
Fibrosis and Hepatocarcinogenesis: Role of Gene-Environment Interactions in Liver Disease Progression


Abstract
1. Introduction
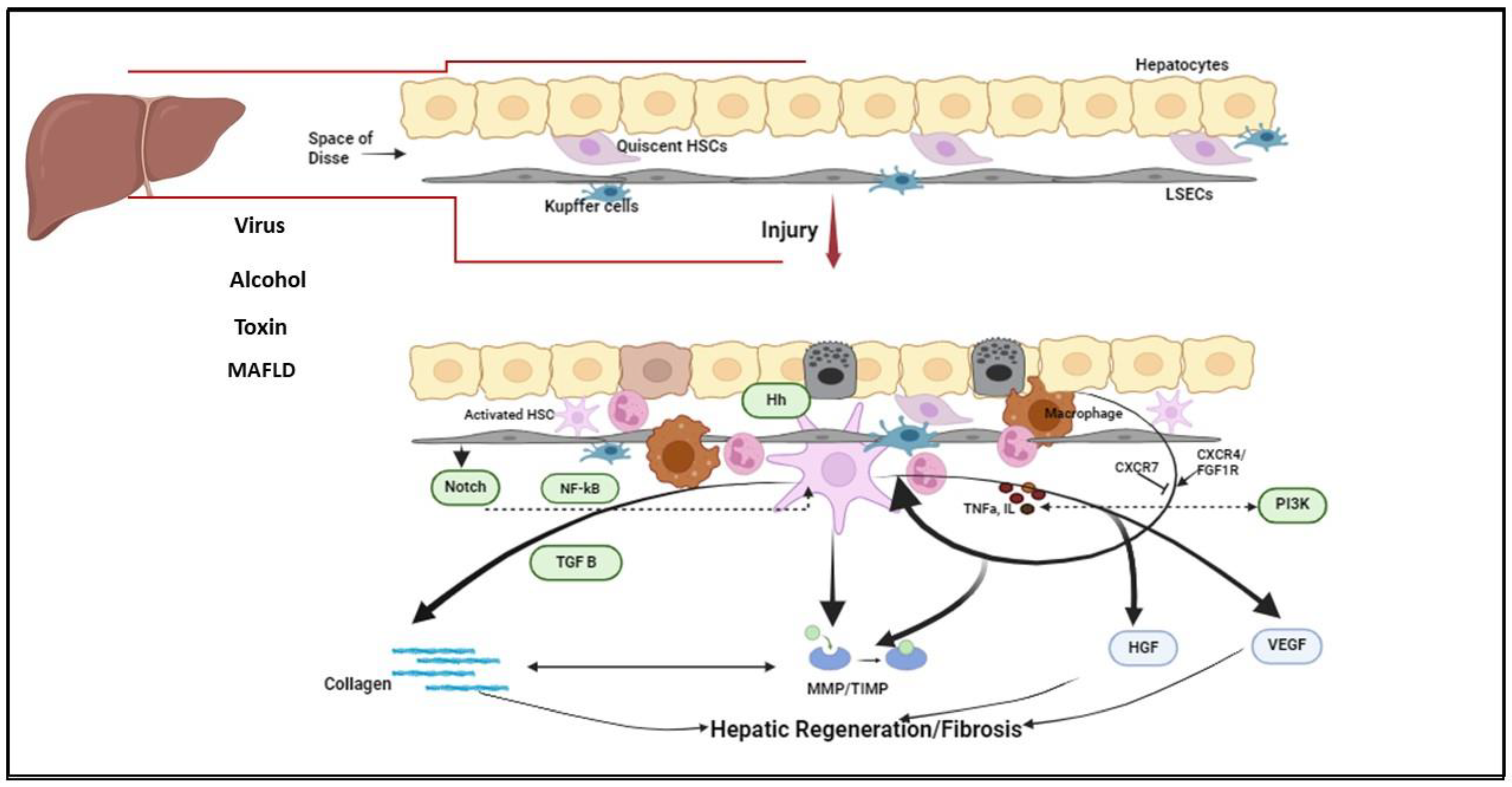
2. Fibrosis Dynamics
3. Major Signaling Pathways in Liver Fibrosis
3.1. NF-κB
3.2. TGF-β/SMAD Pathway
3.3. PI3K/Akt/mTOR Pathway
3.4. Hedgehog Pathway
3.5. Notch Pathway
3.6. Angiogenesis
4. Fibrosis as the Chief Driver of Liver Carcinogenesis
5. Can Genetic Markers Predict Fibrosis Outcome?
5.1. IFN Lambda 4
5.2. Genetic Regulators of Apoptosis: RNF, MERTK, TULP
5.3. PNPLA 3
5.4. TLL1
5.5. TLR
5.6. HLA DQ, HLA-E, HLA-C
5.7. MICA
6. Role of Transcription Factors
6.1. TCF21
6.2. ATF3
6.3. GATA 4/6
6.4. ELF3 and GLIS2
6.5. RAR
6.6. PPARf
6.7. ZNF469
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, J.L.; Xu, D.; Shi, G.; Wan, M.; Goodman, Z.; Tan, D.; Xie, Q.; Chen, C.; Wei, L.; Niu, J.; et al. Long-Term Telbivudine Treatment Results in Resolution of Liver Inflammation and Fibrosis in Patients with Chronic Hepatitis B. Adv. Ther. 2015, 32, 727–741. [Google Scholar] [CrossRef]
- Farci, P.; Roskams, T.; Chessa, L.; Peddis, G.; Mazzoleni, A.P.; Scioscia, R.; Serra, G.; Lai, M.E.; Loy, M.; Caruso, L.; et al. Long-term benefit of interferon alpha therapy of chronic hepatitis D: Regression of advanced hepatic fibrosis. Gastroenterology 2004, 126, 1740–1749. [Google Scholar] [CrossRef]
- Cheng, C.H.; Chu, C.Y.; Chen, H.L.; Lin, I.T.; Wu, C.H.; Lee, Y.K.; Hu, P.J.; Bair, M.J. Direct-acting antiviral therapy of chronic hepatitis C improves liver fibrosis, assessed by histological examination and laboratory markers. J. Formos. Med. Assoc. 2021, 120, 1259–1268. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Anstee, Q.M.; Trauner, M.; Lawitz, E.J.; Abdelmalek, M.F.; Ding, D.; Han, L.; Jia, C.; Huss, R.S.; Chung, C.; et al. Cirrhosis regression is associated with improved clinical outcomes in patients with nonalcoholic steatohepatitis. Hepatology 2022, 75, 1235–1246. [Google Scholar] [CrossRef]
- Lee, M.J. A review of liver fibrosis and cirrhosis regression. J. Pathol. Transl. Med. 2023, 57, 189–195. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [CrossRef]
- Hellerbrand, C. Hepatic stellate cells--the pericytes in the liver. Pflugers Arch. 2013, 465, 775–778. [Google Scholar] [CrossRef]
- Berumen, J.; Baglieri, J.; Kisseleva, T.; Mekeel, K. Liver fibrosis: Pathophysiology and clinical implications. WIREs Mech. Dis. 2021, 13, e1499. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef]
- Deleve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Muhanna, N.; Horani, A.; Doron, S.; Safadi, R. Lymphocyte-hepatic stellate cell proximity suggests a direct interaction. Clin. Exp. Immunol. 2007, 148, 338–347. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix. Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Roderfeld, M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. 2018, 68–69, 452–462. [Google Scholar] [CrossRef]
- Jeong, W.I.; Do, S.H.; Jeong, D.H.; Hong, I.H.; Park, J.K.; Ran, K.M.; Yang, H.J.; Yuan, D.W.; Kim, S.B.; Cha, M.S.; et al. Kinetics of MMP-1 and MMP-3 produced by mast cells and macrophages in liver fibrogenesis of rat. Anticancer Res. 2006, 26, 3517–3526. [Google Scholar]
- Powell, B.; Malaspina, D.C.; Szleifer, I.; Dhaher, Y. Effect of collagenase-gelatinase ratio on the mechanical properties of a collagen fibril: A combined Monte Carlo-molecular dynamics study. Biomech. Model. Mechanobiol. 2019, 18, 1809–1819. [Google Scholar] [CrossRef]
- Iimuro, Y.; Brenner, D.A. Matrix metalloproteinase gene delivery for liver fibrosis. Pharm. Res. 2008, 25, 249–258. [Google Scholar] [CrossRef]
- Takahara, T.; Furui, K.; Yata, Y.; Jin, B.; Zhang, L.P.; Nambu, S.; Sato, H.; Seiki, M.; Watanabe, A. Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology 1997, 26, 1521–1529. [Google Scholar] [CrossRef]
- Lichtinghagen, R.; Bahr, M.J.; Wehmeier, M.; Michels, D.; Haberkorn, C.I.; Arndt, B.; Flemming, P.; Manns, M.P.; Boeker, K.H. Expression and coordinated regulation of matrix metalloproteinases in chronic hepatitis C and hepatitis C virus-induced liver cirrhosis. Clin. Sci. 2003, 105, 373–382. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Rice, A.; Pinato, D.; Rombouts, K.; Del Rio Hernandez, A. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci. Rep. 2019, 9, 7299. [Google Scholar] [CrossRef]
- Prystupa, A.; Boguszewska-Czubara, A.; Bojarska-Junak, A.; Toruń-Jurkowska, A.; Roliński, J.; Załuska, W. Activity of MMP-2, MMP-8 and MMP-9 in serum as a marker of progression of alcoholic liver disease in people from Lublin Region, eastern Poland. Ann. Agric. Environ. Med. 2015, 22, 325–328. [Google Scholar] [CrossRef]
- Arriazu, E.; Ruiz de Galarreta, M.; Cubero, F.J.; Varela-Rey, M.; Pérez de Obanos, M.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid. Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef]
- Thiele, N.D.; Wirth, J.W.; Steins, D.; Koop, A.C.; Ittrich, H.; Lohse, A.W.; Kluwe, J. TIMP-1 is upregulated, but not essential in hepatic fibrogenesis and carcinogenesis in mice. Sci. Rep. 2017, 7, 714. [Google Scholar] [CrossRef]
