
Mechanism of the Oxidative Ring-Closure Reaction during Gliotoxin Biosynthesis by Cytochrome P450 GliF


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
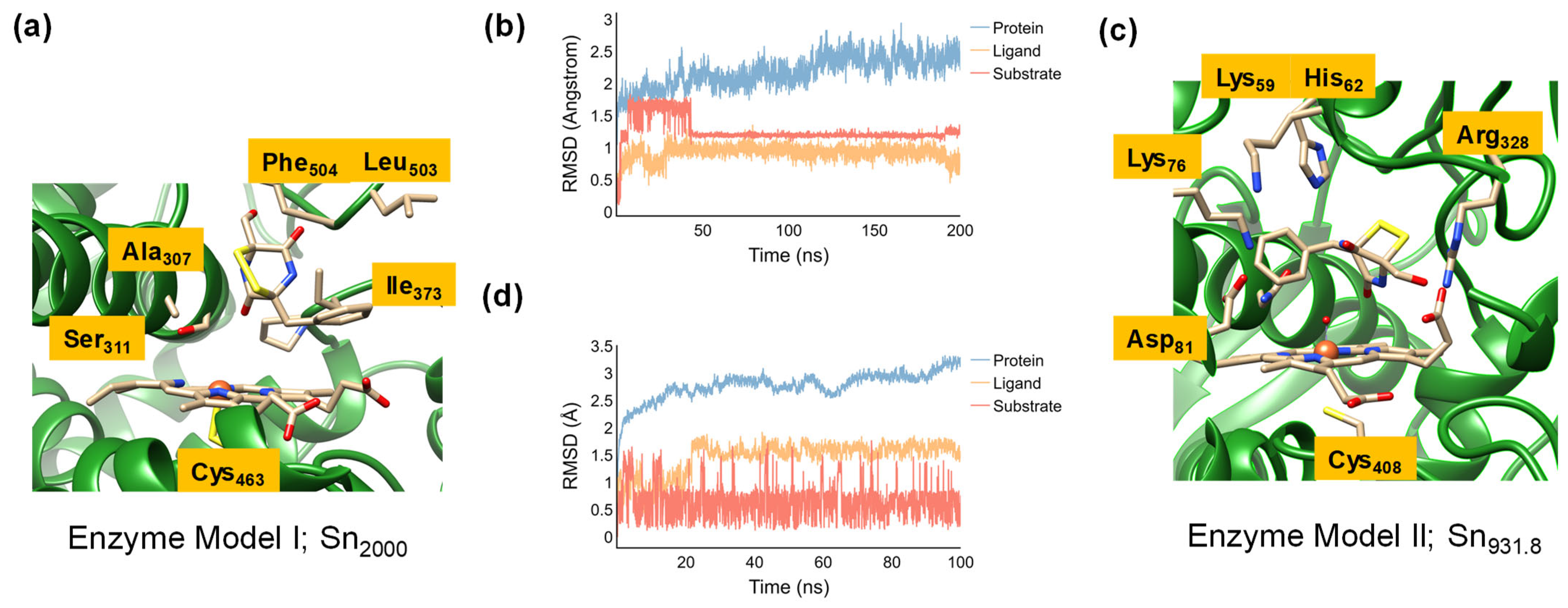
2.1. Model Set-Up and MD Simulations
2.2. CpdI Structure and Electronic Configuration
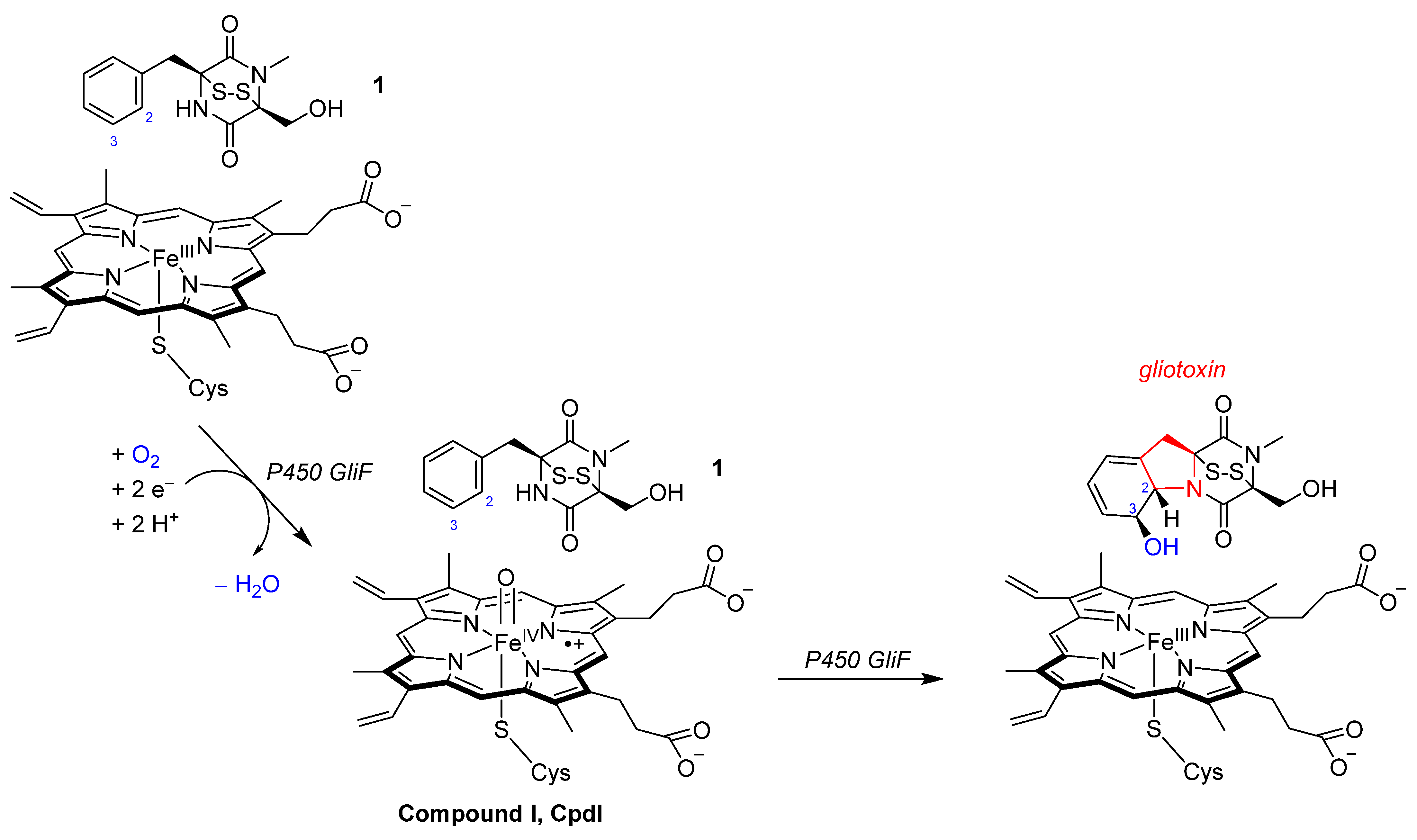
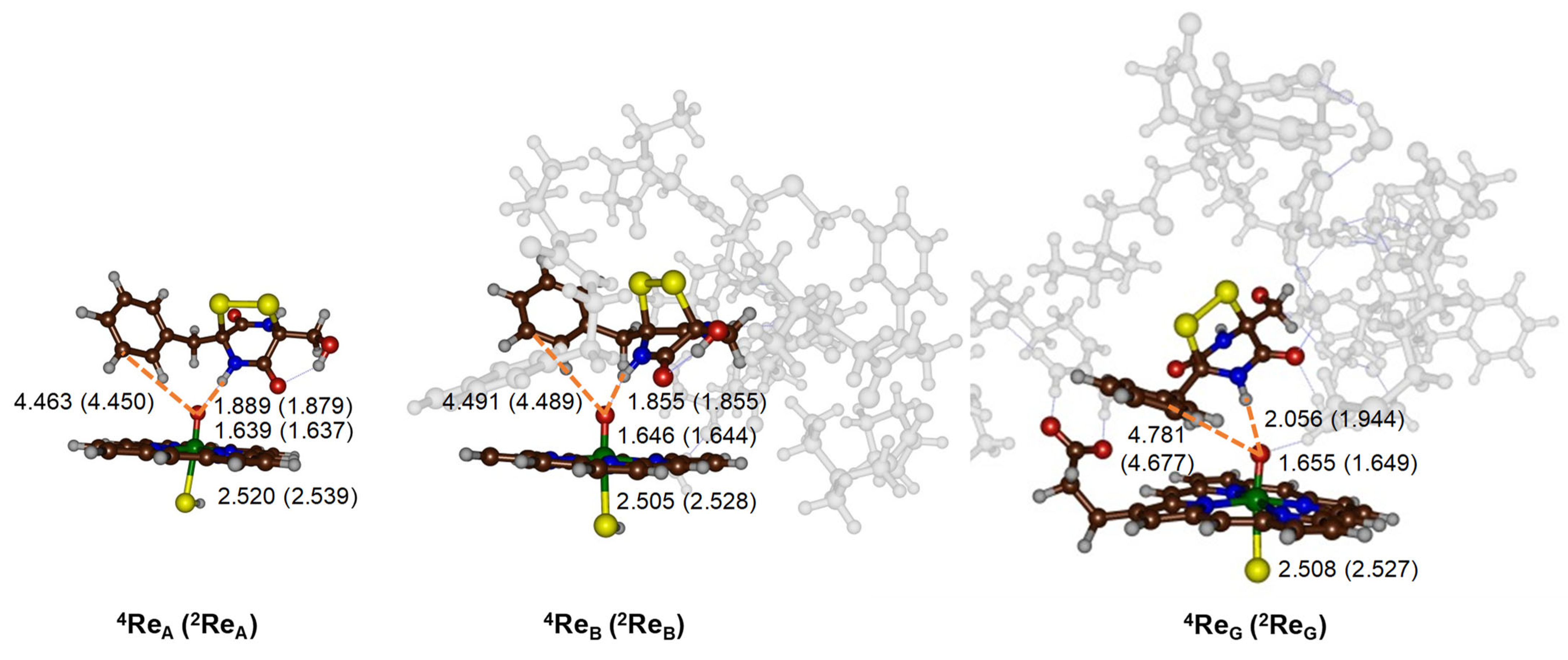
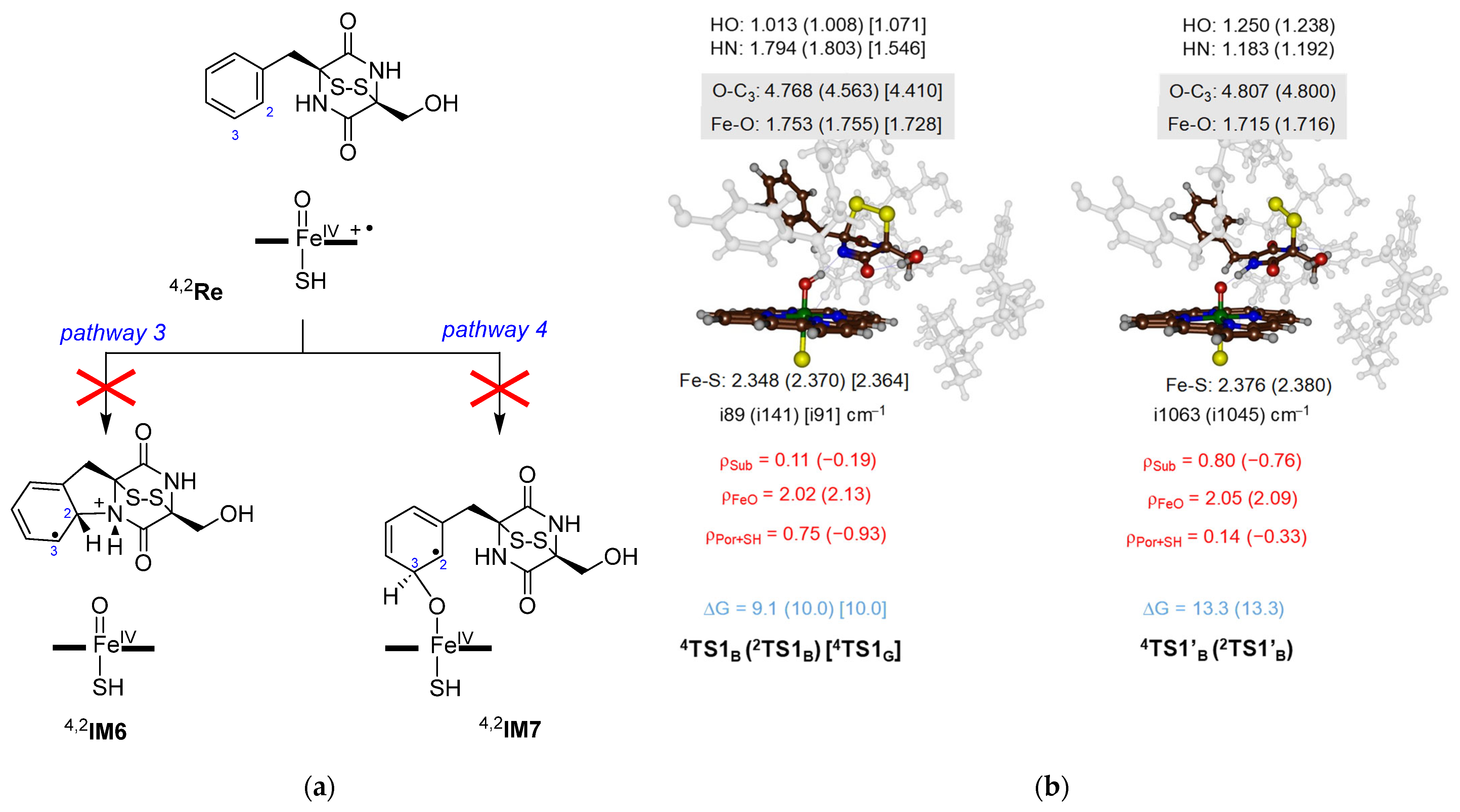
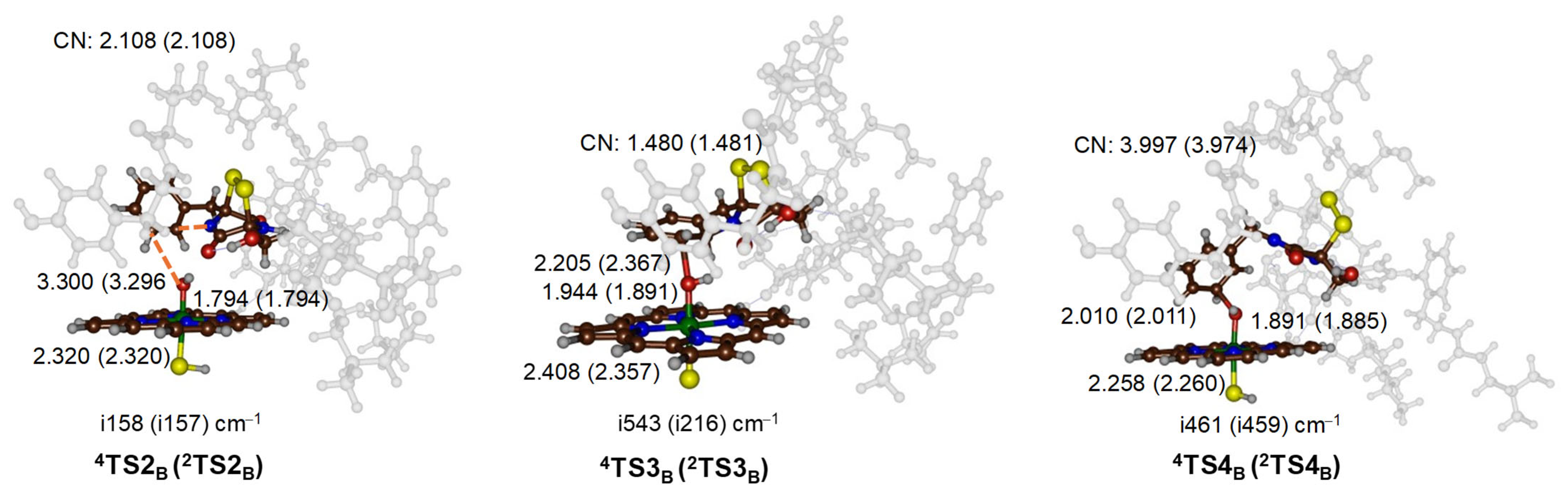
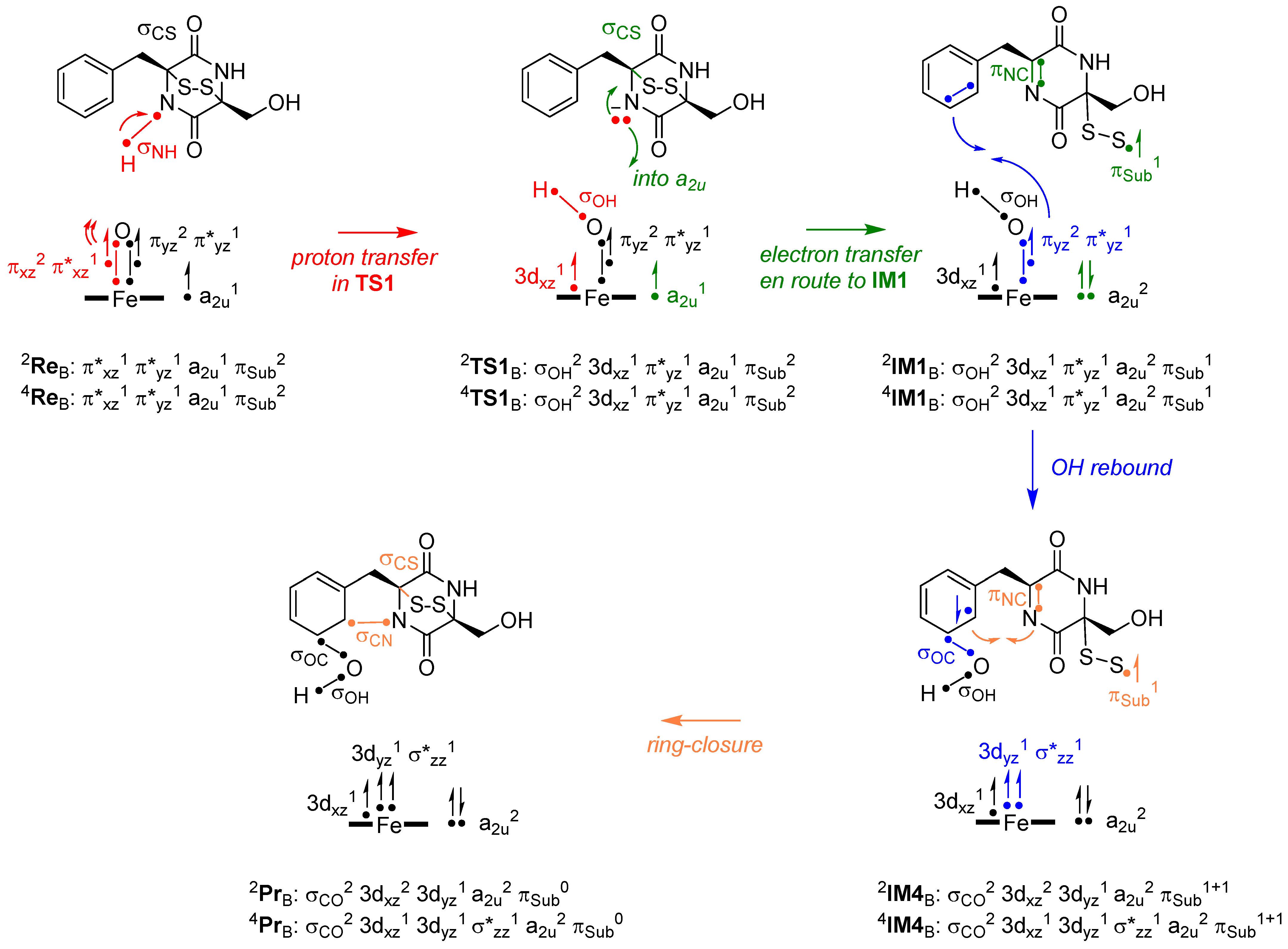
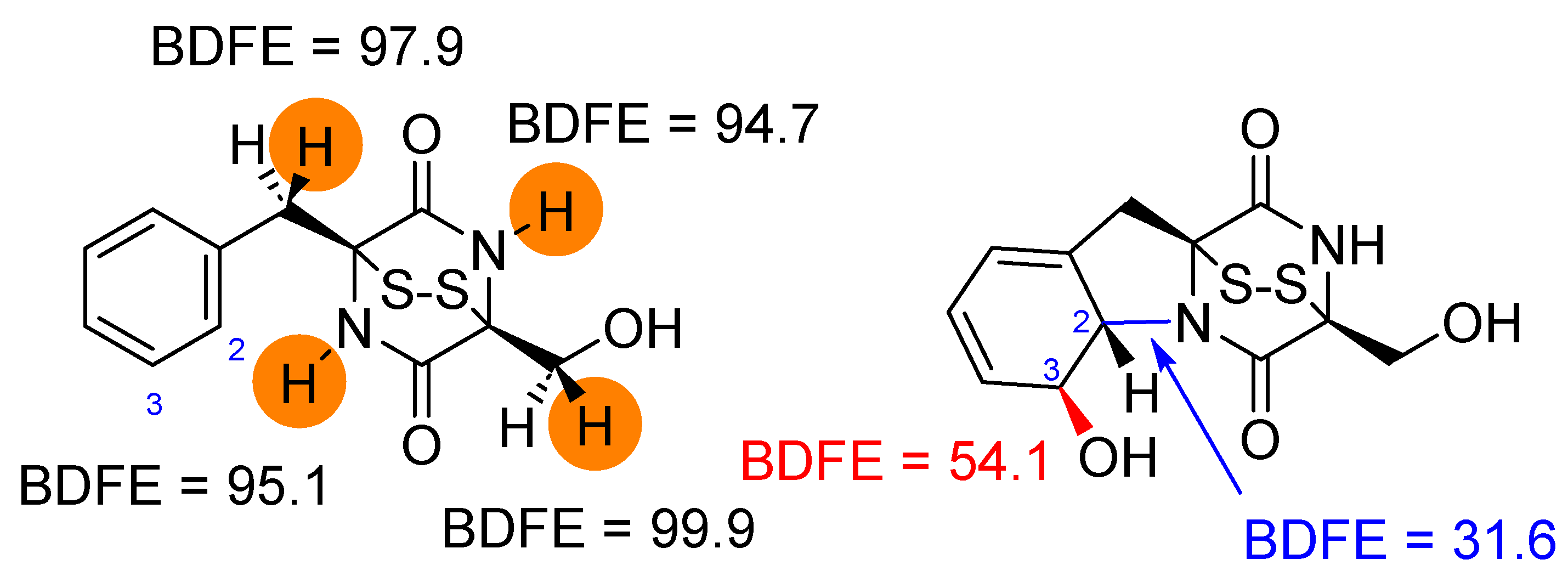
2.3. Oxidative Ring-Closure Mechanism of 1 by CpdI
2.4. Oxidative Ring-Closure Mechanism 1− by CpdI
3. Discussion
4. Materials and Methods
4.1. Structure Preparation
4.2. Substrate Docking
4.3. Molecular Dynamics Simulations
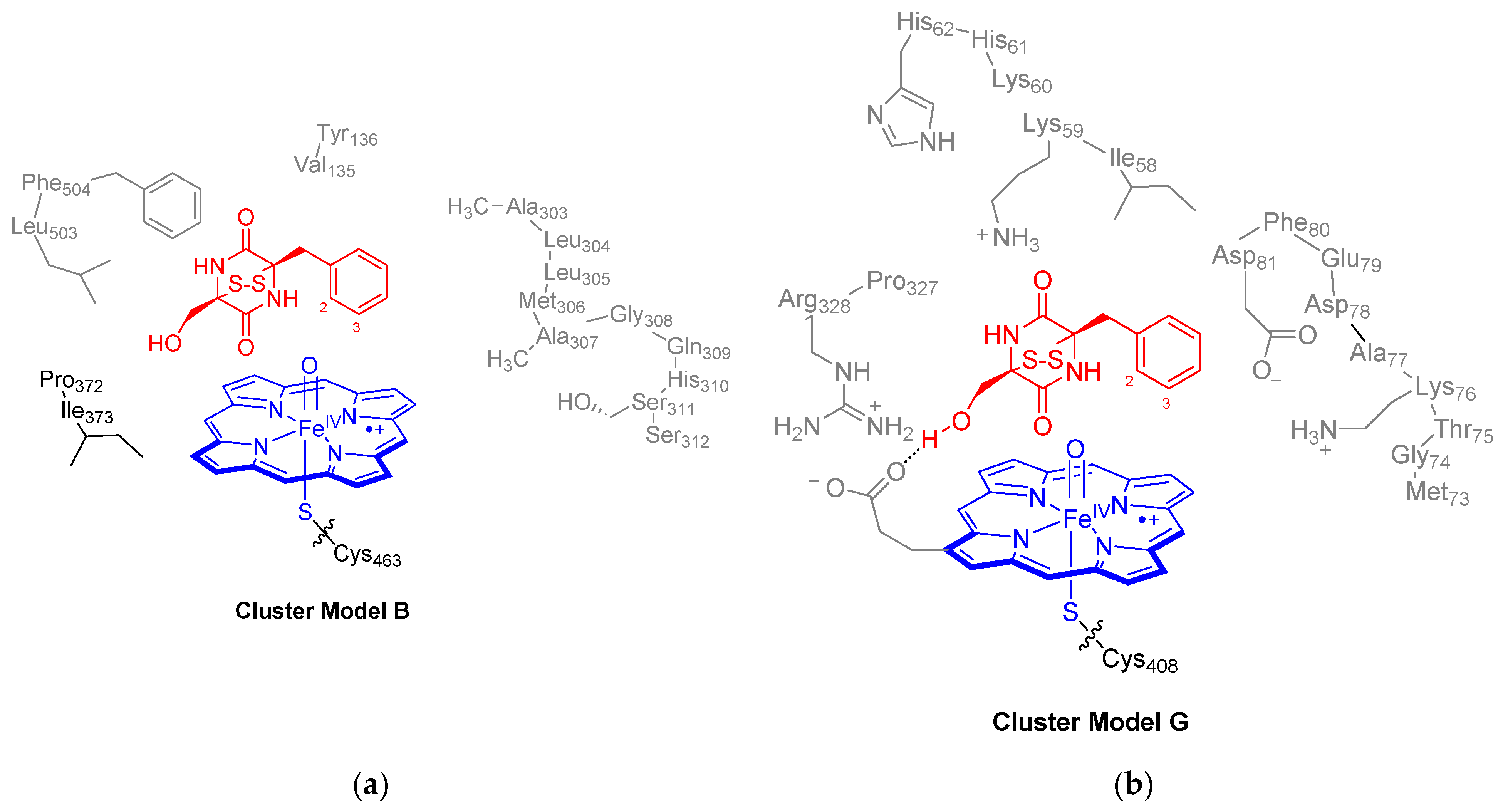
4.4. QM Cluster Model Set-Up
4.5. DFT Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scharf, D.H.; Heinekamp, T.; Remme, N.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. Biosynthesis and Function of Gliotoxin in Aspergillus fumigatus. Appl. Microbiol. Biotechnol. 2012, 93, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Dolan, S.K.; O’Keeffe, G.; Jones, G.W.; Doyle, S. Resistance Is Not Futile: Gliotoxin Biosynthesis, Functionality and Utility. Trends Microbiol. 2015, 23, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Marion, A.; Groll, M.; Scharf, D.H.; Scherlach, K.; Glaser, M.; Sievers, H.; Schuster, M.; Hertweck, C.; Brakhage, A.A.; Antes, I.; et al. Gliotoxin Biosynthesis: Structure, Mechanism, and Metal Promiscuity of Carboxypeptidase GliJ. ACS Chem. Biol. 2017, 12, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Molloy, E.; Hammel, S.; O’Keeffe, G.; Jones, G.W.; Kavanagh, K.; Doyle, S. Gliotoxin Effects on Fungal Growth: Mechanisms and Exploitation. Fungal Genet. Biol. 2012, 49, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Aljofan, M.; Sganga, M.L.; Lo, M.K.; Rootes, C.L.; Porotto, M.; Meyer, A.G.; Saubern, S.; Moscona, A.; Mungall, B.A. Antiviral Activity of Gliotoxin, Gentian Violet and Brilliant Green Against Nipah and Hendra Virus In Vitro. Virol. J. 2009, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.-T.; Lee, J.S.; Qian, Z.-J.; Li, Y.-X.; Kim, K.-N.; Heo, S.-J.; Jeon, Y.-J.; Park, W.S.; Choi, I.-W.; Je, J.-Y.; et al. Gliotoxin Isolated from Marine Fungus Aspergillus sp. Induces Apoptosis of Human Cervical Cancer and Chondrosarcoma Cells. Mar. Drugs 2014, 12, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, C.; Lan, W.; Huang, C.; Lin, M.; Wang, Z.; Liang, W.; Iwamoto, A.; Yang, X.; Liu, H. Gliotoxin Inhibits Proliferation and Induces Apoptosis in Colorectal Cancer Cells. Mar. Drugs 2015, 13, 6259–6273. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.V.M.; Song, Y.W.; Cho, S.K. Effects of the Combination of Gliotoxin and Adriamycin on the Adriamycin-Resistant Non-Small-Cell Lung Cancer A549 Cell Line. Mar. Drugs 2018, 16, 105. [Google Scholar] [CrossRef]
- Park, G.-B.; Jeong, J.-Y.; Kim, D. Gliotoxin Enhances Autophagic Cell Death via the DAPK1-TAp63 Signaling Pathway in Paclitaxel-Resistant Ovarian Cancer Cells. Mar. Drugs 2019, 17, 412. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.M.; Howlett, B.J. Bioinformatic and Expression Analysis of the Putative Gliotoxin Biosynthetic Gene Cluster of Aspergillus fumigatus. FEMS Microbiol. Lett. 2005, 248, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Balibar, C.J.; Walsh, C.T. GliP, a Multimodular Nonribosomal Peptide Synthetase in Aspergillus fumigatus, Makes the Diketopiperazine Scaffold of Gliotoxin. Biochemistry 2006, 45, 15029–15038. [Google Scholar] [CrossRef] [PubMed]
