
TFIIB–Termination Factor Interaction Affects Termination of Transcription on Genome-Wide Scale


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
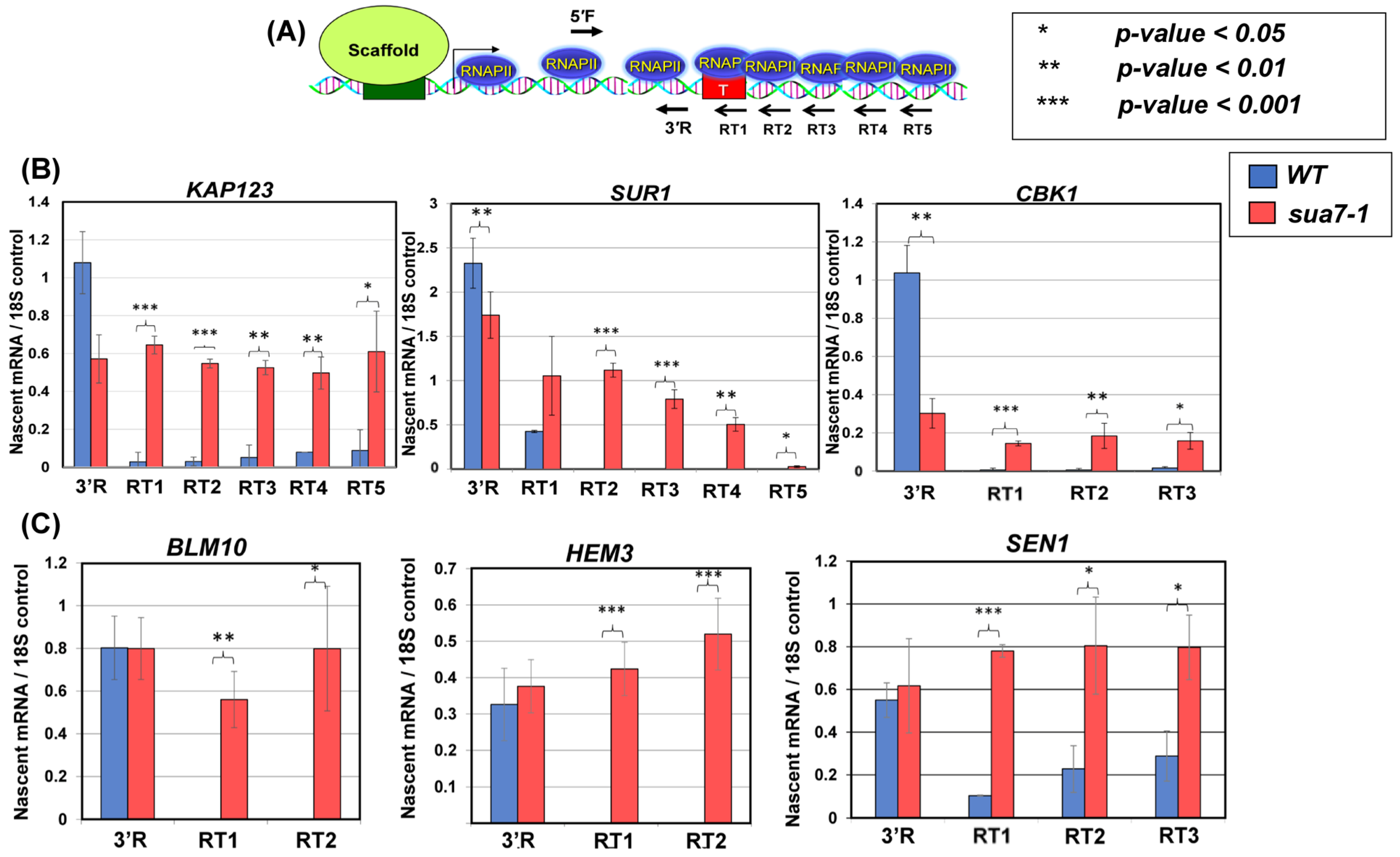
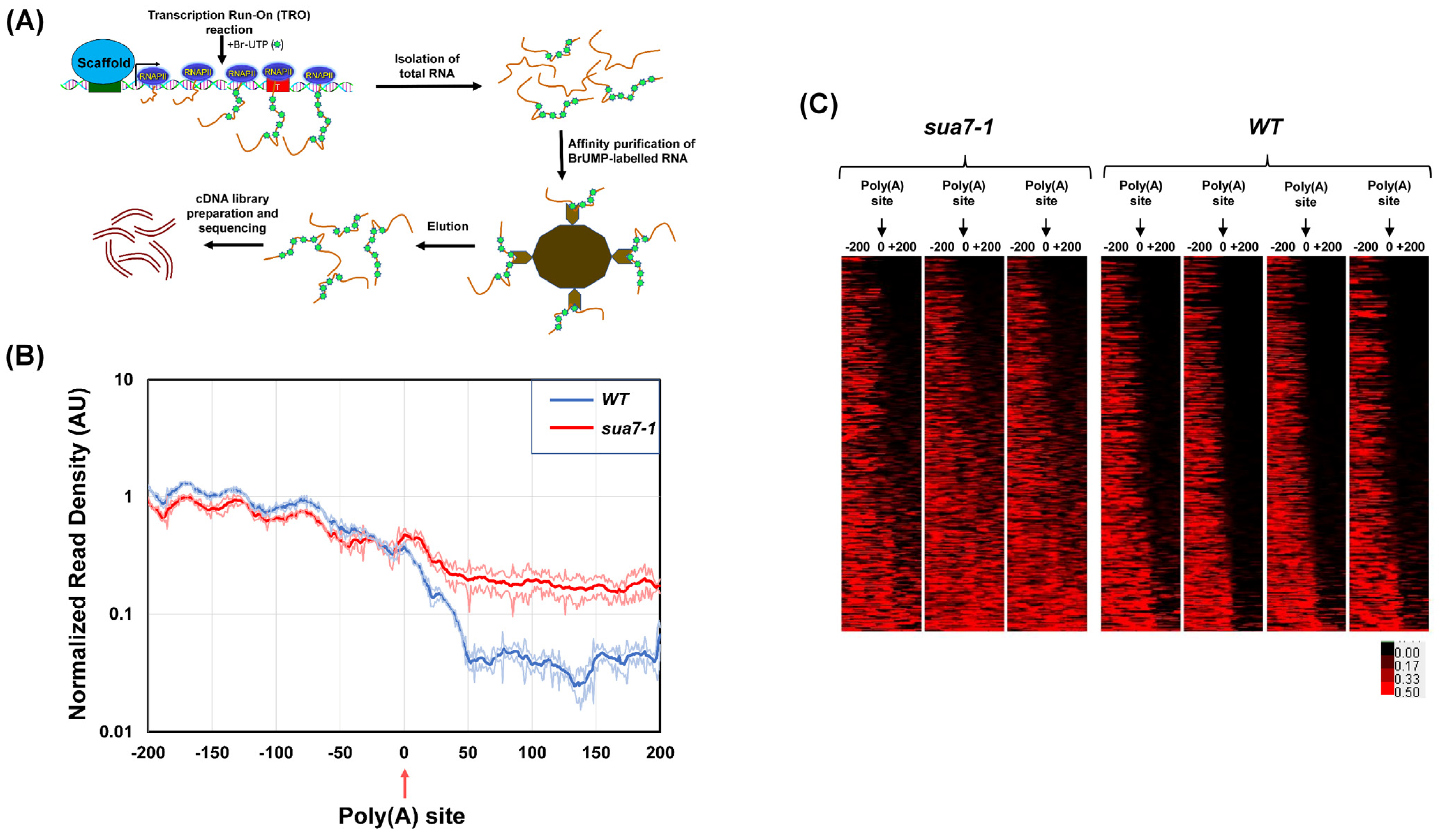
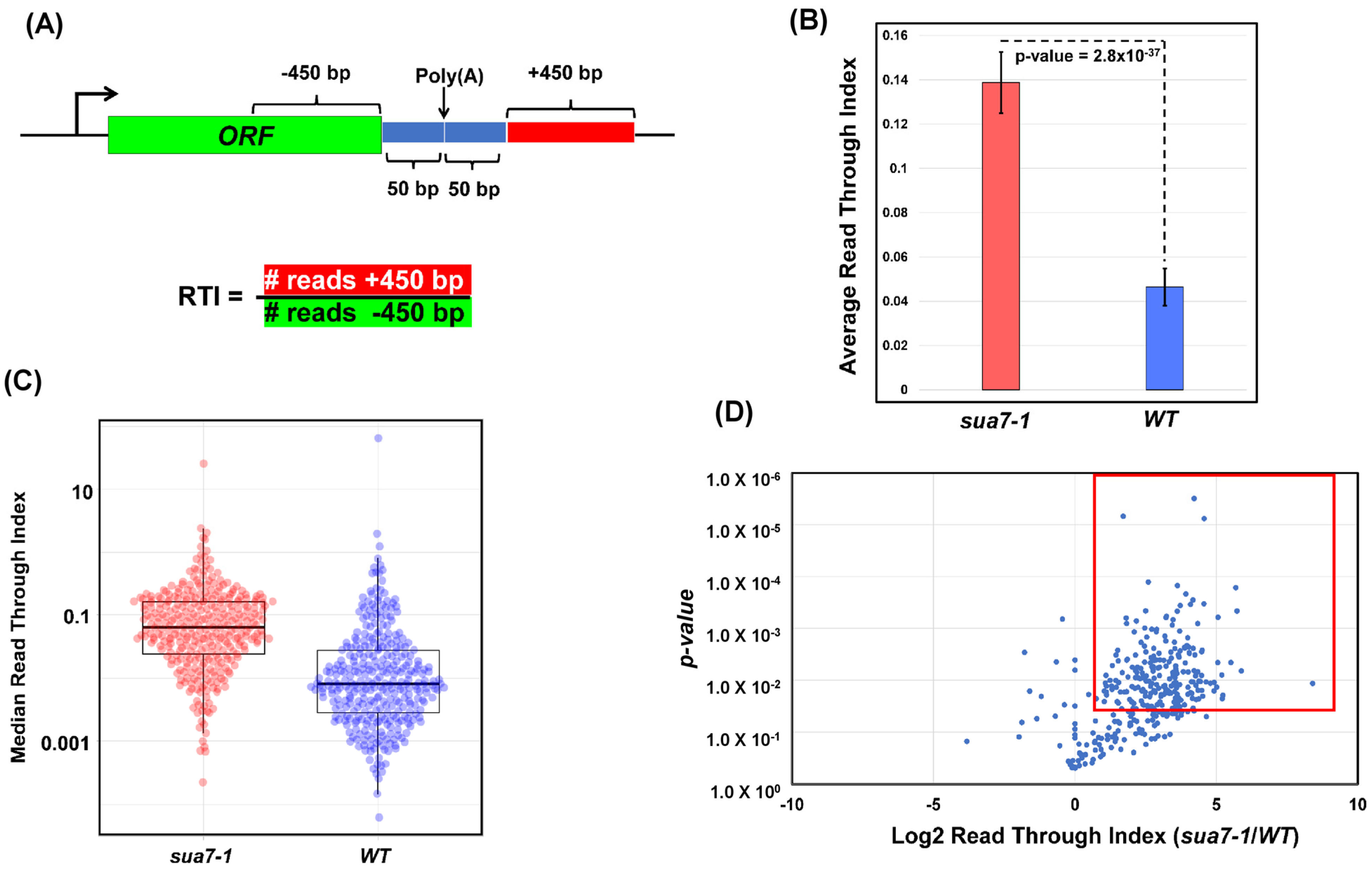
2.1. TFIIB Affects Termination of Transcription on a Genome-Wide Scale
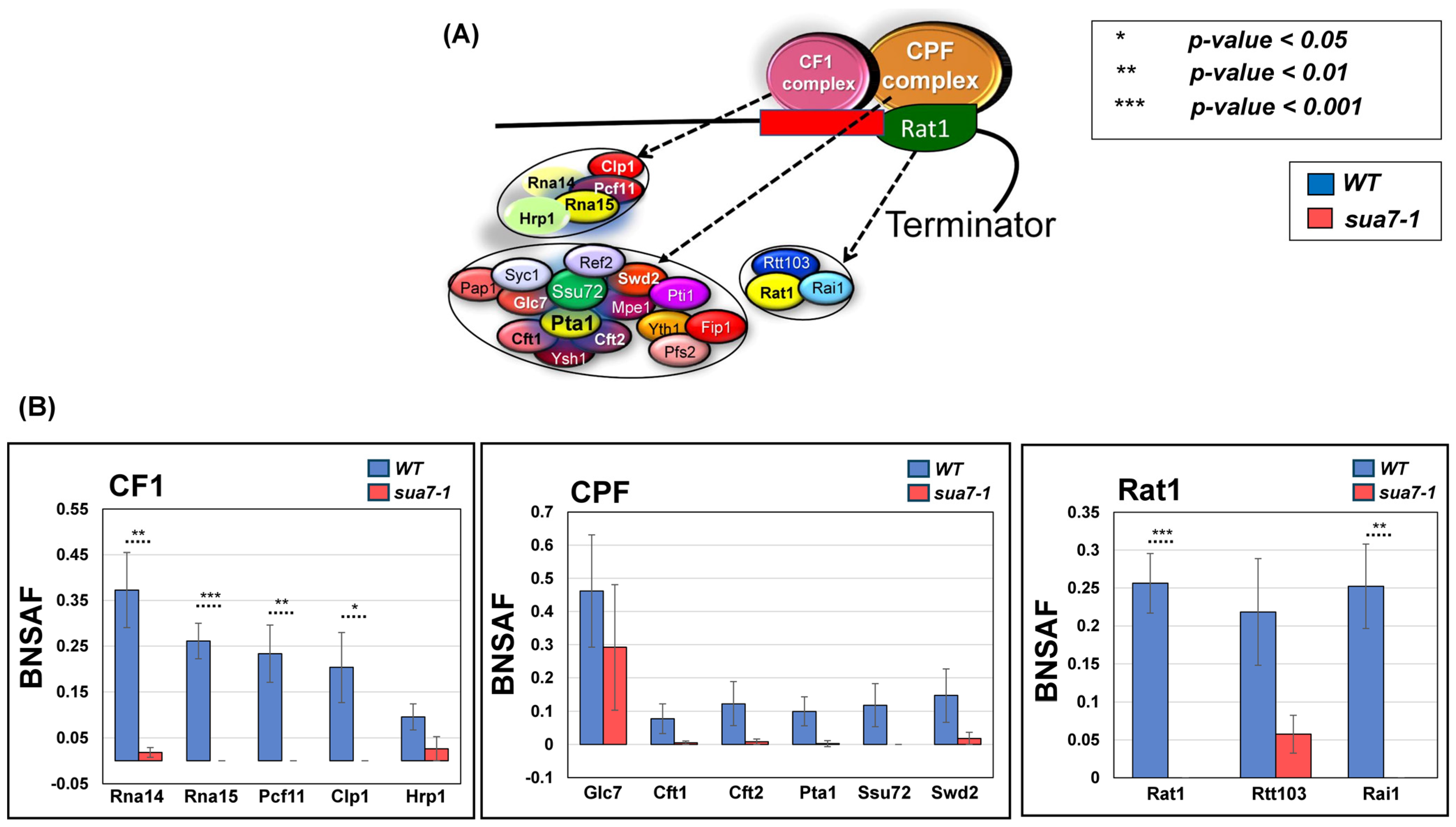
2.2. TFIIB’s Interaction with Termination Factors Is Diminished in the Termination Defective Mutant
2.3. Recruitment of Termination Factors at the 3′ End of Genes Is Compromised in the Termination Defective Mutant
3. Discussion
4. Materials and Methods
4.1. Yeast Strains
4.2. Protein Purification from Soluble and Chromatin Fractions
4.3. Mass Spectrometry and Quantitative Analysis
4.4. GRO-Seq
4.5. GRO-Seq Analysis
4.6. TRO Assay
4.7. ChIP
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dantonel, J.C.; Murthy, K.G.; Manley, J.L.; Tora, L. Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature 1997, 389, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Medler, S.; Al Husini, N.; Raghunayakula, S.; Mukundan, B.; Aldea, A.; Ansari, A. Evidence for a complex of transcription factor IIB (TFIIB) with Poly (A) polymerase and cleavage factor 1 subunits required for gene looping. J. Biol. Chem. 2011, 286, 33709–33718. [Google Scholar] [CrossRef] [PubMed]
- Nagaike, T.; Logan, C.; Hotta, I.; Rozenblatt-Rosen, O.; Meyerson, M.; Manley, J.L. Transcriptional activators enhance polyadenylation of mRNA precursors. Mol. Cell 2011, 41, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Lamas-Maceiras, M.; Singh, B.N.; Hampsey, M.; Freire-Picos, M.A. Promoter-terminator gene loops affect alternative 3′-end processing in yeast. J. Biol. Chem. 2016, 291, 8960–8968. [Google Scholar] [CrossRef] [PubMed]
