
Correlation between Molecular Docking and the Stabilizing Interaction of HOMO-LUMO: Spirostans in CHK1 and CHK2, an In Silico Cancer Approach
 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
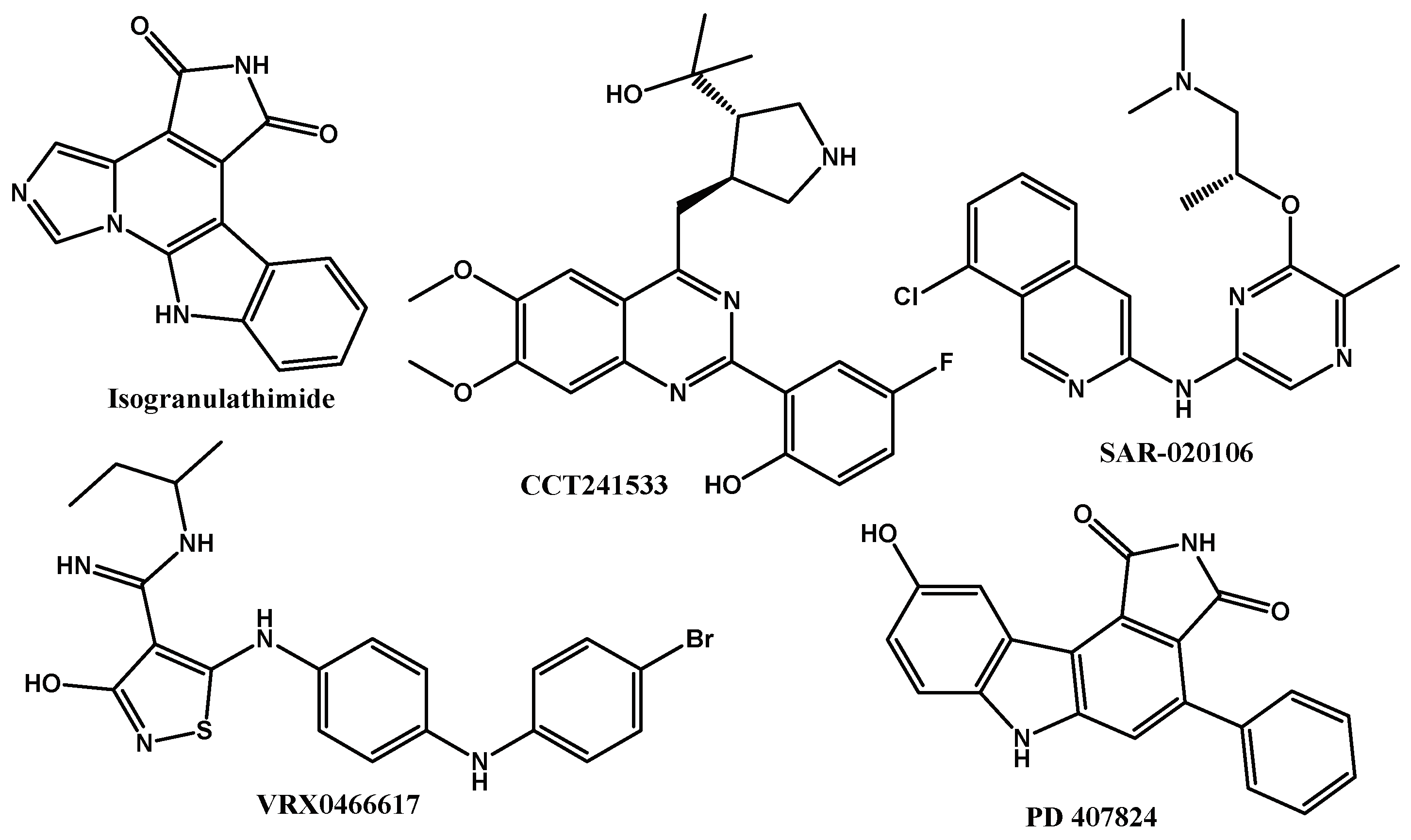
2.1. Reference Docking
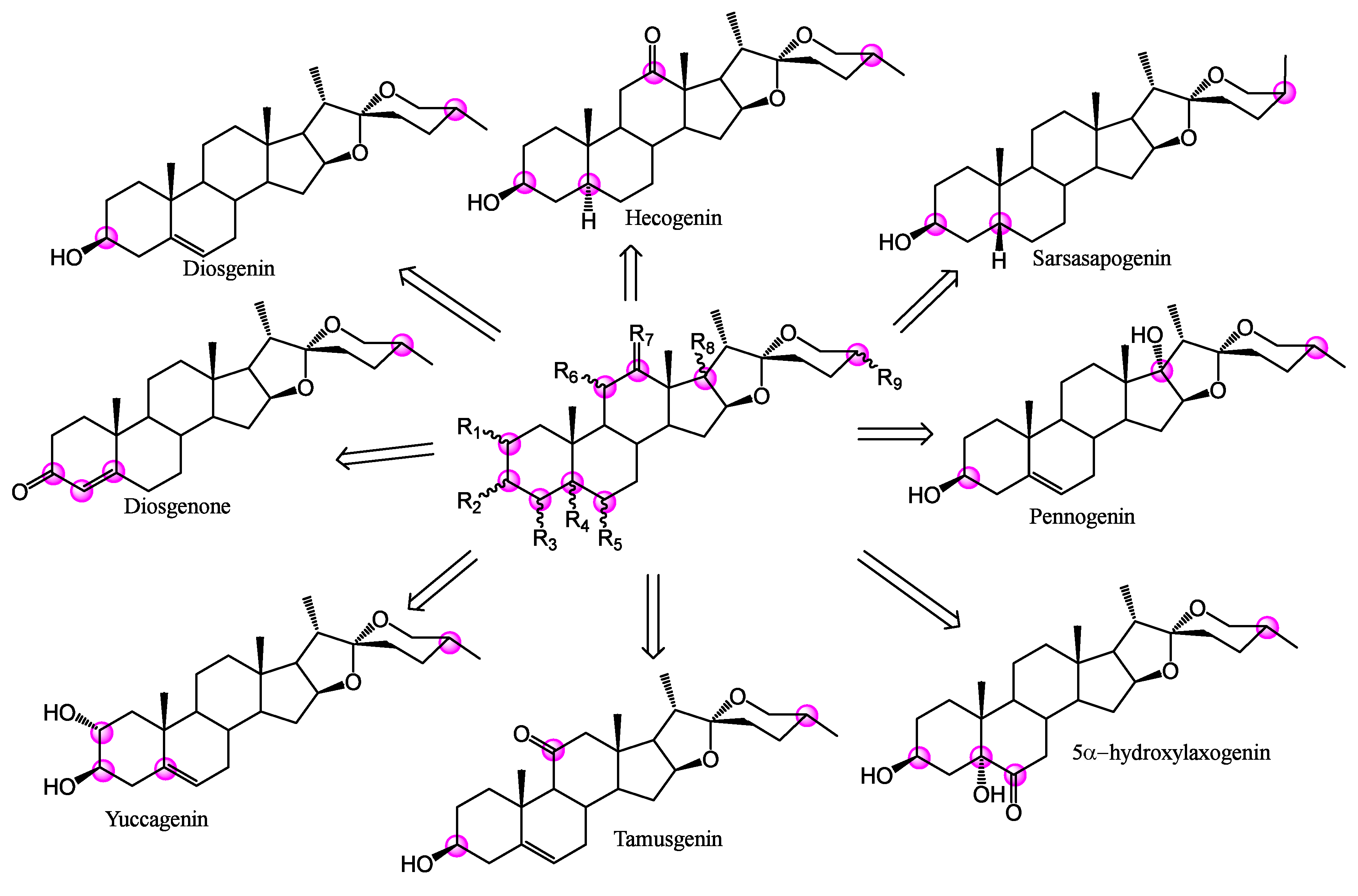
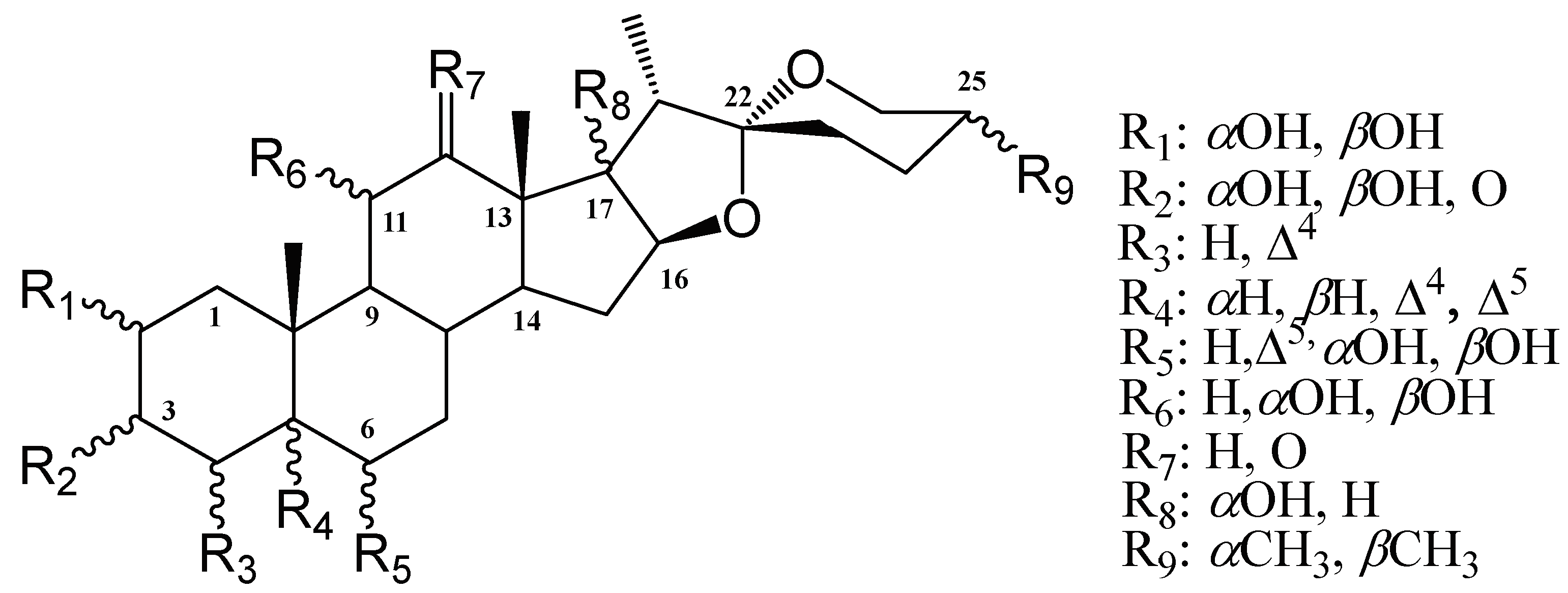
2.2. Spirostans Docking in CHKs Sites
2.3. ADMETx Studies
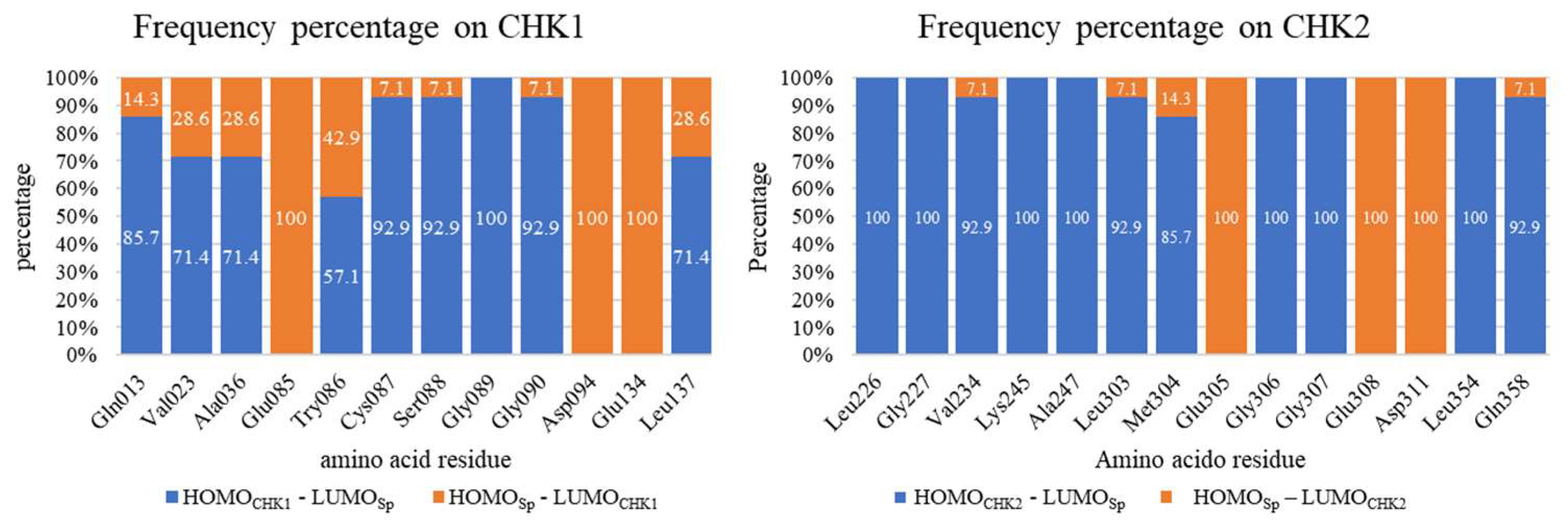
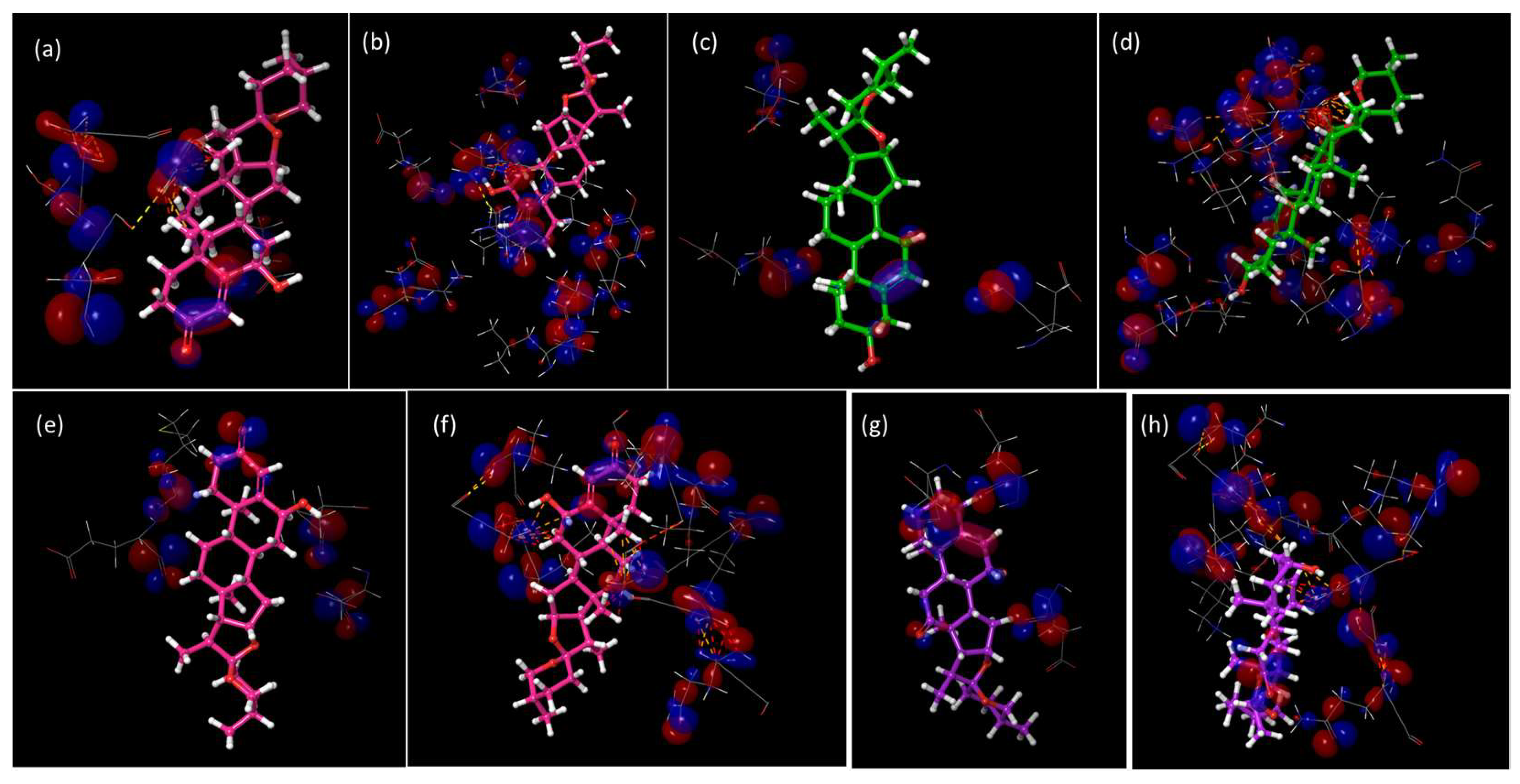
2.4. HOMO vs. LUMO Studies
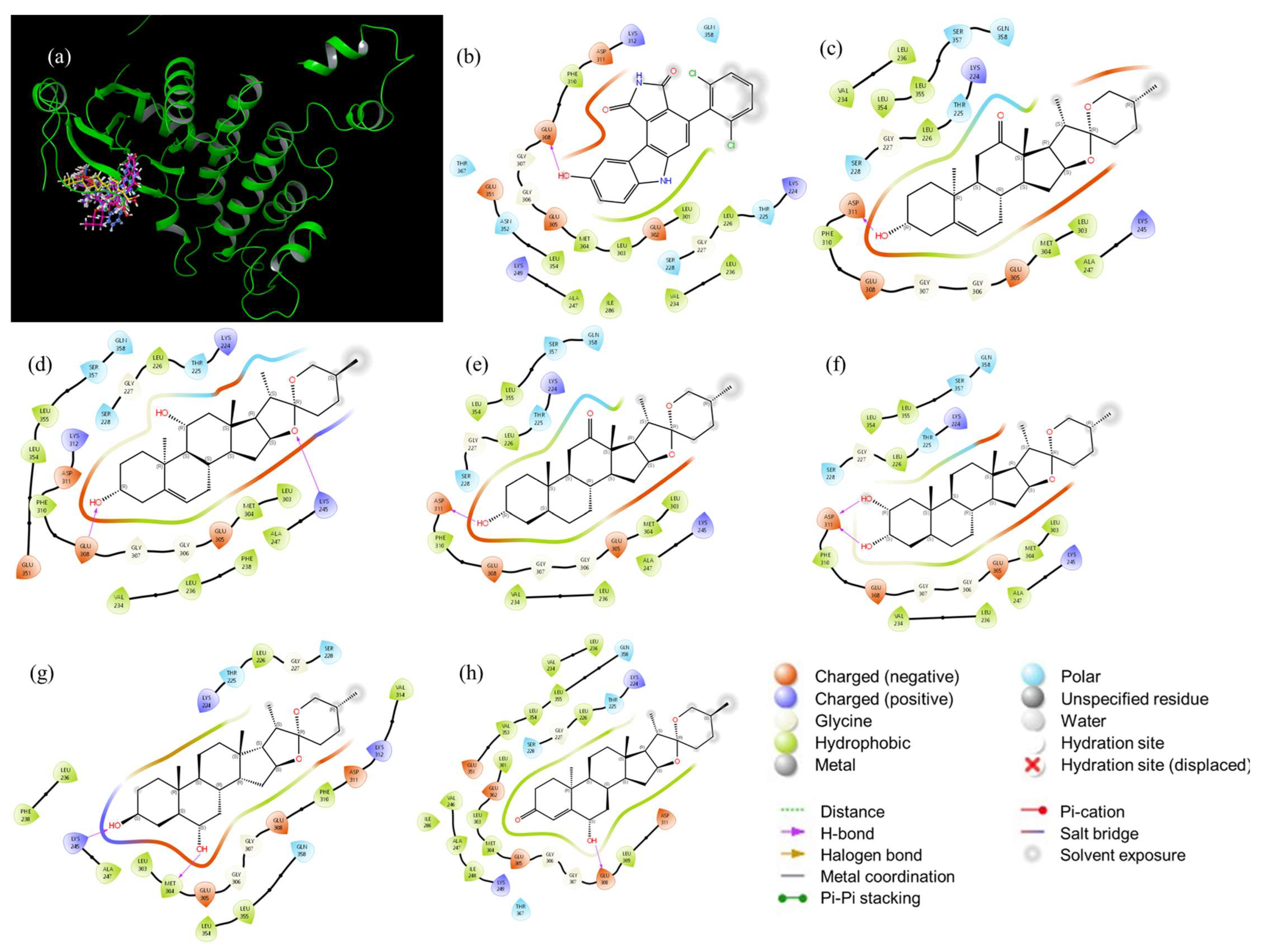
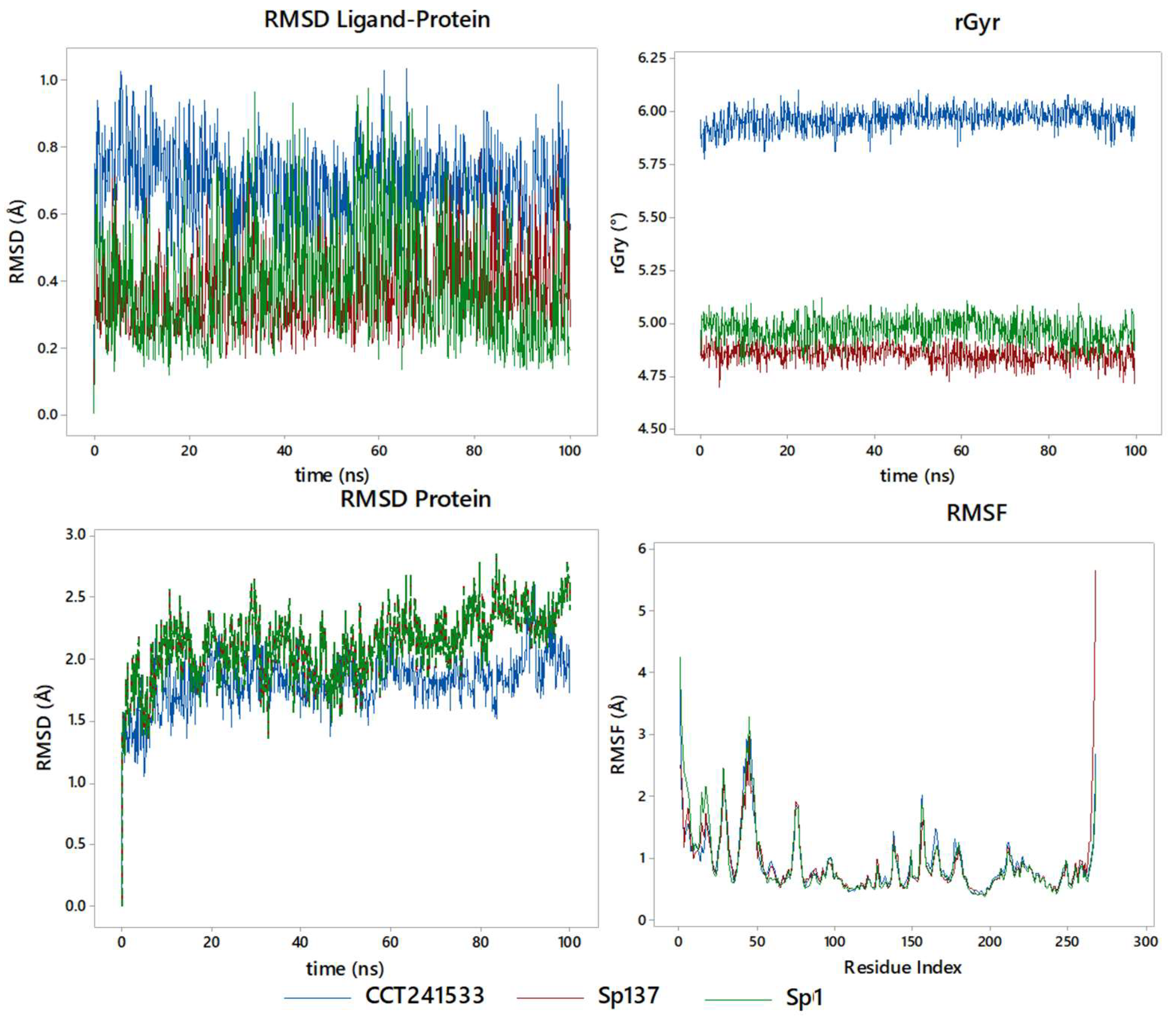
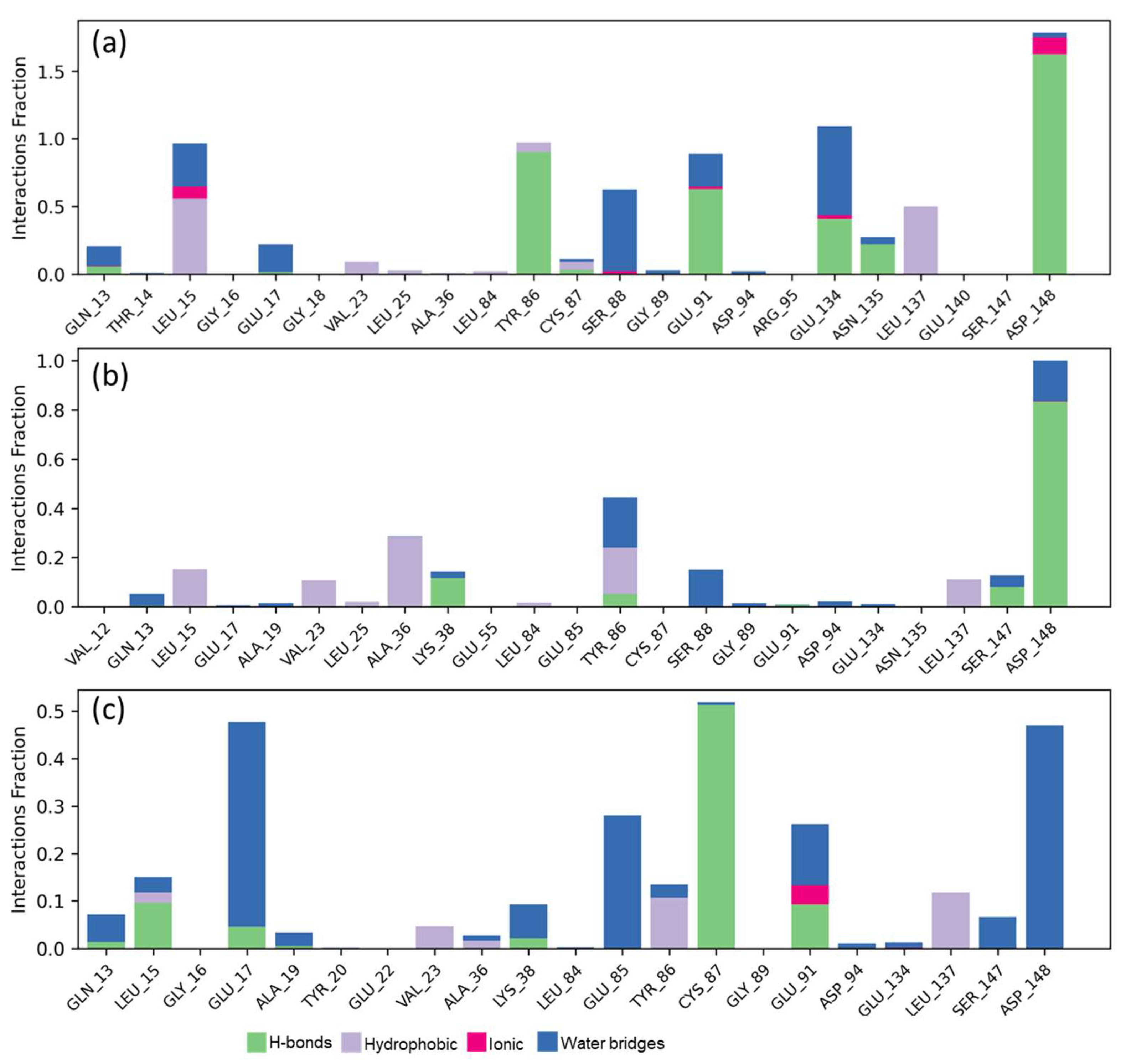
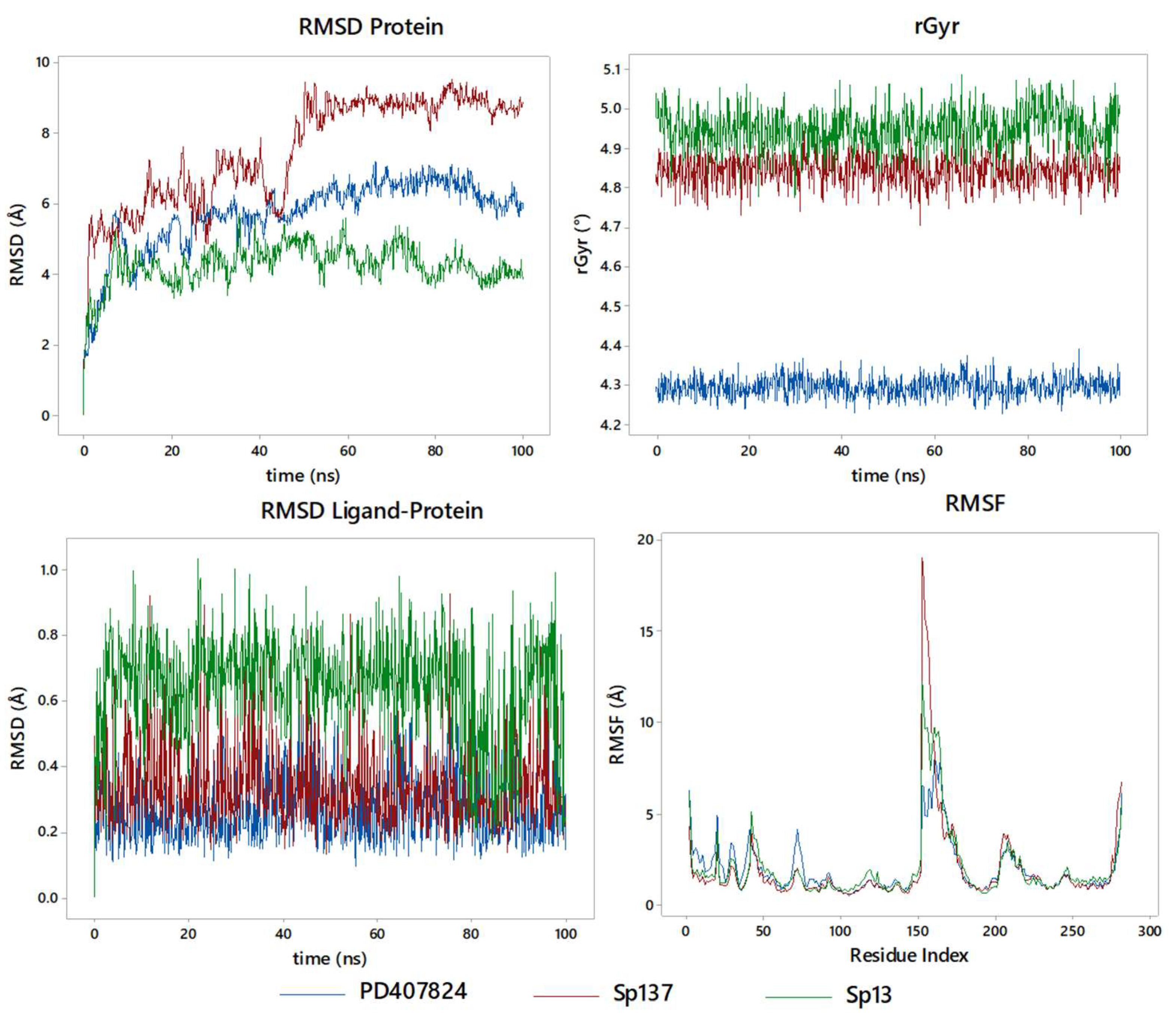
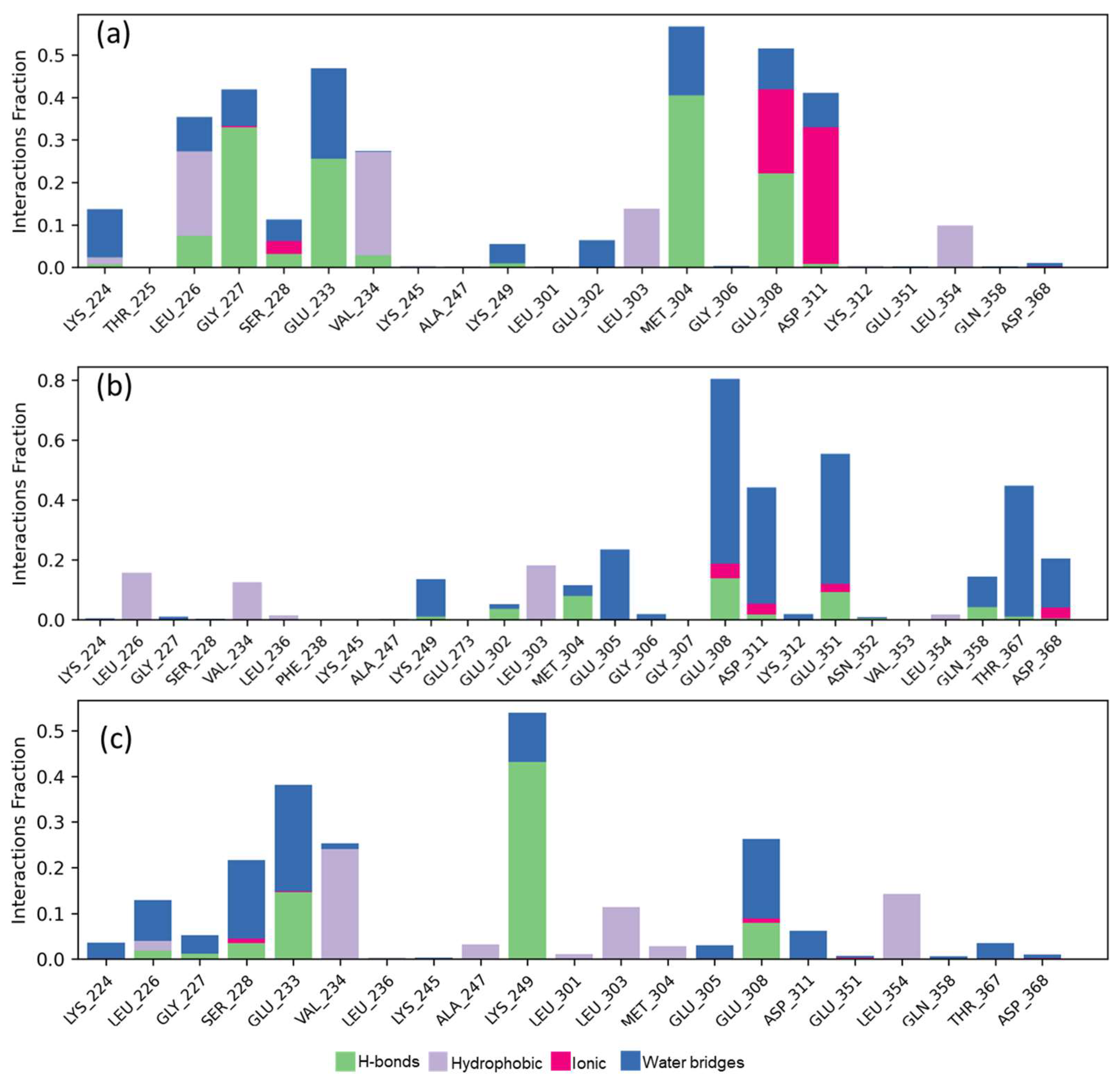
2.5. Molecular Dynamics Studies and Post-MM-GBSA Analysis
3. Materials and Methods
3.1. Protein Preparation and Validation
3.2. Spirostan Preparation
3.3. Density Functional Theory Studies
3.4. Molecular Docking
3.5. ADMETx Studies
3.6. Protein DFT Interaction Site
3.7. Molecular Dynamics
3.8. Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dinoi, G.; Mariani, A.; Martinelli, E.; Ciucci, A.; Zannoni, G.F.; Weaver, A.L.; Keeney, G.L.; Vasmatzis, G.; Anastasiadis, P.Z.; Fanfani, F.; et al. In Search for Biomarkers and Potential Drug Targets for Uterine Serous Endometrial Cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Sofianidi, A.; Dumbrava, E.E.; Syrigos, K.N.; Nasrazadani, A. Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers 2024, 16, 1139. [Google Scholar] [CrossRef] [PubMed]
- Colaco, V.; Goswami, N.; Goel, V.K.; Srivastava, S.K.; Lalrohlua, P.; Senthil Kumar, N.; Borah, P.; Baruah, R.; Varma, A.K. In Silico and Structure-Based Evaluation of Deleterious Mutations Identified in Human Chk1, Chk2, and Wee1 Protein Kinase. J. Cell. Biochem. 2024, 125, e30508. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 Inhibitors and Cancer Therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Mun Tho, L.; Xu, N.; Gillespie, D.A. Chapter 3—The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 108, pp. 73–112. [Google Scholar]
- Jiang, K.; Deng, M.; Du, W.; Liu, T.; Li, J.; Zhou, Y. Functions and Inhibitors of CHK1 in Cancer Therapy. Med. Drug Discov. 2024, 22, 100185. [Google Scholar] [CrossRef]
- Gioia, U.; Tavella, S.; Martínez-Orellana, P.; Cicio, G.; Colliva, A.; Ceccon, M.; Cabrini, M.; Henriques, A.C.; Fumagalli, V.; Paldino, A.; et al. SARS-CoV-2 Infection Induces DNA Damage, through CHK1 Degradation and Impaired 53BP1 Recruitment, and Cellular Senescence. Nat. Cell Biol. 2023, 25, 550–564. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Gao, D.; He, Y.; Ge, N.; Guo, J.; Sun, S.; Wang, C.; Yang, F. Guarding against Digestive-System Cancers: Unveiling the Role of Chk2 as a Potential Therapeutic Target. Genes Dis. 2023, 101191. [Google Scholar] [CrossRef]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 Is a Potent and Selective Inhibitor of CHK2 That Potentiates the Cytotoxicity of PARP Inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Patties, I.; Kallendrusch, S.; Böhme, L.; Kendzia, E.; Oppermann, H.; Gaunitz, F.; Kortmann, R.-D.; Glasow, A. The Chk1 Inhibitor SAR-020106 Sensitizes Human Glioblastoma Cells to Irradiation, to Temozolomide, and to Decitabine Treatment. J. Exp. Clin. Cancer Res. 2019, 38, 420. [Google Scholar] [CrossRef]
- Feng, L.; Cook, B.; Tsai, S.-Y.; Zhou, T.; LaFlamme, B.; Evans, T.; Chen, S. Discovery of a Small-Molecule BMP Sensitizer for Human Embryonic Stem Cell Differentiation. Cell Rep. 2016, 15, 2063–2075. [Google Scholar] [CrossRef]
- Garrett, M.D.; Collins, I. Anticancer Therapy with Checkpoint Inhibitors: What, Where and When? Trends Pharmacol. Sci. 2011, 32, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, L.; Buscemi, G.; Larson, G.; Hong, Z.; Wu, J.Z.; Delia, D. Biochemical and Cellular Characterization of VRX0466617, a Novel and Selective Inhibitor for the Checkpoint Kinase Chk2. Mol. Cancer Ther. 2007, 6, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Semwal, P.; Painuli, S.; Abu-Izneid, T.; Rauf, A.; Sharma, A.; Daştan, S.D.; Kumar, M.; Alshehri, M.M.; Taheri, Y.; Das, R.; et al. Diosgenin: An Updated Pharmacological Review and Therapeutic Perspectives. Oxid. Med. Cell. Longev. 2022, 2022, 1035441. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Gadewar, M.; Tahilyani, V.; Patel, D.K. A Review on Pharmacological and Analytical Aspects of Diosgenin: A Concise Report. Nat. Prod. Bioprospecting 2012, 2, 46–52. [Google Scholar] [CrossRef]
- Liao, W.-L.; Lin, J.-Y.; Shieh, J.-C.; Yeh, H.-F.; Hsieh, Y.-H.; Cheng, Y.-C.; Lee, H.-J.; Shen, C.-Y.; Cheng, C.-W. Induction of G2/M Phase Arrest by Diosgenin via Activation of Chk1 Kinase and Cdc25C Regulatory Pathways to Promote Apoptosis in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 21, 172. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Kuznetsov, A.E.; Pirzada, A.S.; Alsharif, K.F.; Daglia, M.; Khan, H. Computational Pharmacology and Computational Chemistry of 4-Hydroxyisoleucine: Physicochemical, Pharmacokinetic, and DFT-Based Approaches. Front. Chem. 2023, 11, 1145974. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Khan, H.; Serdaroğlu, G. Physicochemical Properties, Drug Likeness, ADMET, DFT Studies, and in Vitro Antioxidant Activity of Oxindole Derivatives. Comput. Biol. Chem. 2023, 104, 107861. [Google Scholar] [CrossRef]
- Lopez-Castillo, G.N.; Alatriste, V.; Sandoval-Ramírez, J.; Luna, F.; Carrasco-Carballo, A. Molecular Docking Studies of Spirostans as MAPK14 (P38α) Inhibitors and Their Potential Use against Cancer. J. Mol. Docking 2021, 1, 59–67. [Google Scholar] [CrossRef]
- Lopez-Castillo, G.N.; Alatriste, V.; Luna, F.; Teran, J.L.; Sandoval, J.; Orea, M.L.; Carrasco-Carballo, A. Spirostans Design as Novel Ligands for CB1 and CB2 Cannabinoid Receptors. Biointerface Res. Appl. Chem. 2022, 13, 418. [Google Scholar]
- Li, S.; Wang, L.; Wang, Y.; Zhang, C.; Hong, Z.; Han, Z. The Synthetic Lethality of Targeting Cell Cycle Checkpoints and PARPs in Cancer Treatment. J. Hematol. Oncol. J. Hematol. Oncol. 2022, 15, 147. [Google Scholar] [CrossRef]