- Latronico, T.; Mascia, C.; Pati, I.; Zuccala, P.; Mengoni, F.; Marocco, R.; Tieghi, T.; Belvisi, V.; Lichtner, M.; Vullo, V.; et al. Liver Fibrosis in HCV Monoinfected and HIV/HCV Coinfected Patients: Dysregulation of Matrix Metalloproteinases (MMPs) and Their Tissue Inhibitors TIMPs and Effect of HCV Protease Inhibitors. Int. J. Mol. Sci. 2016, 17, 455. [Google Scholar] [CrossRef]
- Parsons, C.J.; Bradford, B.U.; Pan, C.Q.; Cheung, E.; Schauer, M.; Knorr, A.; Krebs, B.; Kraft, S.; Zahn, S.; Brocks, B.; et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology 2004, 40, 1106–1115. [Google Scholar] [CrossRef]
- Kitto, L.J.; Henderson, N.C. Hepatic Stellate Cell Regulation of Liver Regeneration and Repair. Hepatol. Commun. 2020, 5, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Ho, C.T.; Liong, E.C.; Nanji, A.A.; Leung, T.M.; Lau, T.Y.; Fung, M.L.; Tipoe, G.L. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur. J. Nutr. 2014, 53, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Gan, F.; Liu, Q.; Liu, Y.; Huang, D.; Pan, C.; Song, S.; Huang, K. Lycium barbarum polysaccharides improve CCl4-induced liver fibrosis, inflammatory response and TLRs/NF-kB signaling pathway expression in wistar rats. Life Sci. 2018, 192, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lu, X.; Zhang, X. Noncanonical NF-κB Signaling Pathway in Liver Diseases. J. Clin. Transl. Hepatol. 2021, 9, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Gardam, S.; Brink, R. Non-Canonical NF-κB Signaling Initiated by BAFF Influences B Cell Biology at Multiple Junctures. Front. Immunol. 2014, 4, 509. [Google Scholar] [CrossRef]
- Feng, J.; Chen, K.; Xia, Y.; Wu, L.; Li, J.; Li, S.; Wang, W.; Lu, X.; Liu, T.; Guo, C. Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-κB and TGF-β1/Smad3 pathways. Drug Des. Dev. Ther. 2018, 12, 1837–1853. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Wang, Y.H.; Suk, F.M.; Liu, C.L.; Chen, T.L.; Twu, Y.C.; Hsu, M.H.; Liao, Y.J. Antifibrotic Effects of a Barbituric Acid Derivative on Liver Fibrosis by Blocking the NF-κB Signaling Pathway in Hepatic Stellate Cells. Front. Pharmacol. 2020, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Cheng, X.; Zeng, F.; Xu, Q.; Hou, L. The Role of TGF-β/SMAD Signaling in Hepatocellular Carcinoma: From Mechanism to Therapy and Prognosis. Int. J. Biol. Sci. 2024, 20, 1436–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, N.; Ping, J.; Xu, L. TGF-β1-induced autophagy activates hepatic stellate cells via the ERK and JNK signaling pathways. Int. J. Mol. Med. 2021, 47, 256–266. [Google Scholar] [CrossRef]
- Gungor, M.Z.; Uysal, M.; Senturk, S. The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers 2022, 14, 940. [Google Scholar] [CrossRef]
- Cao, Q.; Mak, K.M.; Lieber, C.S. DLPC decreases TGF-beta1-induced collagen mRNA by inhibiting p38 MAPK in hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G1051–G1061. [Google Scholar] [CrossRef]
- Popov, Y.; Patsenker, E.; Stickel, F.; Zaks, J.; Bhaskar, K.R.; Niedobitek, G.; Kolb, A.; Friess, H.; Schuppan, D. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J. Hepatol. 2008, 48, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.O.; Gokhan, G.A.; Bozova, S. Thrombospondin-1 expression correlates with angiogenesis in experimental cirrhosis. World J. Gastroenterol. 2008, 14, 2213–2217. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Herrera, B.; Alvarez, A.M.; Beltrán, J.; Valdés, F.; Fabregat, I.; Fernández, M. Resistance to TGF-beta-induced apoptosis in regenerating hepatocytes. J. Cell. Physiol. 2004, 201, 385–392. [Google Scholar] [CrossRef]
- Yuan, B.; Dong, R.; Shi, D.; Zhou, Y.; Zhao, Y.; Miao, M.; Jiao, B. Down-regulation of miR-23b may contribute to activation of the TGF-β1/Smad3 signalling pathway during the termination stage of liver regeneration. FEBS Lett. 2011, 585, 927–934. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef]
- Proell, V.; Carmona-Cuenca, I.; Murillo, M.M.; Huber, H.; Fabregat, I.; Mikulits, W. TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp. Hepatol. 2007, 6, 1. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Copple, B.L. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. 2010, 30, 669–682. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Bottinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad. Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Sancho, P.; Fabregat, I. NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem. 2010, 285, 24815–24824. [Google Scholar] [CrossRef]
- Murillo, M.M.; Carmona-Cuenca, I.; Del Castillo, G.; Ortiz, C.; Roncero, C.; Sánchez, A.; Fernández, M.; Fabregat, I. Activation of NADPH oxidase by transforming growth factor-beta in hepatocytes mediates up-regulation of epidermal growth factor receptor ligands through a nuclear factor-kappaB-dependent mechanism. Biochem. J. 2007, 405, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell. Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jia, X.; Qu, M.; Yang, X.; Fang, Y.; Ying, X.; Zhang, M.; Wei, J.; Pan, Y. Exploring the potential of treating chronic liver disease targeting the PI3K/Akt pathway and polarization mechanism of macrophages. Heliyon 2023, 9, e17116. [Google Scholar] [CrossRef]