- Scharf, D.H.; Chankhamjon, P.; Scherlach, K.; Dworschak, J.; Roth, M.; Brakhage, A.A.; Hertweck, C. N-Heterocyclization in Gliotoxin Biosynthesis is Catalyzed by a Distinct Cytochrome P450 Monooxygenase. ChemBioChem 2021, 22, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wei, P.-L.; Yin, W.-B. Formation of Bridged Disulfide in Epidithiodioxopiperazines. ChemBioChem 2024, 25, e202300770. [Google Scholar] [CrossRef] [PubMed]
- Zerbe, K.; Pylypenko, O.; Vitali, F.; Zhang, W.; Rouset, S.; Heck, M.; Vrijbloed, J.W.; Bischoff, D.; Bister, B.; Süssmuth, R.D.; et al. Crystal Structure of OxyB, a Cytochrome P450 Implicated in an Oxidative Phenol Coupling Reaction during Vancomycin Biosynthesis. J. Biol. Chem. 2002, 277, 47476–47485. [Google Scholar] [CrossRef] [PubMed]
- Cryle, M.J.; Meinhart, A.; Schlichting, I. Structural Characterization of OxyD, a Cytochrome P450 Involved in Beta-Hydroxytyrosine Formation in Vancomycin Biosynthesis. J. Biol. Chem. 2010, 285, 24562–24574. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Cross-linking of aromatic phenolate groups by cytochrome P450 enzymes: A model for the biosynthesis of vancomycin by OxyB. Org. Biomol. Chem. 2020, 18, 4610–4618. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Krauser, J.A.; Guengerich, F.P. Cytochrome P450 3A4-Catalyzed Testosterone 6β-Hydroxylation Stereochemistry, Kinetic Deuterium Isotope Effects, and Rate-Limiting Steps. J. Biol. Chem. 2005, 280, 19496–19506. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (T)Heme—Novel Mechanisms, Redox Partners and Catalytic Functions in the Cytochrome P450 Superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T. C-H Bond Activation in Heme Proteins: The Role of Thiolate Ligation in Cytochrome P450. Curr. Opin. Chem. Biol. 2009, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010. [Google Scholar]
- Ortiz de Montellano, P.R. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A.A. Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme? Chem. Eur. J. 2018, 24, 10840–10849. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Dunham, N.P.; Arnold, F.H. Nature’s Machinery, Repurposed: Expanding the Repertoire of Iron-Dependent Oxygenases. ACS Catal. 2020, 10, 12239–12255. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, J.; Moore, B.S. Minimization of the Thiolactomycin Biosynthetic Pathway Reveals that the Cytochrome P450 Enzyme TlmF Is Required for Five-Membered Thiolactone Ring Formation. ChemBioChem 2017, 18, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Tang, Y. Recent Advances in Enzymatic Complexity Generation: Cyclization Reactions. Biochemistry 2018, 57, 3087–3104. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2019, 58, 6846–6879. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, A.; Sun, D.; Zhao, Y.; Wang, Y. Theoretical Investigation on the Elusive Reaction Mechanism of Spirooxindole Formation Mediated by Cytochrome P450s: A Nascent Feasible Charge-Shift C-O Bond Makes a Difference. J. Phys. Chem. B 2021, 125, 8419–8430. [Google Scholar] [CrossRef] [PubMed]