- Boudny, M.; Trbusek, M. ATR-CHK1 Pathway as a Therapeutic Target for Acute and Chronic Leukemias. Cancer Treat. Rev. 2020, 88, 102026. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Vasbinder, M.M.; Hird, A.W.; Su, Q.; Wang, H.; Yu, Y.; Toader, D.; Lyne, P.D.; Read, J.A.; Breed, J.; et al. Adventures in Scaffold Morphing: Discovery of Fused Ring Heterocyclic Checkpoint Kinase 1 (CHK1) Inhibitors. J. Med. Chem. 2018, 61, 1061–1073. [Google Scholar] [CrossRef]
- Vanderpool, D.; Johnson, T.O.; Ping, C.; Bergqvist, S.; Alton, G.; Phonephaly, S.; Rui, E.; Luo, C.; Deng, Y.-L.; Grant, S.; et al. Characterization of the CHK1 Allosteric Inhibitor Binding Site. Biochemistry 2009, 48, 9823–9830. [Google Scholar] [CrossRef] [PubMed]
- Matijssen, C.; Silva-Santisteban, M.C.; Westwood, I.M.; Siddique, S.; Choi, V.; Sheldrake, P.; Van Montfort, R.L.M.; Blagg, J. Benzimidazole Inhibitors of the Protein Kinase CHK2: Clarification of the Binding Mode by Flexible Side Chain Docking and Protein–Ligand Crystallography. Bioorg. Med. Chem. 2012, 20, 6630–6639. [Google Scholar] [CrossRef] [PubMed]
- Protein Preparation Wizard; Epik, Schrödinger, LLC.; Impact, Schrödinger, LLC.; Prime, Schrödinger, LLC. 2024. Available online: https://www.schrodinger.com/citations/ (accessed on 14 May 2024).