- Czochra, P.; Klopcic, B.; Meyer, E.; Herkel, J.; Garcia-Lazaro, J.F.; Thieringer, F.; Schirmacher, P.; Biesterfeld, S.; Galle, P.R.; Lohse, A.W.; et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J. Hepatol. 2006, 45, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, Z.; Lei, Y.; Han, X.; Tian, D.; Gong, J.; Liu, M. Celastrol Alleviates Autoimmune Hepatitis Through the PI3K/AKT Signaling Pathway Based on Network Pharmacology and Experiments. Front. Pharmacol. 2022, 13, 816350. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Deng, X.; Jiang, Q.; Li, G.; Zhang, J.; Zhang, N.; Xin, S.; Xu, K. Scoparone improves hepatic inflammation and autophagy in mice with nonalcoholic steatohepatitis by regulating the ROS/P38/Nrf2 axis and PI3K/AKT/mTOR pathway in macrophages. Biomed. Pharmacother. 2020, 125, 109895. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, S.M.; Hur, W.; Kang, B.Y.; Lee, H.L.; Nam, H.; Yoo, S.H.; Sung, P.S.; Kwon, J.H.; Jang, J.W.; et al. Tenofovir disoproxil fumarate directly ameliorates liver fibrosis by inducing hepatic stellate cell apoptosis via downregulation of PI3K/Akt/mTOR signaling pathway. PLoS ONE 2021, 16, e0261067. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329.e11. [Google Scholar] [CrossRef]
- Nybakken, K.; Perrimon, N. Hedgehog signal transduction: Recent findings. Curr. Opin. Genet. Dev. 2002, 12, 503–511. [Google Scholar] [CrossRef]
- Choi, S.S.; Omenetti, A.; Witek, R.P.; Moylan, C.A.; Syn, W.K.; Jung, Y.; Yang, L.; Sudan, D.L.; Sicklick, J.K.; Michelotti, G.A.; et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1093–G1106. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Ju, H.L.; Cho, K.J.; Kim, D.Y.; Han, K.H.; Eun, J.W.; Nam, S.W.; Ribback, S.; Dombrowski, F.; et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J. Hepatol. 2016, 64, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.X.; Melhem, A.; Schmelzer, E.; Zdanowicz, M.; Huang, J.; Caballero, M.; Fair, J.H.; Ludlow, J.W.; McClelland, R.E.; et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G859–G870. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; Nash, C.R.N. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, N.; Abo-ElMatty, D.M.; Saleh, S.; Ghattas, M.H.; Omar, N.N. Empagliflozin suppresses hedgehog pathway, alleviates ER stress, and ameliorates hepatic fibrosis in rats. Sci. Rep. 2023, 13, 19046. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Li, H.; Li, J.; Li, X.; Li, Y.; Shi, Y.; Wang, D. Sonic hedgehog signaling pathway mediates development of hepatocellular carcinoma. Tumour Biol. 2016, 37, 16199–16205. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Schulze, K.L.; Bellen, H.J. Introduction to Notch signaling. Methods Mol. Biol. 2014, 1187, 1–14. [Google Scholar] [CrossRef]
- Gérard, C.; Tys, J.; Lemaigre, F.P. Gene regulatory networks in differentiation and direct reprogramming of hepatic cells. Semin. Cell Dev. Biol. 2017, 66, 43–50. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, J.; Li, X.; Li, B.; Tao, K.; Yue, S. Notch signaling pathway regulates cell cycle in proliferating hepatocytes involved in liver regeneration. J. Gastroenterol. Hepatol. 2018, 33, 1538–1547. [Google Scholar] [CrossRef]
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690. [Google Scholar] [CrossRef]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Qiang, L.; Kangsamaksin, T.; Kitajewski, J.; Ginsberg, H.N.; Accili, D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med. 2013, 19, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, Y.; Wang, Y.; Wang, Y.; Zheng, G.; Xu, H. Hepatitis B virus X protein activates Notch signaling by its effects on Notch1 and Notch4 in human hepatocellular carcinoma. Int. J. Oncol. 2016, 48, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Takegami, T.; Shiozaki, T.; Miyazaki, T. Hepatitis C virus NS3 protein can activate the Notch-signaling pathway through binding to a transcription factor, SRCAP. PLoS ONE 2011, 6, e20718. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.C.; Liu, X.; Liu, X.H.; Li, Z.S.; Zhu, G.Z. Notch Signaling Regulates Circulating T Helper 22 Cells in Patients with Chronic Hepatitis, C. Viral Immunol. 2017, 30, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, C.; Fornari, F.; Piscaglia, F.; Gramantieri, L. Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs. Cells 2021, 10, 521. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Ren, Q.G.; Zhang, T.; Pan, X.; Wen, L.; Hu, J.L.; Yu, C.; He, Q.J. Hepatitis B virus X protein and hypoxia-inducible factor-1α stimulate Notch gene expression in liver cancer cells. Oncol. Rep. 2017, 37, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Rodriguez-Carunchio, L.; Solé, M.; Thung, S.; Stanger, B.Z.; et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012, 143, 1660–1669.e7. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Neufeld, G.; Kessler, O. Pro-angiogenic cytokines and their role in tumor angiogenesis. Cancer Metastasis Rev. 2006, 25, 373–385. [Google Scholar] [CrossRef]
- Sanz-Cameno, P.; Trapero-Marugán, M.; Chaparro, M.; Jones, E.A.; Moreno-Otero, R. Angiogenesis: From chronic liver inflammation to hepatocellular carcinoma. J. Oncol. 2010, 2010, 272170. [Google Scholar] [CrossRef] [PubMed]
- Paternostro, C.; David, E.; Novo, E.; Parola, M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J. Gastroenterol. 2010, 16, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Szabo, G. Hypoxia and hypoxia inducible factors: Diverse roles in liver diseases. Hepatology 2012, 55, 622–633. [Google Scholar] [CrossRef]