- Morita, I.; Mori, T.; Abe, I. Enzymatic Formation of Indolactam Scaffold by C-N Bond-Forming Cytochrome P450 Oxidases in Teleocidin Biosynthesis. Chem. Eur. J. 2021, 27, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Q.; He, J.; Li, H.; Pan, X.; Li, Z.; Xu, H.-M.; Rudolf, J.D.; Tantillo, D.J.; Dong, L.-B. Cytochrome P450 Mediated Cyclization in Eunicellane Derived Diterpenoid Biosynthesis. Angew. Chem. Int. Ed. 2023, 62, e202312490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. Oxygen Economy of Cytochrome P450: What Is the Origin of the Mixed Functionality As a Dehydrogenase–Oxidase Enzyme Compared with its Normal Function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Dong, L.; Jiang, X.; Yang, L. Regioselectivity of C-H Activation in Cytochrome P450-Catalyzed Intramolecular Cyclization of Alkylamine Compounds. Int. J. Quantum Chem. 2023, 123, e27193. [Google Scholar] [CrossRef]
- Field, M.J.; Oyala, P.H.; Green, M.T. 17O Electron Nuclear Double Resonance Analysis of Compound I: Inverse Correlation between Oxygen Spin Population and Electron Donation. J. Am. Chem. Soc. 2022, 144, 19272–19283. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active Species of Horseradish Peroxidase (HRP) and Cytochrome P450: Two Electronic Chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Mallinson, S.J.B.; Machovina, M.M.; Silveira, R.L.; Garcia-Borràs, M.; Gallup, N.; Johnson, C.W.; Allen, M.D.; Skaf, M.S.; Crowley, M.F.; Neidle, E.L.; et al. A Promiscuous Cytochrome P450 Aromatic O-Demethylase for Lignin Bioconversion. Nature Commun. 2018, 9, 2487–2499. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Lignin Biodegradation by a Cytochrome P450 Enzyme: A Computational Study into Syringol Activation by GcoA. Chem. Eur. J. 2020, 26, 13093–13102. [Google Scholar] [CrossRef] [PubMed]
- Süssmuth, R.D.; Wohlleben, W. The Biosynthesis of Glycopeptide Antibiotics—A Model for Complex, Non-Ribosomally Synthesized, Peptidic Secondary Metabolites. Appl. Microbiol. Biotechnol. 2004, 63, 344–350. [Google Scholar] [CrossRef]
- Tang, M.-C.; Zou, Y.; Watanabe, K.; Walsh, C.T.; Tang, Y. Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5226–5333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Marschall, E.; Treisman, M.; McKay, A.; Padva, L.; Crüsemann, M.; Nelson, D.R.; Steer, D.L.; Schittenhelm, R.B.; Tailhades, J.; et al. Cytochrome P450Blt Enables Versatile Peptide Cyclisation to Generate Histidine- and Tyrosine-Containing Crosslinked Tripeptide Building Blocks. Angew. Chem. Int. Ed. 2022, 61, e202204957. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Lamb, D.C.; Guengerich, F.P.; Lepesheva, G.I. Structure-Functional Characterization of Cytochrome P450 Sterol 14α-Demethylase (CYP51B) from Aspergillus fumigatus and Molecular Basis for the Development of Antifungal Drugs. J. Biol. Chem. 2015, 290, 23916–23934. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Mokkawes, T.; de Visser, S.P. Caffeine Biodegradation by Cytochrome P450 1A2. What Determines the Product Distributions? Chem. Eur. J. 2023, 29, e202203875. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; de Visser, S.P. Status report on the quantum chemical cluster approach for modeling enzyme reactions. Commun. Chem. 2022, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; de Visser, S.P. Electrostatic perturbations in the substrate-binding pocket of taurine/α-ketoglutarate dioxygenase determine its selectivity. Chem. Eur. J. 2022, 28, e202104167. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Hay, S.; de Visser, S.P. An Active Site Tyr Residue Guides the Regioselectivity of Lysine Hydroxylation by the Nonheme Iron Lysine-4-Hydroxylase enzymes Through Proton-Coupled-Electron-Transfer. J. Am. Chem. Soc. 2024, 146, 11726–11739. [Google Scholar] [CrossRef] [PubMed]
