
Effects of PCNA Stability on the Formation of Mutations


Abstract
:1. Introduction
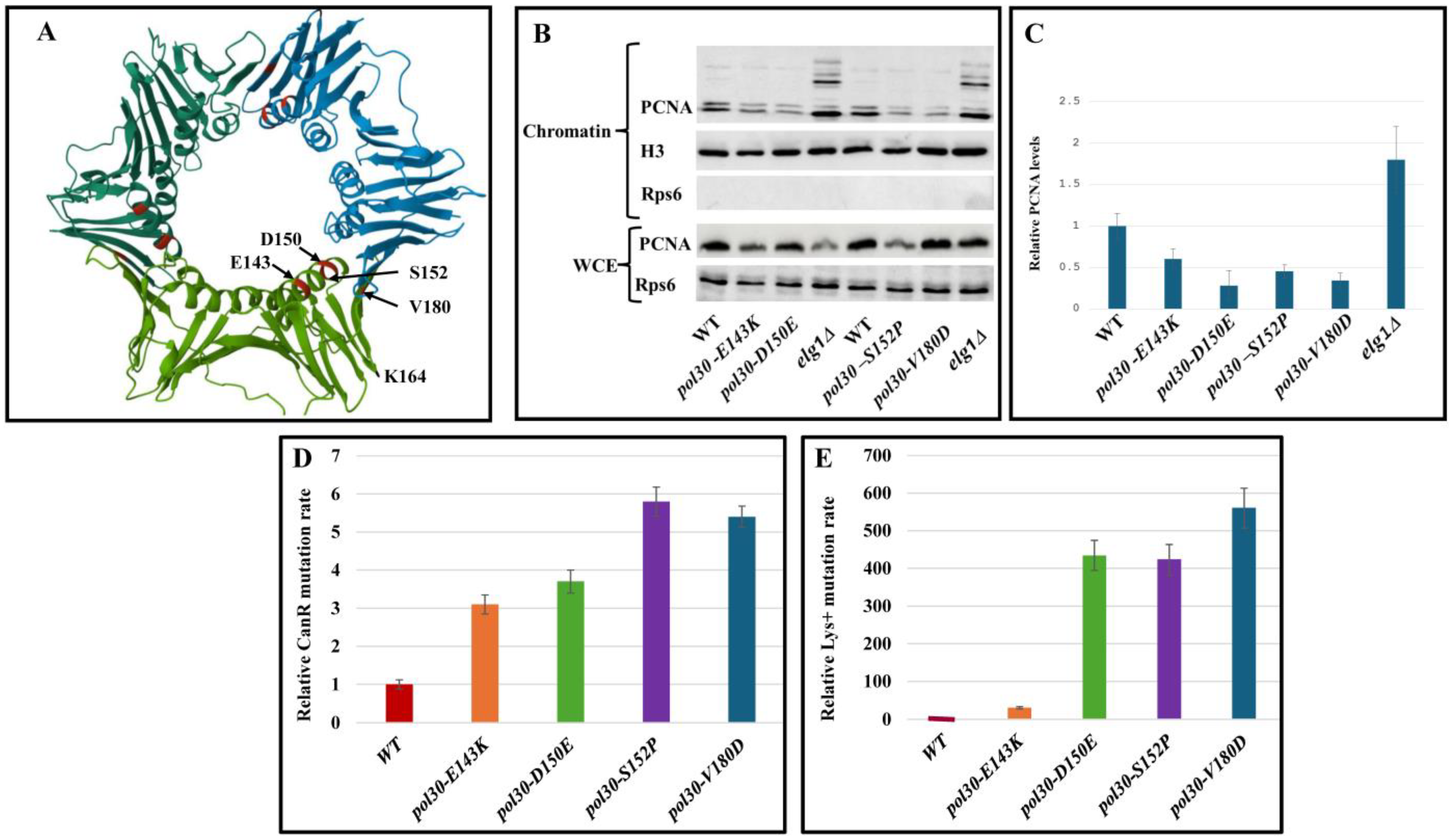
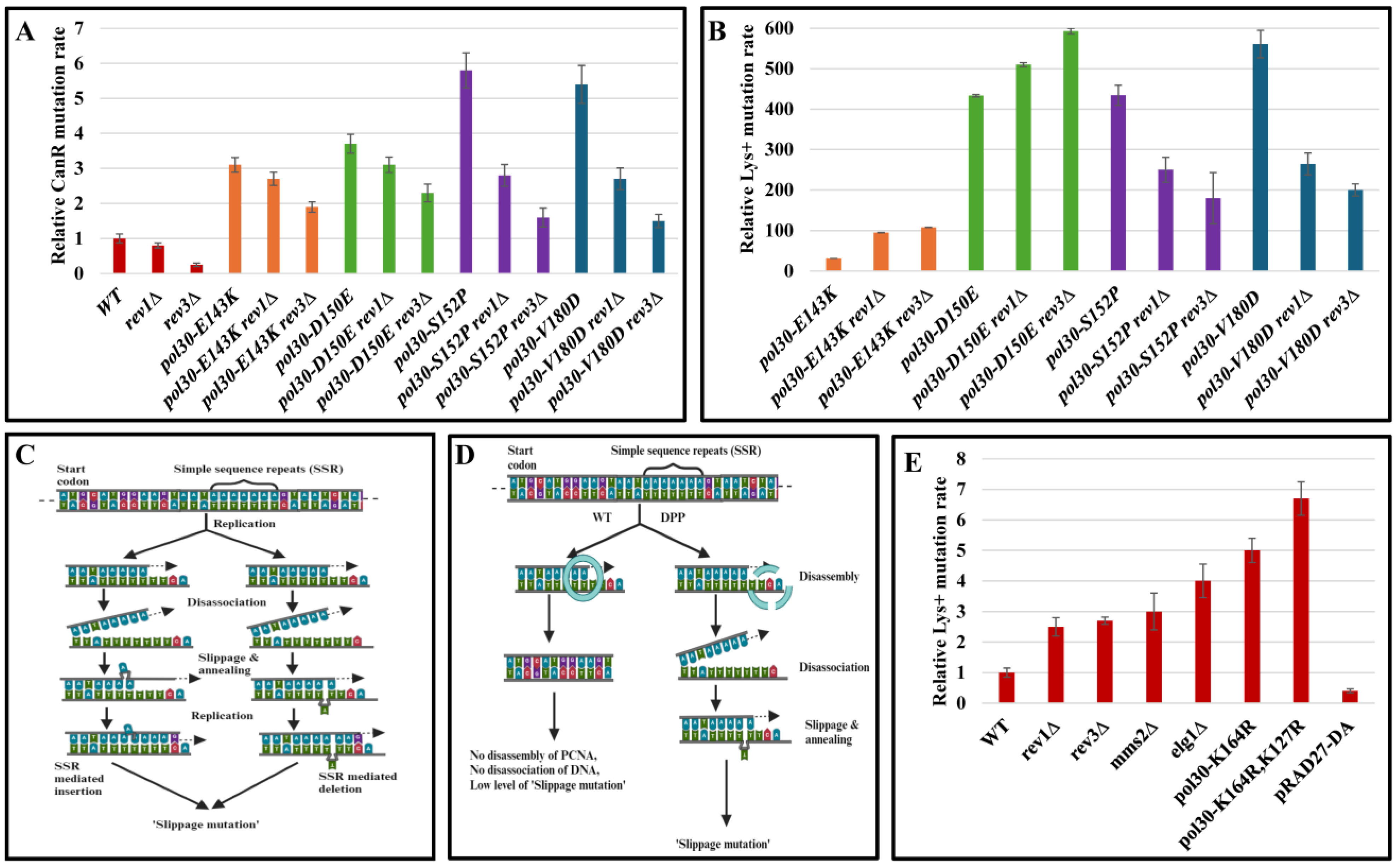
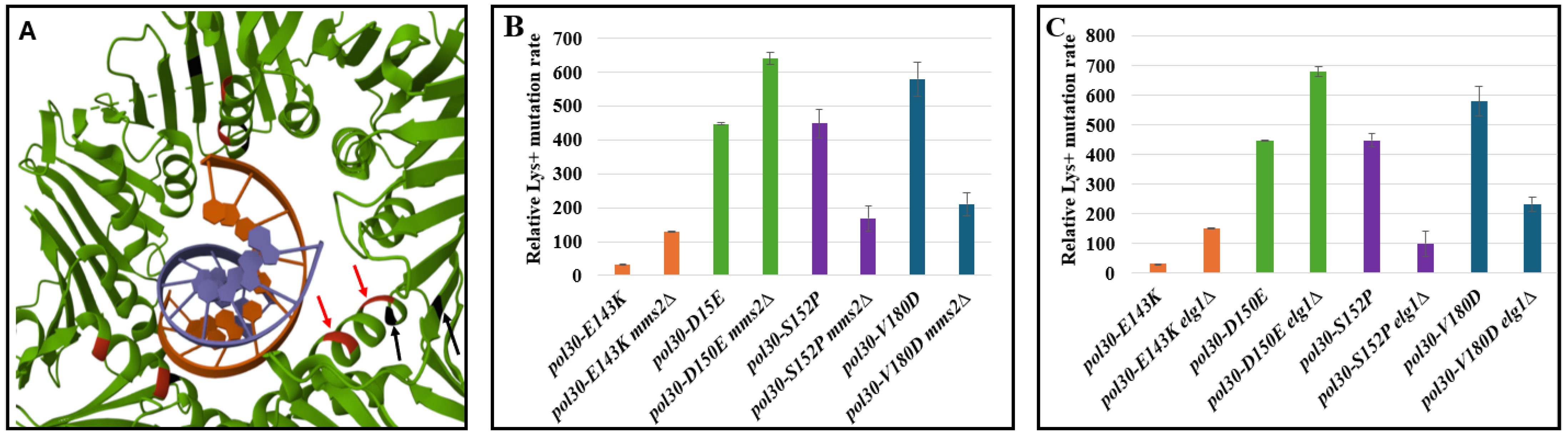
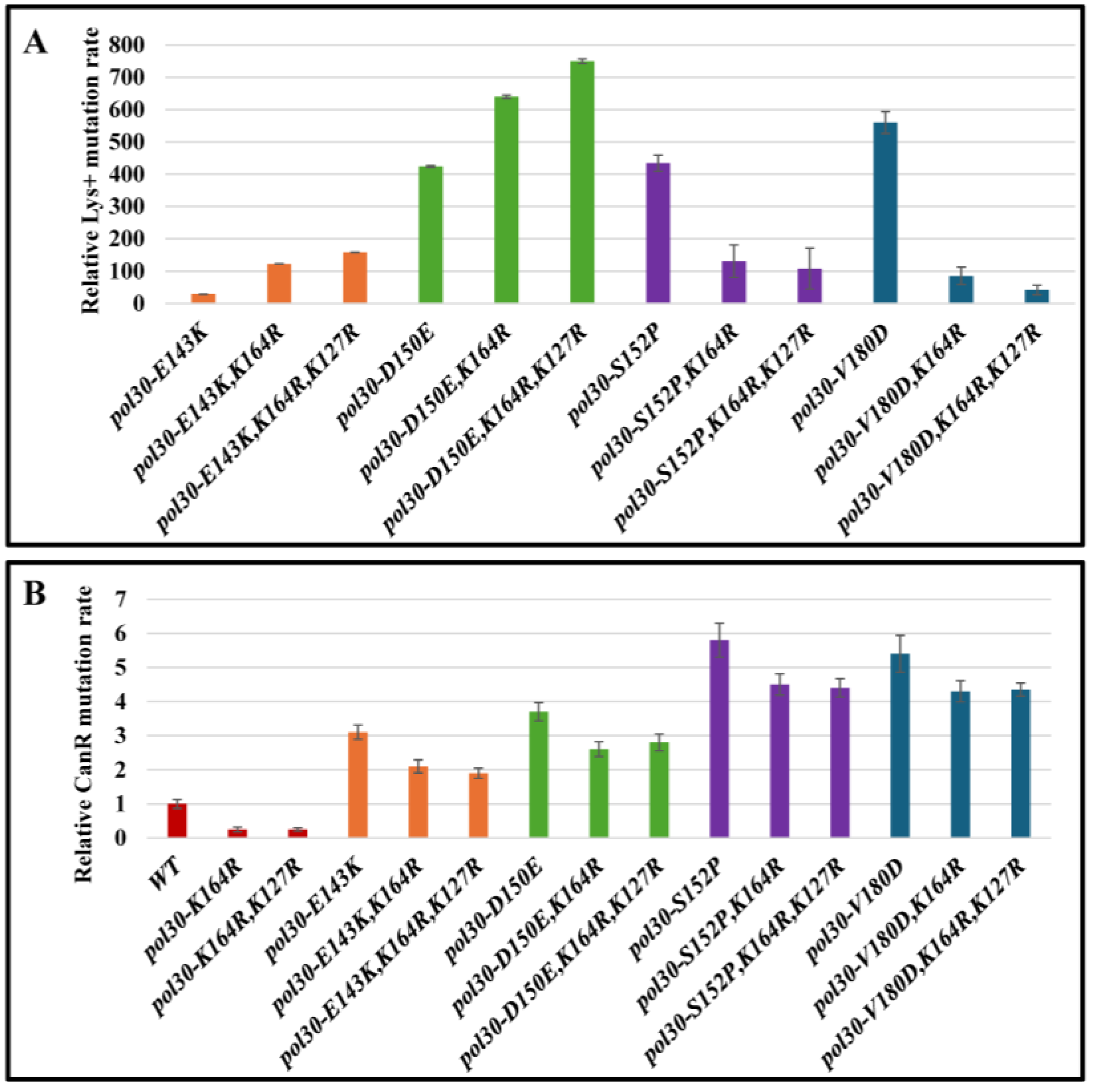
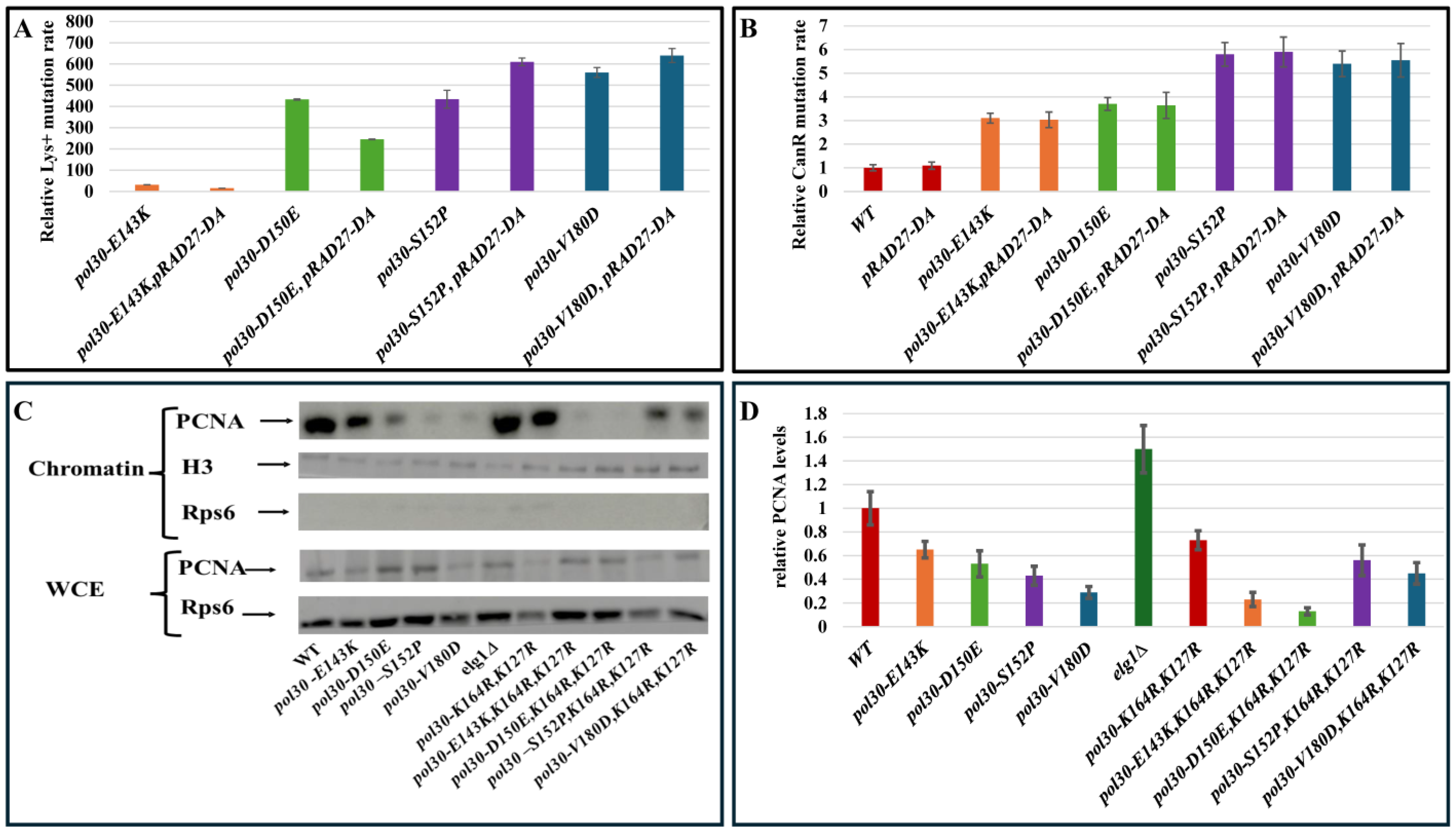
2. Results and Discussion
3. Materials and Methods
3.1. Yeast Strains
3.2. Plasmids
3.3. Growth Media
3.4. Bacteria and Plasmid Extraction
3.5. Determining Yeast Spontaneous Mutation Rate
3.6. Chromatin Fractionation Assay
3.7. Western Blot Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hao, D.; Wang, L.; Di, L.J. Distinct Mutation Accumulation Rates among Tissues Determine the Variation in Cancer Risk. Sci. Rep. 2016, 6, 19458. [Google Scholar] [CrossRef]
- Bębenek, A.; Ziuzia-Graczyk, I. Fidelity of DNA Replication—A Matter of Proofreading. Curr. Genet. 2018, 64, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Barbari, S.R.; Shcherbakova, P.V. Replicative DNA Polymerase Defects in Human Cancers: Consequences, Mechanisms, and Implications for Therapy. DNA Repair 2017, 56, 16–25. [Google Scholar] [CrossRef]