- Thurston, G.; Rudge, J.S.; Ioffe, E.; Zhou, H.; Ross, L.; Croll, S.D.; Glazer, N.; Holash, J.; McDonald, D.M.; Yancopoulos, G.D. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat. Med. 2000, 6, 460–463. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Liu, Z.S.; Sun, Q. Expression of angiopoietins, Tie2 and vascular endothelial growth factor in angiogenesis and progression of hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 4241–4245. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Cameno, P.; Martín-Vílchez, S.; Lara-Pezzi, E.; Borque, M.J.; Salmerón, J.; Muñoz de Rueda, P.; Solís, J.A.; López-Cabrera, M.; Moreno-Otero, R. Hepatitis B virus promotes angiopoietin-2 expression in liver tissue: Role of HBV x protein. Am. J. Pathol. 2006, 169, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127 (Suppl. S1), S35–S50. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the background of liver fibrosis: Implications for the management of hepatocellular cancer. World J. Gastroenterol. 2018, 24, 4436–4447. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6, Erratum in Nat. Rev. Dis. Primers 2024, 10, 10. [Google Scholar] [CrossRef]
- McGovern, B.H.; Golan, Y.; Lopez, M.; Pratt, D.; Lawton, A.; Moore, G.; Epstein, M.; Knox, T.A. The impact of cirrhosis on CD4+ T cell counts in HIV-seronegative patients. Clin. Infect. Dis. 2007, 44, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, H.; Lee, N.Y. Multifaceted roles of TAK1 signaling in cancer. Oncogene 2020, 39, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef]
- Song, I.J.; Yang, Y.M.; Inokuchi-Shimizu, S.; Roh, Y.S.; Yang, L.; Seki, E. The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice. Int. J. Cancer 2018, 142, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Cui, J.; Qin, Q.; Zhang, J.; Liu, L.; Deng, S.; Wu, C.; Yang, M.; Li, S.; Wang, C. Mechanical stiffness of liver tissues in relation to integrin β1 expression may influence the development of hepatic cirrhosis and hepatocellular carcinoma. J. Surg. Oncol. 2010, 102, 482–489. [Google Scholar] [CrossRef]
- Xie, B.; Lin, W.; Ye, J.; Wang, X.; Zhang, B.; Xiong, S.; Li, H.; Tan, G. DDR2 facilitates hepatocellular carcinoma invasion and metastasis via activating ERK signaling and stabilizing SNAIL1. J. Exp. Clin. Cancer Res. 2015, 34, 101. [Google Scholar] [CrossRef]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. 2020, 245, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Roeb, E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018, 68-69, 463–473. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.R. Interferon-alfa, interferon-lambda and hepatitis, C. Nat. Genet. 2009, 41, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Hashem, A.M.; Leung, R.; Romero-Gomez, M.; Berg, T.; Dore, G.J.; Chan, H.L.; Irving, W.L.; Sheridan, D.; Abate, M.L.; et al. Interferon-λ rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease. Nat. Commun. 2015, 6, 6422. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Lin, C.H.; Huang, Y.H.; Huo, T.I.; Su, C.W.; Hou, M.C.; Huang, H.C.; Lee, K.C.; Chan, C.C.; Lin, M.W.; et al. IL28B polymorphism correlates with active hepatitis in patients with HBeAg-negative chronic hepatitis B. PLoS ONE 2013, 8, e58071. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genome-Wide Association Studies and Hepatitis C: Harvesting the Benefits of the Genomic Revolution. Semin. Liver Dis. 2015, 35, 402–420. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Valenti, L.; Tuttolomondo, A.; Dongiovanni, P.; Pipitone, R.M.; Cammà, C.; Cabibi, D.; Di Marco, V.; Fracanzani, A.L.; Badiali, S.; et al. Interferon lambda 4 rs368234815 TT>δG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. Hepatology 2017, 66, 1885–1893. [Google Scholar] [CrossRef]
- Uygun, A.; Ozturk, K.; Demirci, H.; Oztuna, A.; Eren, F.; Kozan, S.; Yilmaz, Y.; Kurt, O.; Turker, T.; Vatansever, S.; et al. The association of nonalcoholic fatty liver disease with genetic polymorphisms: A multicenter study. Eur. J. Gastroenterol. Hepatol. 2017, 29, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Rauff, B.; Amar, A.; Chudhary, S.A.; Mahmood, S.; Tayyab, G.U.N.; Hanif, R. Interferon-λ rs12979860 genotype association with liver fibrosis in chronic hepatitis C (CHC) patients in the Pakistani population. Arch. Virol. 2021, 166, 1047–1056. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients with Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e17. [Google Scholar] [CrossRef]
- Noureddin, M.; Wright, E.C.; Alter, H.J.; Clark, S.; Thomas, E.; Chen, R.; Zhao, X.; Conry-Cantilena, C.; Kleiner, D.E.; Liang, T.J.; et al. Association of IL28B genotype with fibrosis progression and clinical outcomes in patients with chronic hepatitis C: A longitudinal analysis. Hepatology 2013, 58, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.S.; Read, S.A.; Wang, M.; Schibeci, S.; Eslam, M.; Ong, A.; Weltman, M.D.; Douglas, M.W.; Mazzola, A.; Craxì, A.; et al. IFNL3/4 genotype is associated with altered immune cell populations in peripheral blood in chronic hepatitis C infection. Genes Immun. 2016, 17, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; Wijaya, R.; Ramezani-Moghadam, M.; Tay, E.; Schibeci, S.; Liddle, C.; Lam, V.W.T.; Yuen, L.; Douglas, M.W.; Booth, D.; et al. Macrophage Coordination of the Interferon Lambda Immune Response. Front. Immunol. 2019, 10, 2674. [Google Scholar] [CrossRef]
