; Cao, Q.; Zhong, J.; Xie, M.; Ran, Z.; Tang, T.; et al. 775b Olamkicept, an IL-6 Trans-Signaling Inhibitor, is Effective for Induction of Response and Remission in A Randomized, Placebo-Controlled Trial in Moderate to Severe Ulcerative Colitis. Gastroenterology 2021, 161, e28–e29. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, S.; Wang, B.; Chen, H.; Li, Y.; Cao, Q.; Zhong, J.; Xie, M.; Ran, Z.; Tang, T.; et al. DOP01 Efficacy and safety of the IL-6 trans-signalling inhibitor olamkicept: A phase 2 randomized, placebo-controlled trial in moderately to severely active Ulcerative Colitis. J. Crohn’s Colitis 2021, 15 (Suppl. S1), S041–S042. [Google Scholar] [CrossRef]
- Schreiber, S.; Aden, K.; Bernardes, J.P.; Conrad, C.; Tran, F.; Hoper, H.; Volk, V.; Mishra, N.; Blase, J.I.; Nikolaus, S.; et al. Therapeutic Interleukin-6 Trans-signaling Inhibition by Olamkicept (sgp130Fc) in Patients With Active Inflammatory Bowel Disease. Gastroenterology 2021, 160, 2354–2366.e11. [Google Scholar] [CrossRef] [PubMed]
- Schulte, D.M.; Waetzig, G.H.; Schuett, H.; Marx, M.; Schulte, B.; Garbers, C.; Lokau, J.; Vlacil, A.-K.; Schulz, J.; Seoudy, A.K.; et al. Case Report: Arterial Wall Inflammation in Atherosclerotic Cardiovascular Disease is Reduced by Olamkicept (sgp130Fc). Front. Pharmacol. 2022, 13, 758233. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, X.; Chen, X.; Peng, S.; Chen, S.; Zhou, G.; Wei, Y.; Lu, X.; Zhou, C.; Ye, Y.; et al. Selective blockade of interleukin 6 trans-signaling depresses atrial fibrillation. Heart Rhythm. 2023, 20, 1759–1770. [Google Scholar] [CrossRef]
- Rakonczay, Z., Jr.; Hegyi, P.; Takacs, T.; McCarroll, J.; Saluja, A.K. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut 2008, 57, 259–267. [Google Scholar] [CrossRef]
- Diakos, N.A.; Taleb, I.; Kyriakopoulos, C.P.; Shah, K.S.; Javan, H.; Richins, T.J.; Yin, M.Y.; Yen, C.G.; Dranow, E.; Bonios, M.J.; et al. Circulating and Myocardial Cytokines Predict Cardiac Structural and Functional Improvement in Patients with Heart Failure Undergoing Mechanical Circulatory Support. J. Am. Heart Assoc. 2021, 10, e020238. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.; Aromolaran, K.A.; Aromolaran, A.S. STAT4 Mediates IL-6 Trans-Signaling Arrhythmias in High Fat Diet Guinea Pig Heart. Int. J. Mol. Sci. 2024, 25, 7813. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Yin, L.; Ingelsson, E. Cyclooxygenase-2 inhibitors and cardiovascular risk in a nation-wide cohort study after the withdrawal of rofecoxib. Eur. Heart J. 2012, 33, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Christiansen, C.F.; Mehnert, F.; Rothman, K.J.; Sorensen, H.T. Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: Population based case-control study. BMJ 2011, 343, d3450. [Google Scholar] [CrossRef] [PubMed]
- van der Hooft, C.S.; Heeringa, J.; Brusselle, G.G.; Hofman, A.; Witteman, J.C.; Kingma, J.H.; Sturkenboom, M.C.; Stricker, B.H. Corticosteroids and the risk of atrial fibrillation. Arch. Intern. Med. 2006, 166, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Ruigomez, A.; Rodriguez, L.A. Long-term use of anti-inflammatory drugs and risk of atrial fibrillation. Arch. Intern. Med. 2010, 170, 1450–1455. [Google Scholar] [CrossRef]