- Terrone, S.; Valat, J.; Fontrodona, N.; Giraud, G.; Claude, J.-B.; Combe, E.; Lapendry, A.; Polvèche, H.; Ben Ameur, L.; Duvermy, A.; et al. RNA helicase-dependent gene looping impacts messenger RNA processing. Nucleic Acids Res. 2022, 50, 9226–9246. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.; Fansler, M.M.; Patel, N.D.; Lee, J.; Ma, W.; Mayr, C. Enhancers regulate 3′ end processing activity to control expression of alternative 3′ UTR isoforms. Nat. Commun. 2022, 13, 2709. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Gonzalez, C.; Legnini, I.; Holec, S.; Arrigoni, L.; Ozbulut, H.C.; Mateos, F.; Hilgers, V. Sites of transcription initiation drive mRNA isoform selection. Cell 2023, 186, 2438–2455. [Google Scholar] [CrossRef] [PubMed]
- Al-Husini, N.; Medler, S.; Ansari, A. Crosstalk of promoter and terminator during RNA polymerase II transcription cycle. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2020, 1863, 194657. [Google Scholar] [CrossRef] [PubMed]
- Kuehner, J.N.; Pearson, E.L.; Moore, C. Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Biol. 2011, 12, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Proudfoot, N.J. Disengaging polymerase: Terminating RNA polymerase II transcription in budding yeast. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2013, 1829, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Porrua, O.; Boudvillain, M.; Libri, D. Transcription termination: Variations on common themes. Trends Genet. 2016, 32, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Molina, J.B.; West, S.; Passmore, L.A. Knowing when to stop: Transcription termination on protein-coding genes by eukaryotic RNAPII. Mol. Cell 2023, 83, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Woychik, N.A.; Hampsey, M. The RNA polymerase II machinery: Structure illuminates function. Cell 2002, 108, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Luse, D.S. The RNA polymerase II preinitiation complex: Through what pathway is the complex assembled? Transcription 2014, 5, e27050. [Google Scholar] [CrossRef] [PubMed]