- Eslam, M.; McLeod, D.; Kelaeng, K.S.; Mangia, A.; Berg, T.; Thabet, K.; Irving, W.L.; Dore, G.J.; Sheridan, D.; Grønbæk, H.; et al. IFN-λ3, not IFN-λ4, likely mediates IFNL3-IFNL4 haplotype-dependent hepatic inflammation and fibrosis. Nat. Genet. 2017, 49, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Onabajo, O.O.; Wang, F.; Lee, M.H.; Florez-Vargas, O.; Obajemu, A.; Tanikawa, C.; Vargas, J.M.; Liao, S.F.; Song, C.; Huang, Y.H.; et al. Intracellular Accumulation of IFN-λ4 Induces ER Stress and Results in Anti-Cirrhotic but Pro-HCV Effects. Front. Immunol. 2021, 12, 692263. [Google Scholar] [CrossRef]
- Patin, E.; Kutalik, Z.; Guergnon, J.; Bibert, S.; Nalpas, B.; Jouanguy, E.; Munteanu, M.; Bousquet, L.; Argiro, L.; Halfon, P.; et al. Genome-wide association study identifies variants associated with progression of liver fibrosis from HCV infection. Gastroenterology 2012, 143, 1244–1252.e12. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Pauer, G.J.; Marmorstein, A.D.; Crabb, J.W.; Hagstrom, S.A. Tubby-like protein 1 (TULP1) interacts with F-actin in photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4754–4761. [Google Scholar] [CrossRef]
- Jia, D.; Gao, P.; Lv, Y.; Huang, Y.; Reilly, J.; Sun, K.; Han, Y.; Hu, H.; Chen, X.; Zhang, Z.; et al. Tulp1 deficiency causes early-onset retinal degeneration through affecting ciliogenesis and activating ferroptosis in zebrafish. Cell Death Dis. 2022, 13, 962. [Google Scholar] [CrossRef] [PubMed]
- Devane, J.; Ott, E.; Olinger, E.G.; Epting, D.; Decker, E.; Friedrich, A.; Bachmann, N.; Renschler, G.; Eisenberger, T.; Briem-Richter, A.; et al. Progressive liver, kidney, and heart degeneration in children and adults affected by TULP3 mutations. Am. J. Hum. Genet. 2022, 109, 928–943. [Google Scholar] [CrossRef]
- Cai, B.; Kasikara, C.; Doran, A.C.; Ramakrishnan, R.; Birge, R.B.; Tabas, I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci. Signal. 2018, 11, eaar3721. [Google Scholar] [CrossRef]
- Dransfield, I.; Zagórska, A.; Lew, E.D.; Michail, K.; Lemke, G. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 2015, 6, e1646. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, M.; Pan, G.; Nord, H.; Wallén Arzt, E.; Wallerman, O.; Wadelius, C. Genetic prevention of hepatitis C virus-induced liver fibrosis by allele-specific downregulation of MERTK. Hepatol. Res. 2017, 47, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tan, M.; Duan, H.; Swaroop, M. SAG/ROC/Rbx/Hrt, a zinc RING finger gene family: Molecular cloning, biochemical properties, and biological functions. Antioxid. Redox Signal. 2001, 3, 635–650. [Google Scholar] [CrossRef]
- Kupcinskas, J.; Valantiene, I.; Varkalaitė, G.; Steponaitiene, R.; Skieceviciene, J.; Sumskiene, J.; Petrenkiene, V.; Kondrackiene, J.; Kiudelis, G.; Lammert, F.; et al. PNPLA3 and RNF7 Gene Variants are Associated with the Risk of Developing Liver Fibrosis and Cirrhosis in an Eastern European Population. J. Gastrointest. Liver Dis. 2017, 26, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Urabe, Y.; Ochi, H.; Kato, N.; Kumar, V.; Takahashi, A.; Muroyama, R.; Hosono, N.; Otsuka, M.; Tateishi, R.; Lo, P.H.; et al. A genome-wide association study of HCV-induced liver cirrhosis in the Japanese population identifies novel susceptibility loci at the MHC region. J. Hepatol. 2013, 58, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wallace, D.F.; Powell, E.E.; Rahman, T.; Clark, P.J.; Subramaniam, V.N. Gene Variants Implicated in Steatotic Liver Disease: Opportunities for Diagnostics and Therapeutics. Biomedicines 2023, 11, 2809. [Google Scholar] [CrossRef] [PubMed]
- Perttilä, J.; Huaman-Samanez, C.; Caron, S.; Tanhuanpää, K.; Staels, B.; Yki-Järvinen, H.; Olkkonen, V.M. PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1063–E1069. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Claudel, T.; Kumari, P.; Haemmerle, G.; Pollheimer, M.J.; Stojakovic, T.; Scharnagl, H.; Halilbasic, E.; Gumhold, J.; Silbert, D.; et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology 2012, 56, 270–280. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef]
- Rauff, B.; Alzahrani, B.; Chudhary, S.A.; Nasir, B.; Mahmood, S.; Bhinder, M.A.; Faheem, M.; Amar, A. PNPLA3 and TM6SF2 genetic variants and hepatic fibrosis and cirrhosis in Pakistani chronic hepatitis C patients: A genetic association study. BMC Gastroenterol. 2022, 22, 401. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.I.N.; Malta, F.M.; Zitelli, P.M.Y.; Salles, A.P.M.; Gomes-Gouvea, M.S.; Nastri, A.C.S.; Pinho, J.R.R.; Carrilho, F.J.; Oliveira, C.P.; Mendes-Corrêa, M.C.; et al. The role of PNPLA3 and TM6SF2 polymorphisms on liver fibrosis and metabolic abnormalities in Brazilian patients with chronic hepatitis C. BMC Gastroenterol. 2021, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Rady, B.; Nishio, T.; Dhar, D.; Liu, X.; Erion, M.; Kisseleva, T.; Brenner, D.A.; Pocai, A. PNPLA3 downregulation exacerbates the fibrotic response in human hepatic stellate cells. PLoS ONE 2021, 16, e0260721. [Google Scholar] [CrossRef]