- Frustaci, A.; Chimenti, C.; Bellocci, F.; Morgante, E.; Russo, M.A.; Maseri, A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 1997, 96, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Granier, M.; Massin, F.; Pasquie, J.L. Pro- and anti-arrhythmic effects of anti-inflammatory drugs. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2013, 12, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef]
- Shiratori, I.; Matsumoto, M.; Tsuji, S.; Nomura, M.; Toyoshima, K.; Seya, T. Molecular cloning and functional characterization of guinea pig IL-12. Int. Immunol. 2001, 13, 1129–1139. [Google Scholar] [CrossRef]
- Xia, R.; Tomsits, P.; Loy, S.; Zhang, Z.; Pauly, V.; Schuttler, D.; Clauss, S. Cardiac Macrophages and Their Effects on Arrhythmogenesis. Front. Physiol. 2022, 13, 900094. [Google Scholar] [CrossRef] [PubMed]
- Aprikian, O.; Reynaud, D.; Pace-Asciak, C.; Leone, P.; Blancher, F.; Monnard, I.; Darimont, C.; Mace, K. Neonatal dietary supplementation of arachidonic acid increases prostaglandin levels in adipose tissue but does not promote fat mass development in guinea pigs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R2006–R2012. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Freake, H.C.; Fernandez, M.L. Gender and hormonal status affect the regulation of hepatic cholesterol 7alpha-hydroxylase activity and mRNA abundance by dietary soluble fiber in the guinea pig. Atherosclerosis 2002, 163, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Torres-Gonzalez, M.; Volek, J.S.; Sharman, M.; Contois, J.H.; Fernandez, M.L. Dietary carbohydrate and cholesterol influence the number of particles and distributions of lipoprotein subfractions in guinea pigs. J. Nutr. Biochem. 2006, 17, 773–779. [Google Scholar] [CrossRef]
- Fernandez, M.L.; Volek, J.S. Guinea pigs: A suitable animal model to study lipoprotein metabolism, atherosclerosis and inflammation. Nutr. Metab. 2006, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Johnson, D.G. cDNA cloning and expression of guinea pig neutrophil attractant protein-1 (NAP-1). NAP-1 is highly conserved in guinea pig. J. Immunol. 1993, 151, 6225–6236. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shryock, J.C.; Song, Y.; Li, Y.; Antzelevitch, C.; Belardinelli, L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J. Pharmacol. Exp. Ther. 2004, 310, 599–605. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Schuttler, D.; Bapat, A.; Kaab, S.; Lee, K.; Tomsits, P.; Clauss, S.; Hucker, W.J. Animal Models of Atrial Fibrillation. Circ. Res. 2020, 127, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Joukar, S. A comparative review on heart ion channels, action potentials and electrocardiogram in rodents and human: Extrapolation of experimental insights to clinic. Lab. Anim. Res. 2021, 37, 25. [Google Scholar] [CrossRef]
- Souidi, M.; Combettes-Souverain, M.; Milliat, F.; Eckhardt, E.R.; Audas, O.; Dubrac, S.; Parquet, M.; Férézou, J.; Lutton, C. Hamsters predisposed to sucrose-induced cholesterol gallstones (LPN strain) are more resistant to excess dietary cholesterol than hamsters that are not sensitive to cholelithiasis induction. J. Nutr. 2001, 131, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, S.E.; Smith, D.E.; Osgood, D.P.; Lichtenstein, A.H. Study of diet-induced changes in lipoprotein metabolism in two strains of Golden-Syrian hamsters. J. Nutr. 2003, 133, 4183–4188. [Google Scholar] [CrossRef] [PubMed]
- Nistor, A.; Bulla, A.; Filip, D.A.; Radu, A. The hyperlipidemic hamster as a model of experimental atherosclerosis. Atherosclerosis 1987, 68, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Lodge, N.J.; Normandin, D.E. Alterations in Ito1, IKr and Ik1 density in the BIO TO-2 strain of syrian myopathic hamsters. J. Mol. Cell Cardiol. 1997, 29, 3211–3221. [Google Scholar] [CrossRef]