- Lagrange, T.; Kapanidis, A.N.; Tang, H.; Reinberg, D.; Ebright, R.H. New core promoter element in RNA polymerase II-dependent transcription: Sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998, 12, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Roberts, S.G. A core promoter element downstream of the TATA box that is recognized by TFIIB. Genes Dev. 2005, 19, 2418–2423. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.S.; Pugh, B.F. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 2012, 483, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Yochum, G.S.; Rajaraman, V.; Cleland, R.; McWeeney, S. Localization of TFIIB binding regions using serial analysis of chromatin occupancy. BMC Mol. Biol. 2007, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Pugh, B.F.; Venters, B.J. Genomic organization of human transcription initiation complexes. PLoS ONE 2016, 11, e0149339. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.J.; Kuntala, P.K.; Lai, W.K.; Yamada, N.; Badjatia, N.; Mittal, C.; Pugh, B.F. A high-resolution protein architecture of the budding yeast genome. Nature 2021, 592, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Hampsey, M. A transcription-independent role for TFIIB in gene looping. Mol. Cell 2007, 27, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Ioshikhes, I.P.; Venters, B.J.; Jiang, C.; Tomsho, L.P.; Qi, J.; Pugh, B.F. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008, 18, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- El Kaderi, B.; Medler, S.; Raghunayakula, S.; Ansari, A. Gene looping is conferred by activator-dependent interaction of transcription initiation and termination machineries. J. Biol. Chem. 2009, 284, 25015–25025. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Lidschreiber, M.; Siebert, M.; Leike, K.; Söding, J.; Cramer, P. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 2010, 17, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.C.; Serra Barros, A.; Brown, D.A.; Dudek, P.; Ayling, J.; Mellor, J. A pre-initiation complex at the 3′-end of genes drives antisense transcription independent of divergent sense transcription. Nucleic Acids Res. 2012, 40, 2432–2444. [Google Scholar] [CrossRef] [PubMed]
- Mapendano, C.K.; Lykke-Andersen, S.; Kjems, J.; Bertrand, E.; Jensen, T.H. Crosstalk between mRNA 3′ end processing and transcription initiation. Mol. Cell 2010, 40, 410–422. [Google Scholar]
- Pinto, I.; Wu, W.H.; Na, J.G.; Hampsey, M. Characterization of sua7 mutations defines a domain of TFIIB involved in transcription start site selection in yeast. J. Biol. Chem. 1994, 269, 30569–30573. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.W.; Hampsey, M. Synthetic enhancement of a TFIIB defect by a mutation in SSU72, an essential yeast gene encoding a novel protein that affects transcription start site selection in vivo. Mol. Cell Biol. 1996, 16, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Pinto, I.; Chen, B.S.; Hampsey, M. Mutational analysis of yeast TFIIB: A functional relationship between Ssu72 and Sub1/Tsp1 defined by allele-specific interactions with TFIIB. Genetics 1999, 153, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Allepuz-Fuster, P.; O’Brien, M.J.; González-Polo, N.; Pereira, B.; Dhoondia, Z.; Ansari, A.; Calvo, O. RNA polymerase II plays an active role in the formation of gene loops through the Rpb4 subunit. Nucleic Acids Res. 2019, 47, 8975–8987. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fairley, J.A.; Roberts, S.G. Phosphorylation of TFIIB links transcription initiation and termination. Curr. Biol. 2010, 20, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Bharatula, V.; Elfving, N.; Blomberg, J.; Larsson, M.; Morozov, A.V.; Björklund, S. Mediator binds to boundaries of chromosomal interaction domains and to proteins involved in DNA looping, RNA metabolism, chromatin remodeling, and actin assembly. Nucleic Acids Res. 2017, 45, 8806–8821. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.J.; Ansari, A. Protein interaction network revealed by quantitative proteomic analysis links TFIIB to multiple aspects of the transcription cycle. Biochim. Biophys. Acta BBA-Proteins Proteom. 2024, 1872, 140968. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Chanfreau, G.F. A global function for transcription factors in assisting RNA polymerase II termination. Transcription 2018, 9, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Henriques, T.; Ji, Z.; Tan-Wong, S.M.; Carmo, A.M.; Tian, B.; Proudfoot, N.J.; Moreira, A. Transcription termination between polo and snap, two closely spaced tandem genes of D. melanogaster. Transcription 2012, 3, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.J.; Buratowski, S. Evidence that transcription factor IIB is required for a post-assembly step in transcription initiation. J. Biol. Chem. 1999, 274, 25807–25813. [Google Scholar] [CrossRef] [PubMed]
- Core, L.J.; Waterfall, J.J.; Lis, J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Baejen, C.; Andreani, J.; Torkler, P.; Battaglia, S.; Schwalb, B.; Lidschreiber, M.; Cramer, P. Genome-wide analysis of RNA polymerase II termination at protein-coding genes. Mol. Cell 2017, 66, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, A.C.; Parmely, T.J.; Tomomori-Sato, C.; Sato, S.; Zhu, D.; Conaway, R.C.; Washburn, M.P. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. USA 2006, 103, 18928–18933. [Google Scholar] [CrossRef] [PubMed]