- Sandhu, B.; Perez Matos, M.C.; Tran, S.; Zhong, A.; Csizmadia, E.; Kim, M.; Herman, M.A.; Nasser, I.; Lai, M.; Jiang, Z.G. Quantitative digital pathology reveals association of cell-specific PNPLA3 transcription with NAFLD disease activity. JHEP Rep. 2019, 1, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.C.; Blitz, I.L.; Pappano, W.N.; Imamura, Y.; Clark, T.G.; Steiglitz, B.M.; Thomas, C.L.; Maas, S.A.; Takahara, K.; Cho, K.W.; et al. Mammalian BMP-1/Tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev. Biol. 1999, 213, 283–300. [Google Scholar] [CrossRef]
- Matsuura, K.; Sawai, H.; Ikeo, K.; Ogawa, S.; Iio, E.; Isogawa, M.; Shimada, N.; Komori, A.; Toyoda, H.; Kumada, T.; et al. Genome-Wide Association Study Identifies TLL1 Variant Associated With Development of Hepatocellular Carcinoma after Eradication of Hepatitis C Virus Infection. Gastroenterology 2017, 152, 1383–1394. [Google Scholar] [CrossRef]
- John, M.; Metwally, M.; Mangia, A.; Romero-Gomez, M.; Berg, T.; Sheridan, D.; George, J.; Eslam, M. TLL1 rs17047200 Increases the Risk of Fibrosis Progression in Caucasian Patients with Chronic Hepatitis, C. Gastroenterology 2017, 153, 1448–1449. [Google Scholar] [CrossRef]
- Fu, S.; Karim, D.; Prieto, J.; Balderramo, D.; Ferrer, J.D.; Mattos, A.Z.; Arrese, M.; Carrera, E.; Oliveira, J.; Debes, J.D.; et al. Assessment of TLL1 variant and risk of hepatocellular carcinoma in Latin Americans and Europeans. Ann. Hepatol. 2024, 29, 101181. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, E.; Galmozzi, E.; Facchetti, F.; Farina, E.; D’Ambrosio, R.; Soffredini, R.; Iavarone, M.; Lampertico, P. TLL1 variants do not predict hepatocellular carcinoma development in HCV cirrhotic patients treated with direct-acting antivirals. J. Viral Hepat. 2019, 26, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Mohsin, A.K.; Matta, C.; Ghazy, R.M.; Elhadidi, A.; Tahoun, M.; Rahal, A.; Domiaty, D.; Mohamed, N. Tolloid-like 1 gene variant rs17047200, pretreatment FIB-4, ALBI and PALBI scores as predictors of hepatocellular carcinoma occurrence after directly acting antivirals. Clin. Exp. Hepatol. 2022, 8, 330–334. [Google Scholar] [CrossRef]
- Huebener, P.; Schwabe, R.F. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim. Biophys. Acta 2013, 1832, 1005–1017. [Google Scholar] [CrossRef]
- Zhu, Q.; Zou, L.; Jagavelu, K.; Simonetto, D.A.; Huebert, R.C.; Jiang, Z.D.; DuPont, H.L.; Shah, V.H. Intestinal decontamination inhibits TLR4 dependent fibronectin-mediated cross-talk between stellate cells and endothelial cells in liver fibrosis in mice. J. Hepatol. 2012, 56, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Isayama, F.; Hines, I.N.; Kremer, M.; Milton, R.J.; Byrd, C.L.; Perry, A.W.; McKim, S.E.; Parsons, C.; Rippe, R.A.; Wheeler, M.D. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1318–G1328. [Google Scholar] [CrossRef]
- Jagavelu, K.; Routray, C.; Shergill, U.; O’Hara, S.P.; Faubion, W.; Shah, V.H. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 2010, 52, 590–601. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164.e10. [Google Scholar] [CrossRef]
- Huang, H.; Shiffman, M.L.; Friedman, S.; Venkatesh, R.; Bzowej, N.; Abar, O.T.; Rowland, C.M.; Catanese, J.J.; Leong, D.U.; Sninsky, J.J.; et al. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology 2007, 46, 297–306. [Google Scholar] [CrossRef]
- Guo, J.; Loke, J.; Zheng, F.; Hong, F.; Yea, S.; Fukata, M.; Tarocchi, M.; Abar, O.T.; Huang, H.; Sninsky, J.J.; et al. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology 2009, 49, 960–968. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef]
- Zhou, Z.; Kim, J.W.; Qi, J.; Eo, S.K.; Lim, C.W.; Kim, B. Toll-Like Receptor 5 Signaling Ameliorates Liver Fibrosis by Inducing Interferon β-Modulated IL-1 Receptor Antagonist in Mice. Am. J. Pathol. 2020, 190, 614–629. [Google Scholar] [CrossRef]
- Wang, Z.D.; Qiao, Y.L.; Tian, X.F.; Zhang, X.Q.; Zhou, S.X.; Liu, H.X.; Chen, Y. Toll-like receptor 5 agonism protects mice from radiation pneumonitis and pulmonary fibrosis. Asian Pac. J. Cancer Prev. 2012, 13, 4763–4767. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef]
- Coenen, M.; Nischalke, H.D.; Krämer, B.; Langhans, B.; Glässner, A.; Schulte, D.; Körner, C.; Sauerbruch, T.; Nattermann, J.; Spengler, U. Hepatitis C virus core protein induces fibrogenic actions of hepatic stellate cells via toll-like receptor 2. Lab. Investig. 2011, 91, 1375–1382. [Google Scholar] [CrossRef]
- Tan, H.L.; Zain, S.M.; Eng, H.S.; Mohamed, Z.; Mahadeva, S.; Chan, W.K.; Lau, P.C.; Basu, R.C.; Mohamed, R. Allele HLA-DQB1*06 reduces fibrosis score in patients with non-alcoholic fatty liver disease. Hepatol. Res. 2020, 50, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Karrar, A.; Hariharan, S.; Fazel, Y.; Moosvi, A.; Houry, M.; Younoszai, Z.; Jeffers, T.; Zheng, L.; Munkhzul, O.; Hunt, S.; et al. Analysis of human leukocyte antigen allele polymorphism in patients with non alcoholic fatty liver disease. Medicine 2019, 98, e16704. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Gea-Mallorquí, E.; Colin-York, H.; Fritzsche, M.; Gillespie, G.M.; Brackenridge, S. Intracellular trafficking of HLA-E and its regulation. J. Exp. Med. 2023, 220, e20221941. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Araújo, R.C.; Bertol, B.C.; César Dias, F.; Debortoli, G.; Almeida, P.H.; Fernandes Souza, F.; Villanova, M.G.; Ramalho, L.N.Z.; Candolo Martinelli, A.L.; Cruz Castelli, É.D.; et al. HLA-E gene polymorphisms in chronic hepatitis C: Impact on HLA-E liver expression and disease severity. Hum. Immunol. 2021, 82, 177–185. [Google Scholar] [CrossRef]