- Li, R.; Miao, J.; Tabaran, A.F.; O’Sullivan, M.G.; Anderson, K.J.; Scott, P.M.; Wang, Z.; Cormier, R.T. A novel cancer syndrome caused by KCNQ1-deficiency in the golden Syrian hamster. J. Carcinog. 2018, 17, 6. [Google Scholar] [PubMed]
- Wu, S.H.; Chen, Y.C.; Higa, S.; Lin, C.I. Oscillatory transient inward currents in ventricular myocytes of healthy versus myopathic Syrian hamster. Clin. Exp. Pharmacol. Physiol. 2004, 31, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.; Blythe, H.; Wood, E.G.; Sandhar, B.; Sarker, S.J.; Balmforth, D.; Ambekar, S.G.; Yap, J.; Edmondson, S.J.; Di Salvo, C.; et al. Obesity and diabetes are major risk factors for epicardial adipose tissue inflammation. JCI Insight 2021, 6, e145495. [Google Scholar] [CrossRef]
- Conte, M.; Petraglia, L.; Poggio, P.; Valerio, V.; Cabaro, S.; Campana, P.; Comentale, G.; Attena, E.; Russo, V.; Pilato, E.; et al. Inflammation and Cardiovascular Diseases in the Elderly: The Role of Epicardial Adipose Tissue. Front. Med. 2022, 9, 844266. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Konwerski, M.; Gąsecka, A.; Opolski, G.; Grabowski, M.; Mazurek, T. Role of Epicardial Adipose Tissue in Cardiovascular Diseases: A Review. Biology 2022, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Ernault, A.C.; Verkerk, A.O.; Bayer, J.D.; Aras, K.; Montanes-Agudo, P.; Mohan, R.A.; Veldkamp, M.; Rivaud, M.R.; de Winter, R.; Kawasaki, M.; et al. Secretome of atrial epicardial adipose tissue facilitates reentrant arrhythmias by myocardial remodeling. Heart Rhythm. 2022, 19, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Ramos Muniz, M.G.; Palfreeman, M.; Setzu, N.; Sanchez, M.A.; Saenz Portillo, P.; Garza, K.M.; Gosselink, K.L.; Spencer, C.T. Obesity Exacerbates the Cytokine Storm Elicited by Francisella tularensis Infection of Females and Is Associated with Increased Mortality. Biomed. Res. Int. 2018, 2018, 3412732. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Nakagawa, M.; Watanabe, N.; Nakamura, M. Ubiquitin-like protein MNSFbeta covalently binds to Bcl-G and enhances lipopolysaccharide/interferon gamma-induced apoptosis in macrophages. FEBS J. 2013, 280, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Garg, V.; Shrestha, R.; Sanguinetti, M.C.; Kamp, T.J.; Wu, J.C. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes as Models for Cardiac Channelopathies: A Primer for Non-Electrophysiologists. Circ. Res. 2018, 123, 224–243. [Google Scholar] [CrossRef]
- Pourrier, M.; Fedida, D. The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases. Int. J. Mol. Sci. 2020, 21, 657. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aromolaran, K.A.; Corbin, A.; Aromolaran, A.S. Obesity Arrhythmias: Role of IL-6 Trans-Signaling. Int. J. Mol. Sci. 2024, 25, 8407. https://doi.org/10.3390/ijms25158407
Aromolaran KA, Corbin A, Aromolaran AS. Obesity Arrhythmias: Role of IL-6 Trans-Signaling. International Journal of Molecular Sciences. 2024; 25(15):8407. https://doi.org/10.3390/ijms25158407
Chicago/Turabian StyleAromolaran, Kelly A., Andrea Corbin, and Ademuyiwa S. Aromolaran. 2024. "Obesity Arrhythmias: Role of IL-6 Trans-Signaling" International Journal of Molecular Sciences 25, no. 15: 8407. https://doi.org/10.3390/ijms25158407