- Carrasco-Carballo, A.; Mendoza-Lara, D.F.; Rojas-Morales, J.A.; Alatriste, V.; Merino-Montiel, P.; Luna, F.; Sandoval-Ramirez, J. In Silico Study of Coumarins Derivatives With Potential Use in Systemic Diseases. Biointerface Res. Appl. Chem. 2022, 13, 240. [Google Scholar] [CrossRef]
- Schrödinger Release 2024-1: MacroModel 2024. Available online: https://www.schrodinger.com/citations/ (accessed on 14 May 2024).
- Schrödinger Release 2024-1: LigPrep 2024. Available online: https://www.schrodinger.com/citations/ (accessed on 14 May 2024).
- Schrödinger LLC, Schrödinger Release 2024-1: Jaguar, Schrödinger Inc. 2024. Available online: https://www.schrodinger.com/citations/ (accessed on 14 May 2024).
- Genc, M.; Shehu, A.; Servi, S. Synthesis, Spectroscopic Characterization, Solvatochromic Properties and DFT Quantum Chemical Calculations of 3β-Acetoxy-17β-(1-Acetyl-5-(3-Pyridinyl)-3-Pyrazolinyl) Androat-5-Ene. J. Mol. Struct. 2019, 1193, 195–206. [Google Scholar] [CrossRef]
- Solomon, R.V.; Veerapandian, P.; Vedha, S.A.; Venuvanalingam, P. Tuning Nonlinear Optical and Optoelectronic Properties of Vinyl Coupled Triazene Chromophores: A Density Functional Theory and Time-Dependent Density Functional Theory Investigation. J. Phys. Chem. A 2012, 116, 4667–4677. [Google Scholar] [CrossRef] [PubMed]
- Kathiravan, A.; Panneerselvam, M.; Sundaravel, K.; Pavithra, N.; Srinivasan, V.; Anandan, S.; Jaccob, M. Unravelling the Effect of Anchoring Groups on the Ground and Excited State Properties of Pyrene Using Computational and Spectroscopic Methods. Phys. Chem. Chem. Phys. 2016, 18, 13332–13345. [Google Scholar] [CrossRef] [PubMed]
- Bella, A.P.; Panneerselvam, M.; Vedha, S.A.; Jaccob, M.; Solomon, R.V.; Merlin, J.P. DFT-TDDFT Framework of Diphenylamine Based Mixed Valence Compounds for Optoelectronic Applications—Structural Modification of π-Acceptors. Comput. Mater. Sci. 2019, 162, 359–369. [Google Scholar] [CrossRef]
- Schrödinger Release 2024-1: Glide, Schrödinger, LLC, New York, NY, 2024. Available online: https://www.schrodinger.com/citations/ (accessed on 14 May 2024).
- Schrödinger LLC, Schrödinger Release 2024-1: QikProp, Schrödinger Inc. 2024. Available online: https://www.schrodinger.com/citations/ (accessed on 14 May 2024).
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Dudev, T.; Lin, Y.; Dudev, M.; Lim, C. First−Second Shell Interactions in Metal Binding Sites in Proteins: A PDB Survey and DFT/CDM Calculations. J. Am. Chem. Soc. 2003, 125, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Lever, G.; Cole, D.J.; Hine, N.D.M.; Haynes, P.D.; Payne, M.C. Electrostatic Considerations Affecting the Calculated HOMO–LUMO Gap in Protein Molecules. J. Phys. Condens. Matter 2013, 25, 152101. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2024-2: Jaguar. 2024.
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2024-2: Desmond Molecular Dynamics System, D.E. Schrödinger Release 2024-2: Desmond Molecular Dynamics System, D.E. Shaw Research. Maestro-Desmond Interoperability Tools. 2024.
- Kalath, H.; Koshy, A.J.; Banjan, B.; Soman, S.; Hosadevasthana, G.; Raju, R.; Rehman, N.; Revikumar, A. In-Silico Studies of Brassica Oleracea Active Compounds and Their Role in Thyroid Peroxidase Activity. J. Biomol. Struct. Dyn. 2023, 41, 1–17. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preclinical Inhibitor | DS with CHK1 | DS with CHK2 | |
|---|---|---|---|
| Catalytic | Allosteric | Catalytic | |
| Isogranulathimide | −6.041 | - | −5.684 |
| CCT241533 | −3.993 | - | −5.145 |
| Sar-020106 | −6.041 | - | −6.021 |
| PD 407824 | −4.754 | - | −3.261 |
| VRX0466617 | −5.900 | - | −6.592 |
| Molecule co-crystallized with the protein | - | −6.342 | - |
| R(C-#) | CHK1 (Cat) Better DS → | CHK1(Allo) Better DS → | CHK2 (Cat) Better DS → | ||||
|---|---|---|---|---|---|---|---|
| 1(C-2) | NSD | αOH, βOH | H | NSD | |||
| 2(C-3) | αOH | βOH, O | αOH | βOH, O | αOH | ||
| 3(C-4) | NSD | H | Δ4 | H | Δ4 | ||
| 4(C-5) | NSD | Δ4, αH, Δ5 | αOH | βH | Δ4, βH, Δ5 | αOH, αH | |
| 5(C-6) | NSD | Δ5, O, H | αOH, βOH | NSD | |||
| 6(C-11) | NSD | αOH, O | βOH, H | NSD | |||
| 7(C-12) | O | H | NSD | NSD | |||
| 8(C-17) | NSD | NSD | NSD | ||||
| 9(C-25) | NSD | NSD | R | S | |||
| Spirostan | CHK | CNS 1 | Molecular Weight | Hydrogen BondD/A 2 | QPlog Po/w 3 | #metab 4 | QPlogHERG 5 | QPlogKp 6 | Lipinski’s Rule Violations |
|---|---|---|---|---|---|---|---|---|---|
| Sp137 | 1, 2 | 0 | 428.611 | 1.00/5.20 | 4.893 | 2 | −4.233 | −3.155 | 0 |
| Sp3 | 1 | 1 | 414.627 | 1.00/3.20 | 6.074 | 3 | −4.3 | −2.219 | 1 |
| Sp24 | 1 | 0 | 430.626 | 2.00/4.90 | 5.071 | 4 | −4.447 | −2.913 | 1 |