- de Oliveira Crispim, J.C.; Silva, T.G.; Souto, F.J.; Souza, F.F.; Bassi, C.L.; Soares, C.P.; Zucoloto, S.; Moreau, P.; Martinelli Ade, L.; Donadi, E.A. Upregulation of soluble and membrane-bound human leukocyte antigen G expression is primarily observed in the milder histopathological stages of chronic hepatitis C virus infection. Hum. Immunol. 2012, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Souto, F.J.; Crispim, J.C.; Ferreira, S.C.; da Silva, A.S.; Bassi, C.L.; Soares, C.P.; Zucoloto, S.; Rouas-Freiss, N.; Moreau, P.; Martinelli, A.L.; et al. Liver HLA-G expression is associated with multiple clinical and histopathological forms of chronic hepatitis B virus infection. J. Viral Hepat. 2011, 18, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Chen, H.X.; Zhu, C.C.; Zhang, X.; Xu, H.H.; Zhang, J.G.; Wang, Q.; Zhou, W.J.; Yan, W.H. Aberrant human leucocyte antigen-G expression and its clinical relevance in hepatocellular carcinoma. J. Cell. Mol. Med. 2010, 14, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Amiot, L.; Vu, N.; Rauch, M.; L’Helgoualc’h, A.; Chalmel, F.; Gascan, H.; Turlin, B.; Guyader, D.; Samson, M. Expression of HLA-G by mast cells is associated with hepatitis C virus-induced liver fibrosis. J. Hepatol. 2014, 60, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bahram, S.; Bresnahan, M.; Geraghty, D.E.; Spies, T. A second lineage of mammalian major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. USA 1994, 91, 6259–6263. [Google Scholar] [CrossRef]
- Mizuki, N.; Ando, H.; Kimura, M.; Ohno, S.; Miyata, S.; Yamazaki, M.; Tashiro, H.; Watanabe, K.; Ono, A.; Taguchi, S.; et al. Nucleotide sequence analysis of the HLA class I region spanning the 237-kb segment around the HLA-B and -C genes. Genomics 1997, 42, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Kumar, V.; Kato, N.; Urabe, Y.; Takahashi, A.; Muroyama, R.; Hosono, N.; Otsuka, M.; Tateishi, R.; Omata, M.; Nakagawa, H.; et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat. Genet. 2011, 43, 455–458. [Google Scholar] [CrossRef]
- Sharkawy, R.E.; Bayoumi, A.; Metwally, M.; Mangia, A.; Berg, T.; Romero-Gomez, M.; Abate, M.L.; Irving, W.L.; Sheridan, D.; Dore, G.J.; et al. A variant in the MICA gene is associated with liver fibrosis progression in chronic hepatitis C through TGF-β1 dependent mechanisms. Sci. Rep. 2019, 9, 1439. [Google Scholar] [CrossRef]
- Liu, X.; Xu, J.; Rosenthal, S.; Zhang, L.J.; McCubbin, R.; Meshgin, N.; Shang, L.; Koyama, Y.; Ma, H.Y.; Sharma, S.; et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 2020, 158, 1728–1744.e14. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, S.; Estoppey, D.; Buehler, D.P.; Xiong, Y.; Pizzato, N.; Rietsch, A.; Wu, F.; Leroy, N.; Wunderlin, T.; Claerr, I.; et al. The transcription factor ZNF469 regulates collagen production in liver fibrosis. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed Central]
- Ramos-Lopez, O.; Martinez-Lopez, E.; Roman, S.; Fierro, N.A.; Panduro, A. Genetic, metabolic and environmental factors involved in the development of liver cirrhosis in Mexico. World J. Gastroenterol. 2015, 21, 11552–11566. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Rametta, R.; Dongiovanni, P. Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. Int. J. Mol. Sci. 2018, 19, 3857. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, K.; Chen, T.; Zhang, Y.; Du, X.; Zhao, Y.; Shao, S.; Zheng, L.; Han, T.; Hong, W. Transcriptional factor ATF3 promotes liver fibrosis via activating hepatic stellate cells. Cell Death Dis. 2020, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.; Sanyal, A.J. Gene-Environmental Interactions as Metabolic Drivers of Nonalcoholic Steatohepatitis. Front. Endocrinol. 2021, 12, 665987. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, X.; Cheng, Q.N.; Wu, J.F.; Ai, W.B.; Ma, L. Analysis of key genes and related transcription factors in liver fibrosis based on bioinformatic technology. Int. J. Clin. Exp. Pathol. 2021, 14, 444–454. [Google Scholar] [PubMed]
- Loft, A.; Alfaro, A.J.; Schmidt, S.F.; Pedersen, F.B.; Terkelsen, M.K.; Puglia, M.; Chow, K.K.; Feuchtinger, A.; Troullinaki, M.; Maida, A.; et al. Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell Metab. 2021, 33, 1685–1700.e9. [Google Scholar] [CrossRef]
- Steensels, S.; Qiao, J.; Ersoy, B.A. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites 2020, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Zahra, M.; Azzazy, H.; Moustafa, A. Transcriptional Regulatory Networks in Hepatitis C Virus-induced Hepatocellular Carcinoma. Sci. Rep. 2018, 8, 14234. [Google Scholar] [CrossRef]
- Blagotinšek Cokan, K.; Urlep, Ž.; Moškon, M.; Mraz, M.; Kong, X.Y.; Eskild, W.; Rozman, D.; Juvan, P.; Režen, T. Common Transcriptional Program of Liver Fibrosis in Mouse Genetic Models and Humans. Int. J. Mol. Sci. 2021, 22, 832. [Google Scholar] [CrossRef]
- Nakano, Y.; Kamiya, A.; Sumiyoshi, H.; Tsuruya, K.; Kagawa, T.; Inagaki, Y. A Deactivation Factor of Fibrogenic Hepatic Stellate Cells Induces Regression of Liver Fibrosis in Mice. Hepatology 2020, 71, 1437–1452. [Google Scholar] [CrossRef]