- Hardy, F.G.; Wong, H.P.H.; de Visser, S.P. Computational Study into the Oxidative Ring-Closure Mechanism During the Biosynthesis of Deoxypodophyllotoxin. Chem. Eur. J. 2024, 30, e202400019. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T. Evidence for Sulfur-Based Radicals in Thiolate Compound I Intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Ogliaro, F.; Cohen, S.; de Visser, S.P.; Shaik, S. Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: What kind of a radical is it really? J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar] [CrossRef]
- Schöneboom, J.C.; Lin, H.; Reuter, N.; Thiel, W.; Cohen, S.; Ogliaro, F.; Shaik, S. The Elusive Oxidant Species of Cytochrome P450 Enzymes: Characterization by Combined Quantum Mechanical/Molecular Mechanical (QM/MM) Calculations. J. Am. Chem. Soc. 2002, 124, 8142–8151. [Google Scholar] [CrossRef] [PubMed]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic Structure of Compound I in Human Isoforms of Cytochrome P450 from QM/MM Modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Tahsini, L.; de Visser, S.P.; Kang, H.Y.; Kim, S.J.; Nam, W. The effect of porphyrin ligands on the regioselective dehydrogenation versus epoxidation of olefins by oxoiron(IV) mimics of cytochrome P450. J. Phys. Chem. A 2009, 113, 11713–11722. [Google Scholar] [CrossRef] [PubMed]
- Radoń, M.; Broclawik, E.; Pierloot, K. DFT and Ab Initio Study of Iron-Oxo Porphyrins: May They Have a Low-Lying Iron(V)-Oxo Electromer? J. Chem. Theory Comput. 2011, 7, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Oláh, J.; Mulholland, A.J.; Harvey, J.N. Does Compound I Vary Significantly between Isoforms of Cytochrome P450? J. Am. Chem. Soc. 2011, 133, 15464–15474. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the Enhanced Reactivity of μ-Nitrido-Bridged Diiron(IV)-Oxo Porphyrinoid Complexes Over Cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef]
- Kepp, K.P. Heme Isomers Substantially Affect Heme’s Electronic Structure and Function. Phys. Chem. Chem. Phys. 2017, 19, 22355–22362. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; DeYonker, N.J. QM-Cluster Model Study of the Guaiacol Hydrogen Atom Transfer and Oxygen Rebound with Cytochrome P450 Enzyme GcoA. J. Phys. Chem. B 2021, 125, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Ma, G.; Liu, Y. Theoretical Insights into the Mechanism and Stereoselectivity of Olefin Cyclopropanation Catalyzed by Two Engineered Cytochrome P450 Enzymes. Inorg. Chem. 2018, 57, 11738–11745. [Google Scholar] [CrossRef]