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical analysis of membrane proteome expression changes in Saccharomyces c erevisiae. J. Proteome Res. 2006, 5, 2339–2347. [Google Scholar] [CrossRef] [PubMed]
- Santana, J.F.; Collins, G.S.; Parida, M.; Luse, D.S.; Price, D.H. Differential dependencies of human RNA polymerase II promoters on TBP, TAF1, TFIIB and XPB. Nucleic Acids Res. 2022, 50, 9127–9148. [Google Scholar] [CrossRef]
- Sansó, M.; Levin, R.S.; Lipp, J.J.; Wang, V.Y.-F.; Greifenberg, A.K.; Quezada, E.M.; Ali, A.; Ghosh, A.; Larochelle, S.; Rana, T.M.; et al. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev. 2016, 30, 117–131. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.J.; Gurdziel, K.; Ansari, A. Global Run-On sequencing to measure nascent transcription in Saccharomyces cerevisiae. Methods 2023, 217, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Dhoondia, Z.; Tarockoff, R.; Alhusini, N.; Medler, S.; Agarwal, N.; Ansari, A. Analysis of termination of transcription using BrUTP-strand-specific transcription run-on (TRO) approach. JoVE J. Vis. Exp. 2017, 121, e55446. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Brien, M.J.; Schrader, J.M.; Ansari, A. TFIIB–Termination Factor Interaction Affects Termination of Transcription on Genome-Wide Scale. Int. J. Mol. Sci. 2024, 25, 8643. https://doi.org/10.3390/ijms25168643
O’Brien MJ, Schrader JM, Ansari A. TFIIB–Termination Factor Interaction Affects Termination of Transcription on Genome-Wide Scale. International Journal of Molecular Sciences. 2024; 25(16):8643. https://doi.org/10.3390/ijms25168643
Chicago/Turabian StyleO’Brien, Michael J., Jared M. Schrader, and Athar Ansari. 2024. "TFIIB–Termination Factor Interaction Affects Termination of Transcription on Genome-Wide Scale" International Journal of Molecular Sciences 25, no. 16: 8643. https://doi.org/10.3390/ijms25168643