- Li, L.; Diao, S.; Chen, Z.; Zhang, J.; Chen, W.; Wang, T.; Chen, X.; Zhao, Y.; Xu, T.; Huang, C.; et al. DNMT3a-mediated methylation of TCF21/hnRNPA1 aggravates hepatic fibrosis by regulating the NF-κB signaling pathway. Pharmacol. Res. 2023, 193, 106808. [Google Scholar] [CrossRef]
- Arroyo, N.; Villamayor, L.; Díaz, I.; Carmona, R.; Ramos-Rodríguez, M.; Muñoz-Chápuli, R.; Pasquali, L.; Toscano, M.G.; Martín, F.; Cano, D.A.; et al. GATA4 induces liver fibrosis regression by deactivating hepatic stellate cells. JCI Insight 2021, 6, e150059. [Google Scholar] [CrossRef]
- Winkler, M.; Staniczek, T.; Kürschner, S.W.; Schmid, C.D.; Schönhaber, H.; Cordero, J.; Kessler, L.; Mathes, A.; Sticht, C.; Neßling, M.; et al. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J. Hepatol. 2021, 74, 380–393. [Google Scholar] [CrossRef]
- Xie, Z.; Li, Y.; Xiao, P.; Ke, S. GATA3 promotes the autophagy and activation of hepatic stellate cell in hepatic fibrosis via regulating miR-370/HMGB1 pathway. Gastroenterol. Hepatol. 2024, 47, 219–229, (In English and Spanish). [Google Scholar] [CrossRef]
- Jiang, X.; Yang, Z. Multiple biological functions of transcription factor 21 in the development of various cancers. Onco Targets Ther. 2018, 11, 3533–3539. [Google Scholar] [CrossRef]
- Arceci, R.J.; King, A.A.; Simon, M.C.; Orkin, S.H.; Wilson, D.B. Mouse GATA-4: A retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol. Cell. Biol. 1993, 13, 2235–2246. [Google Scholar] [CrossRef]
- Herzig, T.C.; Jobe, S.M.; Aoki, H.; Molkentin, J.D.; Cowley, A.W., Jr.; Izumo, S.; Markham, B.E. Angiotensin II type1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc. Natl. Acad. Sci. USA 1997, 94, 7543–7548. [Google Scholar] [CrossRef]
- Delgado, I.; Carrasco, M.; Cano, E.; Carmona, R.; García-Carbonero, R.; Marín-Gómez, L.M.; Soria, B.; Martín, F.; Cano, D.A.; Muñoz-Chápuli, R.; et al. GATA4 loss in the septum transversum mesenchyme promotes liver fibrosis in mice. Hepatology 2014, 59, 2358–2370. [Google Scholar] [CrossRef]
- Wang, L.; Tankersley, L.R.; Tang, M.; Potter, J.J.; Mezey, E. Regulation of the murine alpha(2)(I) collagen promoter by retinoic acid and retinoid X receptors. Arch. Biochem. Biophys. 2002, 401, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Zardi, E.M.; Navarini, L.; Sambataro, G.; Piccinni, P.; Sambataro, F.M.; Spina, C.; Dobrina, A. Hepatic PPARs: Their role in liver physiology, fibrosis and treatment. Curr. Med. Chem. 2013, 20, 3370–3396. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Guo, C.; Wu, J. Therapeutic potential of PPARγ natural agonists in liver diseases. J. Cell. Mol. Med. 2020, 24, 2736–2748. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; He, J.; Zhang, S.; Wang, H.; Jin, G.; Jin, H.; Cheng, Z.; Tao, X.; Yu, C.; Li, B.; et al. PPARγ Coactivator-1α Suppresses Metastasis of Hepatocellular Carcinoma by Inhibiting Warburg Effect by PPARγ-Dependent WNT/β-Catenin/Pyruvate Dehydrogenase Kinase Isozyme 1 Axis. Hepatology 2021, 73, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, M.; Spencer, H.L.; Porter, L.F.; Burkitt-Wright, E.M.; Bürer, C.; Janecke, A.; Bakshi, M.; Sillence, D.; Al-Hussain, H.; Baumgartner, M.; et al. ZNF469 frequently mutated in the brittle cornea syndrome (BCS) is a single exon gene possibly regulating the expression of several extracellular matrix components. Mol. Genet. Metab. 2013, 109, 289–295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
| Environmental Factor | Genetic Factors Involved | Transcription Factors Activated/Suppressed |
|---|---|---|
| Alcohol | PNPLA3, TM6SF2, MBOAT7, DRD2, CYP2E1 [183,184] | ATF3 [185] |
| MAFLD | PNPLA3, TM6SF2, MTTP, PPAR [186] COL1A1, IGFBP7, DCN, AEBP1, LGALS3BP, THBS2, FBLN2, FBLN5, FBN1, SERPINE1, ADAMTS2, and LUM [182,187] | LXR, FXR, SREBP, PPAR δ, ATF4, ELF3 and GLIS2 ZNF469, RUNX1 and TBX3, ATF3 [182,185,188,189] |
| Hepatotrpic virus infection | IL-28B, IFNL4, APOE, LDLr MMP-1, MMP-3 and MMP9. TLR4, MTTP, MICA [180] | AP-1, PPARγ, and NF-κB [190] |
| Animal models of liver fibrosis | COL1A1, FBN1, BGN, COL6A3, MMP2, FBLN5, LUM, PDGFRB, LOXL1, SMAD2, SMAD4, YAP1, NOTCH1, EP300, p63, NCOR [191] | STAT3, NF-κB1, Sp1 PPAR, HIF1, FOXO3, HDAC2, STAT5b and STAT6 [187] TCF21 [190,192,193] GATA 3,4 [194,195,196] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, A.; Farci, P. Fibrosis and Hepatocarcinogenesis: Role of Gene-Environment Interactions in Liver Disease Progression. Int. J. Mol. Sci. 2024, 25, 8641. https://doi.org/10.3390/ijms25168641
Banerjee A, Farci P. Fibrosis and Hepatocarcinogenesis: Role of Gene-Environment Interactions in Liver Disease Progression. International Journal of Molecular Sciences. 2024; 25(16):8641. https://doi.org/10.3390/ijms25168641
Chicago/Turabian StyleBanerjee, Anindita, and Patrizia Farci. 2024. "Fibrosis and Hepatocarcinogenesis: Role of Gene-Environment Interactions in Liver Disease Progression" International Journal of Molecular Sciences 25, no. 16: 8641. https://doi.org/10.3390/ijms25168641
APA StyleBanerjee, A., & Farci, P. (2024). Fibrosis and Hepatocarcinogenesis: Role of Gene-Environment Interactions in Liver Disease Progression. International Journal of Molecular Sciences, 25(16), 8641. https://doi.org/10.3390/ijms25168641