- Phung, Q.M.; Pierloot, K. Low-Lying Electromeric States in Chloro-Ligated Iron(IV)-Oxo Porphyrin as a Model for Compound I, Studied with Second-Order Perturbation Theory Based on Density Matrix Renormalization Group. J. Chem. Theory Comput. 2019, 15, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-selective formation of an iron(IV)-oxo species at the more electron-rich iron atom of heteroleptic µ-nitrido diiron phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.; Gergel, S.; Klaus, C.; Hammer, S.C.; Garcia-Borras, M. Enzymatic Control over Reactive Intermediates Enables Direct Oxidation of Alkenes to Carbonyls by a P450 Iron-Oxo Species. J. Am. Chem. Soc. 2022, 144, 15954–15968. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.H.S.; Yadav, R.; Mokkawes, T.; Kumar, A.; Skaf, M.S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Biotransformation of Bisphenol by Human Cytochrome P450 2C9 Enzymes: A Density Functional Theory Study. Inorg. Chem. 2023, 62, 2244–2256. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Shaik, S. The “push” effect of the thiolate ligand in cytochrome P450: A theoretical gauging. J. Inorg. Biochem. 2002, 91, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Idle, J.; Krausz, K.; Gonzalez, F. Metabolism of Melatonin by Human Cytochromes P450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, Y.; Powell, M.M.; Hylton, S.M.; Hiller, N.W.; Loria, R.; Ding, Y. A Promiscuous Cytochrome P450 Hy-droxylates Aliphatic and Aromatic C-H Bonds of Aromatic 2,5-Diketopiperazines. ChemBioChem 2019, 20, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Coleman, T.; Kirk, A.M.; Lee, J.H.Z.; Doherty, D.Z.; Bruning, J.B.; Krenske, E.H.; De Voss, J.J.; Bell, S.G. Different Geometric Requirements for Cytochrome P450-Catalyzed Aliphatic Versus Aromatic Hydroxylation Results in Chemoselective Oxidation. ACS Catal. 2022, 12, 1258–1267. [Google Scholar] [CrossRef]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Shaik, S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Kamachi, T.; Yoshizawa, K. A Theoretical Study on the Mechanism of Camphor Hydroxylation by Compound I of Cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Schöneboom, S.; Cohen, S.; Lin, H.; Shaik, S.; Thiel, W. Quantum Mechanical/Molecular Mechanical Investigation of the Mechanism of C-H Hydroxylation of Camphor by Cytochrome P450cam: Theory Supports a Two-State Rebound Mechanism. J. Am. Chem. Soc. 2004, 126, 4017–4034. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Lai, W.; Chen, H.; Wang, Y. The Valence Bond Way: Reactivity Patterns of Cytochrome P450 Enzymes and Synthetic Analogs. Acc. Chem. Res. 2010, 43, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Isobe, H.; Yamaguchi, K.; Okumura, M.; Shimada, J. Role of Perferryl—Oxo Oxidant in Alkane Hydroxylation Catalyzed by Cytochrome P450: A Hybrid Density Functional Study. J. Phys. Chem. B 2012, 116, 4713–4730. [Google Scholar] [CrossRef] [PubMed]
- Hirao, H.; Cheong, Z.H.; Wang, X. Pivotal Role of Water in Terminating Enzymatic Function: A Density Functional Theory Study of the Mechanism-Based Inactivation of Cytochromes P450. J. Phys. Chem. B 2012, 116, 7787–7794. [Google Scholar] [CrossRef] [PubMed]
- Taxak, N.; Desai, P.V.; Patel, B.; Mohutsky, M.; Klimkowski, V.J.; Gombar, V.; Bharatam, P.V. Metabolic-intermediate Complex Formation with Cytochrome P450: Theoretical Studies in Elucidating the Reaction Pathway for the Generation of Reactive Nitroso Intermediate. J. Comput. Chem. 2012, 33, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Houghton, K.T.; Żurek, J.; Bathelt, C.M.; Foloppe, N.; de Groot, M.J.; Harvey, J.N.; Mulholland, A.J. Quantum Mechanics/Molecular Mechanics Modeling of Regioselectivity of Drug Metabolism in Cytochrome P450 2C9. J. Am. Chem. Soc. 2013, 135, 8001–8015. [Google Scholar] [CrossRef] [PubMed]