| Sp97 | 1 | 0 | 430.626 | 2.00/4.90 | 5.071 | 4 | −4.447 | −2.913 | 1 |
| Sp1 | 1 | 1 | 414.627 | 1.00/3.20 | 6.123 | 3 | −4.407 | −2.212 | 1 |
| Sp131 | 1 | −1 | 448.642 | 3.00/5.65 | 4.574 | 3 | −4.062 | −2.937 | 0 |
| Sp155 | 1 | 0 | 428.611 | 1.00/4.25 | 5.381 | 3 | −4.282 | −2.787 | 1 |
| Sp142 | 1 | 0 | 428.611 | 1.00/5.20 | 4.909 | 2 | −4.458 | −3.215 | 0 |
| Sp154 | 1 | 0 | 428.611 | 1.00/4.25 | 5.437 | 3 | −4.417 | −2.793 | 1 |
| Sp101 | 1 | 0 | 430.626 | 1.00/5.20 | 4.835 | 3 | −3.765 | −3.034 | 0 |
| Sp53 | 1 | 0 | 432.642 | 2.00/4.90 | 5.112 | 2 | −4.174 | −2.847 | 1 |
| Sp67 | 1 | 0 | 432.642 | 2.00/4.90 | 5.055 | 2 | −4.042 | −2.843 | 1 |
| Sp37 | 1 | 0 | 428.611 | 1.00/5.20 | 5.064 | 5 | −4.146 | −2.702 | 1 |
| Sp13 | 2 | 0 | 428.611 | 1.00/5.20 | 5.125 | 4 | −4.26 | −2.692 | 1 |
| Sp28 | 2 | 0 | 430.626 | 2.00/4.90 | 4.945 | 4 | −4.072 | −2.645 | 0 |
| Sp45 | 2 | 0 | 432.642 | 2.00/4.90 | 5.060 | 2 | −4.215 | −2.911 | 1 |
| Sp41 | 2 | 0 | 430.626 | 1.00/5.20 | 5.155 | 2 | −4.144 | −2.794 | 1 |
| Sp14 | 2 | 0 | 430.626 | 2.00/4.90 | 5.142 | 4 | −4.15 | −2.59 | 1 |
| Sp5 | 2 | 1 | 416.643 | 1.00/3.20 | 6.219 | 1 | −4.267 | −2.188 | 1 |
| Sp44 | 2 | 0 | 430.626 | 1.00/5.20 | 5.134 | 3 | −4.097 | −2.662 | 1 |
| Sp16 | 2 | 0 | 428.611 | 1.00/5.20 | 5.120 | 5 | −4.144 | −2.575 | 1 |
| Sp65 | 2 | 0 | 430.626 | 1.00/5.20 | 5.095 | 3 | −4.036 | −2.813 | 1 |
| Sp139 | 2 | 0 | 428.611 | 1.00/5.20 | 4.909 | 2 | −4.35 | −3.061 | 0 |
| Sp35 | 2 | 0 | 430.626 | 2.00/4.90 | 5.080 | 4 | −4.147 | −2.726 | 1 |
| Sp106 | 2 | 0 | 432.642 | 2.00/4.90 | 5.028 | 2 | −4.164 | −2.977 | 1 |
| CHK1 | CHK2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| aa | Sp137 | Sp1 | aa | Sp137 | Sp13 | ||||
| HOMOCHK1-LUMOSp137 | HOMOSp137-LUMOCHK1 | HOMOCHK1-LUMOSp1 | HOMOSp1-LUMOCHK1 | HOMOCHK2-LUMOSp137 | HOMOSp137-LUMOCHK2 | HOMOCHK2-LUMOSp13 | HOMOSp13-LUMOCHK2 | ||
| Gln013 | −0.1990 | −0.1953 | −0.2687 | −0.1966 | Leu226 | −0.2151 | 0.0184 | −0.2483 | −0.0147 |
| Val023 | −0.1924 | −0.1990 | −0.2621 | −0.2003 | Gly227 | −0.2302 | −0.2104 | −0.2634 | −0.2138 |
| Ala036 | −0.1969 | −0.1982 | −0.2666 | −0.1994 | Val234 | −0.2145 | −0.2018 | −0.2477 | −0.2052 |
| Glu085 | 0.0166 | −0.3112 | −0.0530 | −0.3125 | Lys245 | −0.3067 | −0.0342 | −0.3399 | −0.0376 |
| Try086 | −0.1825 | −0.2030 | −0.2522 | −0.2042 | Ala247 | −0.2201 | −0.2024 | −0.2533 | −0.2058 |
| Cys087 | −0.2101 | −0.1946 | −0.2798 | −0.1958 | Leu303 | −0.2168 | −0.2059 | −0.2500 | −0.2093 |
| Ser088 | −0.2186 | −0.2075 | −0.2883 | −0.2087 | Met304 | −0.1864 | −0.1977 | −0.2197 | −0.2010 |
| Gly089 | −0.2214 | −0.1850 | −0.2911 | −0.1862 | Glu305 | 0.0184 | −0.2999 | −0.0148 | −0.3033 |
| Gly090 | −0.2289 | −0.2085 | −0.2986 | −0.2098 | Gly306 | −0.2215 | −0.1850 | −0.2547 | −0.1884 |
| Asp094 | 0.0021 | −0.3360 | −0.0675 | −0.3372 | Gly307 | −0.2304 | −0.2124 | −0.2636 | −0.2158 |
| Glu134 | 0.0053 | −0.3339 | −0.0644 | −0.3351 | Glu308 | 0.0184 | −0.3023 | −0.0147 | −0.3057 |
| Leu137 | −0.1945 | −0.2023 | −0.2642 | −0.2036 | Asp311 | 0.0104 | −0.3091 | −0.0228 | −0.3125 |
| Leu354 | −0.2124 | −0.1968 | −0.2456 | −0.2001 | |||||
| Gln358 | −0.2147 | −0.2010 | −0.2480 | −0.2044 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosales-López, A.; López-Castillo, G.N.; Sandoval-Ramírez, J.; Terán, J.L.; Carrasco-Carballo, A. Correlation between Molecular Docking and the Stabilizing Interaction of HOMO-LUMO: Spirostans in CHK1 and CHK2, an In Silico Cancer Approach. Int. J. Mol. Sci. 2024, 25, 8588. https://doi.org/10.3390/ijms25168588
Rosales-López A, López-Castillo GN, Sandoval-Ramírez J, Terán JL, Carrasco-Carballo A. Correlation between Molecular Docking and the Stabilizing Interaction of HOMO-LUMO: Spirostans in CHK1 and CHK2, an In Silico Cancer Approach. International Journal of Molecular Sciences. 2024; 25(16):8588. https://doi.org/10.3390/ijms25168588
Chicago/Turabian StyleRosales-López, Antonio, Guiee N. López-Castillo, Jesús Sandoval-Ramírez, Joel L. Terán, and Alan Carrasco-Carballo. 2024. "Correlation between Molecular Docking and the Stabilizing Interaction of HOMO-LUMO: Spirostans in CHK1 and CHK2, an In Silico Cancer Approach" International Journal of Molecular Sciences 25, no. 16: 8588. https://doi.org/10.3390/ijms25168588
APA StyleRosales-López, A., López-Castillo, G. N., Sandoval-Ramírez, J., Terán, J. L., & Carrasco-Carballo, A. (2024). Correlation between Molecular Docking and the Stabilizing Interaction of HOMO-LUMO: Spirostans in CHK1 and CHK2, an In Silico Cancer Approach. International Journal of Molecular Sciences, 25(16), 8588. https://doi.org/10.3390/ijms25168588