- Elenewski, J.E.; Hackett, J.C. Ab Initio Dynamics of the Cytochrome P450 Hydroxylation Reaction. J. Chem. Phys. 2015, 142, 064307. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Li, H. Hydrogen Abstraction of Camphor Catalyzed by Cytochrome P450cam: A QM/MM Study. J. Phys. Chem. B 2016, 120, 12312–12320. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Shaik, S.; Dubey, K.D. On the Engineering of Reductase-Based Monooxygenase Activity in CYP450 Peroxygenases. Chem. Sci. 2024, 15, 5174–5186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kumar, D.; Yang, C.L.; Han, K.L.; Shaik, S. Theoretical Study of N-Demethylation of Substituted N,N-Dimethylanilines by Cytochrome P450: The Mechanistic Significance of Kinetic Isotope Effect Profiles. J. Phys. Chem. B 2007, 111, 7700–7710. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Tan, L.S. Is the Bound Substrate in Nitric Oxide Synthase Protonated or Neutral and What Is the Active Oxidant That Performs Substrate Hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Ryde, U.; Olsen, L. Sulfoxide, Sulfur, and Nitrogen Oxidation and Dealkylation by Cytochrome P450. J. Chem. Theory Comput. 2008, 4, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Yang, C.; Han, K. Theoretical Study of N-Dealkylation of N-Cyclopropyl-N-Methylaniline Catalyzed by Cytochrome P450: Insight into the Origin of the Regioselectivity. Dalton Trans. 2009, 38, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Hirao, H.; Thellamurege, N.M.; Chuanprasit, P.; Xu, K. Importance of H-Abstraction in the Final Step of Nitrosoalkane Formation in the Mechanism-Based Inactivation of Cytochrome P450 by Amine-Containing Drugs. Int. J. Mol. Sci. 2013, 14, 24692–24705. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Schüürman, G. Model and Mechanism: N-Hydroxylation of Primary Aromatic Amines by Cytochrome P450. Angew. Chem. Int. Ed. 2013, 52, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Li, Z.; Wang, S.; Wang, B. Unravelling the C-C and C-N Coupling Mechanism for the CYP96T1-Catalyzed Biosynthesis of Amaryllidaceae Alkaloids. Mol. Catal. 2023, 550, 113609. [Google Scholar] [CrossRef]
- Satpathy, J.K.; Yadav, R.; Bagha, U.K.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Enhanced Reactivity through Equatorial Sulfur Coordination in Nonheme Iron(IV)-Oxo Complexes: Insights from Experiment and Theory. Inorg. Chem. 2024, 63, 6752–6766. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.-P.; Feng, J.; Wang, Y.; Li, S.; Wang, B. Substrate Conformational Switch Enables the Stereoselective Dimerization in P450 NascB: Insights from Molecular Dynamics Simulations and Quantum Mechanical/Molecular Mechanical Calculations. JACS Au 2024, 4, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Shaik, S. A Proton-Shuttle Mechanism Mediated by the Porphyrin in Benzene Hydroxylation by Cytochrome P450 Enzymes. J. Am. Chem. Soc. 2003, 125, 7413–7424. [Google Scholar] [CrossRef] [PubMed]
- Bathelt, C.M.; Mulholland, A.J.; Harvey, J.N. QM/MM Modelling of Benzene Hydroxylation in Human Cytochrome P450 2C9. J. Phys. Chem. A 2008, 112, 13149–13156. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Milko, P.; Schyman, P.; Usharani, D.; Chen, H. Trends in Aromatic Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes: A Valence Bond Modeling. J. Chem. Theory Comput. 2011, 7, 327–339. [Google Scholar] [CrossRef]
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of Oxidative Activation of Fluorinated Aromatic Compounds by N-Bridged Diiron-Phthalocyanine. What Determines the Reactivity? Chem. Eur. J. 2019, 25, 14320–14331. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P. Substitution of Hydrogen by Deuterium Changes the Regioselectivity of Ethylbenzene Hydroxylation by an Oxo-Iron-Porphyrin Catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Axial Ligand Effect On the Rate Constant of Aromatic Hydroxylation By Iron(IV)-Oxo Complexes Mimicking Cytochrome P450 Enzymes. J. Phys. Chem. B 2012, 116, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A Systematic Account on Aromatic Hydroxylation by a Cytochrome P450 Model Compound I: A Low-Pressure Mass Spectrometry and Computational Study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, J.P.T.; Cummins, D.C.; Mubarak, M.Q.E.; Siegler, M.A.; de Visser, S.P.; Goldberg, D.P. Hydrogen atom abstraction by high-valent Fe(OH) versus Mn(OH) porphyrinoid complexes: Mechanistic insights from experimental and computational studies. Inorg. Chem. 2019, 58, 16761–16770. [Google Scholar] [CrossRef]
- Gérard, E.F.; Yadav, V.; Goldberg, D.P.; de Visser, S.P. What Drives Radical Halogenation Versus Hydroxylation in Mono-nuclear Nonheme Iron Complexes? A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2022, 144, 10752–10767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Diao, W.; Wu, P.; Li, J.; Fu, Y.; Guo, Z.; Cao, Z.; Shaik, S.; Wang, B. How the Conformational Movement of the Substrate Drives the Regioselective C-N Bond Formation in P450 TleB: Insights from Molecular Dynamics Simulations and Quantum Mechanical/Molecular Mechanical Calculations. J. Am. Chem. Soc. 2023, 145, 7252–7267. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y. Multiscale Study on the Intramolecular C-S Bond Formation Catalyzed by P450 Monooxygenase CxnD Involved in the Biosynthesis of Chuangxinmycin: The Critical Roles of Noncrystal Water Molecule and Conformational Change. Inorg. Chem. 2024, 63, 4086–4098. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.; Upadhyay, P.; Faponle, A.S.; Kumar, J.; Nag, S.S.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Deformylation reaction by a nonheme manganese(III)-peroxo complex via initial hydrogen atom abstraction. Angew. Chem. Int. Ed. 2016, 55, 11091–11095. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Barman, P.; Mukherjee, G.; Kumar, J.; Kumar, D.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Keto-enol tautomerization triggers an electrophilic aldehyde deformylation reaction by a nonheme manganese(III)-peroxo complex. J. Am. Chem. Soc. 2017, 139, 18328–18338. [Google Scholar] [CrossRef] [PubMed]
- Bagha, U.K.; Yadav, R.; Mokkawes, T.; Satpathy, J.K.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Defluorination of Fluorophenols by a Nonheme Iron(IV)-Oxo Species: Observation of a New Intermediate Along the Reaction. Chem. Eur. J. 2023, 29, e202300478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mokkawes, T.; de Visser, S.P. Insights into Cytochrome P450 Enzyme Catalyzed Defluorination of Aromatic Fluorides. Angew. Chem. Int. Ed. 2023, 62, e202310785. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The Toxic Exposome Database. Nucl. Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3658. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. University of California San Fransisco (USA) Chimera–A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Träg, J.; Zahn, D. Improved GAFF2 Parameters for Fluorinated Alkanes and Mixed Hydro- and Fluorocarbons. J. Mol. Model. 2019, 25, 39. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Yan, S.; Zhang, X.; Liao, L.; Zhang, J.; Shaik, S.; Wang, B. How Do Preorganized Electric Fields Function in Catalytic Cycles? The Case of the Enzyme Tyrosine Hydroxylase. J. Am. Chem. Soc. 2022, 144, 20484–20494. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; Warwicker, J.; de Visser, S.P. How Does the Nonheme Iron Enzyme NapI React Through L-Arginine Desaturation Rather Than Hydroxylation? A QM/MM Study. ACS Catal. 2023, 13, 10705–10721. [Google Scholar] [CrossRef]
- Louka, S.; Barry, S.M.; Heyes, D.J.; Mubarak, M.Q.E.; Ali, H.S.; Alkhalaf, L.M.; Munro, A.W.; Scrutton, N.S.; Challis, G.L.; de Visser, S.P. The Catalytic Mechanism of Aromatic Nitration by Cytochrome P450 TxtE: Involvement of a Ferric-Peroxynitrite Intermediate. J. Am. Chem. Soc. 2020, 142, 15764–15779. [Google Scholar] [CrossRef] [PubMed]
- Nandy, A.; Adamji, H.; Kastner, D.W.; Vennelakanti, V.; Nazemi, A.; Liu, M.; Kulik, H.J. Using Computational Chemistry To Reveal Nature’s Blueprints for Single-Site Catalysis of C–H Activation. ACS Catal. 2022, 12, 9281–9306. [Google Scholar] [CrossRef]
- Sheng, X.; Himo, F. The Quantum Chemical Cluster Approach in Biocatalysis. Acc. Chem. Res. 2023, 56, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Agbaglo, D.A.; Summers, T.J.; Cheng, Q.; DeYonker, N.J. The Influence of Model Building Schemes and Molecular Dynamics Sampling on QM-Cluster Models: The Chorismate Mutase Case Study. Phys. Chem. Chem. Phys. 2024, 26, 12467–12482. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parameterization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods Without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Yang, T.; Quesne, M.G.; Neu, H.M.; Cantú Reinhard, F.G.; Goldberg, D.P.; de Visser, S.P. Singlet versus Triplet Reactivity in an Mn(V)-Oxo Species: Testing Theoretical Predictions Against Experimental Evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate Sulfoxidation by an Iron(IV)-Oxo Complex: Benchmarking Computationally Calculated Barrier Heights to Experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; Saint-André, M.; de Visser, S.P. Understanding How Prolyl-4-Hydroxylase Structure Steers a Ferryl Oxidant Toward Scission of a Strong C–H Bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qureshi, M.; Mokkawes, T.; Cao, Y.; de Visser, S.P. Mechanism of the Oxidative Ring-Closure Reaction during Gliotoxin Biosynthesis by Cytochrome P450 GliF. Int. J. Mol. Sci. 2024, 25, 8567. https://doi.org/10.3390/ijms25168567
Qureshi M, Mokkawes T, Cao Y, de Visser SP. Mechanism of the Oxidative Ring-Closure Reaction during Gliotoxin Biosynthesis by Cytochrome P450 GliF. International Journal of Molecular Sciences. 2024; 25(16):8567. https://doi.org/10.3390/ijms25168567
Chicago/Turabian StyleQureshi, Muizz, Thirakorn Mokkawes, Yuanxin Cao, and Sam P. de Visser. 2024. "Mechanism of the Oxidative Ring-Closure Reaction during Gliotoxin Biosynthesis by Cytochrome P450 GliF" International Journal of Molecular Sciences 25, no. 16: 8567. https://doi.org/10.3390/ijms25168567