- Iyer, D.R.; Rhind, N. Replication Fork Slowing and Stalling Are Distinct, Checkpoint-Independent Consequences of Replicating Damaged DNA. PLoS Genet. 2017, 13, e1006958. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Boiteux, S.; Jinks-Robertson, S. DNA Repair Mechanisms and the Bypass of DNA Damage in Saccharomyces Cerevisiae. Genetics 2013, 193, 1025–1064. [Google Scholar] [CrossRef] [PubMed]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Arbel, M.; Choudhary, K.; Tfilin, O.; Kupiec, M. PCNA Loaders and Unloaders—One Ring That Rules Them All. Genes 2021, 12, 1812. [Google Scholar] [CrossRef]
- Goellner, E.M.; Smith, C.E.; Campbell, C.S.; Hombauer, H.; Desai, A.; Putnam, C.D.; Kolodner, R.D. PCNA and Msh2-Msh6 Activate an Mlh1-Pms1 Endonuclease Pathway Required for Exo1-Independent Mismatch Repair. Mol. Cell 2014, 55, 291–304. [Google Scholar] [CrossRef]
- Nesta, A.V.; Tafur, D.; Beck, C.R. Hotspots of Human Mutation. Trends Genet. 2021, 37, 717–729. [Google Scholar] [CrossRef]
- Bzymek, M.; Lovett, S.T. Instability of Repetitive DNA Sequences: The Role of Replication in Multiple Mechanisms. Proc. Natl. Acad. Sci. USA 2001, 98, 8319–8325. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R.; Richards, R.I. Simple Tandem DNA Repeats and Human Genetic Disease. Proc. Natl. Acad. Sci. USA 1995, 92, 3636–3641. [Google Scholar] [CrossRef] [PubMed]
- Krishna, T.S.R.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal Structure of the Eukaryotic DNA Polymerase Processivity Factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.S.; Nguyen, M.-N.; Oh, S.; Kolodner, R.D. Exo1-Dependent Mutator Mutations: Model System for Studying Functional Interactions in Mismatch Repair. Mol. Cell. Biol. 2001, 21, 5142–5155. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 Replication Factor C-like Complex Functions in PCNA Unloading during DNA Replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, K.; Sebesta, M.; Pacesa, M.; Sau, S.; Bronstein, A.; Parnas, O.; Liefshitz, B.; Venclovas, Č.; Krejci, L.; Kupiec, M. A Structure-Function Analysis of the Yeast Elg1 Protein Reveals the Importance of PCNA Unloading in Genome Stability Maintenance. Nucleic Acids Res. 2017, 45, 3189–3203. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.J.; Cimprich, K.A. DNA Damage Tolerance: When It’s OK to Make Mistakes. Nat. Chem. Biol. 2009, 5, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Wittschieben, J.P.; Wittschieben, B.; Wood, R.D. DNA Polymerase Zeta (Polζ) in Higher Eukaryotes. Cell Res. 2008, 18, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A Four-Subunit DNA Polymerase ζ Complex Containing Pol δ Accessory Subunits Is Essential for PCNA-Mediated Mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Johnson, R.E.; Prakash, S.; Prakash, L. Complex Formation with Rev1 Enhances the Proficiency of Saccharomyces Cerevisiae DNA Polymerase ζ for Mismatch Extension and for Extension Opposite from DNA Lesions. Mol. Cell. Biol. 2006, 26, 9555–9563. [Google Scholar] [CrossRef]
- Aksenova, A.; Volkov, K.; Maceluch, J.; Pursell, Z.F.; Rogozin, I.B.; Kunkel, T.A.; Pavlov, Y.I.; Johansson, E. Mismatch Repair-Independent Increase in Spontaneous Mutagenesis in Yeast Lacking Non-Essential Subunits of DNA Polymerase ε. PLoS Genet. 2010, 6, 1001209. [Google Scholar] [CrossRef] [PubMed]
- Streisinger, G.; Owen, J. Mechanisms of Spontaneous and Induced Frameshift Mutation in Bacteriophage T4. Genetics 1985, 109, 633–659. [Google Scholar] [CrossRef] [PubMed]
- McNally, R.; Bowman, G.D.; Goedken, E.R.; O’Donnell, M.; Kuriyan, J. Analysis of the Role of PCNA-DNA Contacts during Clamp Loading. BMC Struct. Biol. 2010, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-Dependent DNA Repair Is Linked to Modification of PCNA by Ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Myung, K. PCNA Modifications for Regulation of Post-Replication Repair Pathways. Mol. Cells 2008, 26, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.C.Y.; Li, Y.; Zhang, Q.; Huen, M.S.Y. Molecular Architecture of the Ub-PCNA/Pol ν Complex Bound to DNA. Sci. Rep. 2015, 5, 15759. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Kitano, K.; Yamaguchi, H.; Hamada, K.; Okada, K.; Fukuda, K.; Uchida, M.; Ohtsuka, E.; Morioka, H.; Hakoshima, T. Structural Basis for Recruitment of Human Flap Endonuclease 1 to PCNA. EMBO J. 2005, 24, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, S.; Chow, B.L.; Xiao, W. MMS 2, Encoding a Ubiquitin-Conjugating-Enzyme-like Protein, Is a Member of the Yeast Error-Free Postreplication Repair Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5678–5683. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of Spontaneous and Damage-Induced Mutagenesis by SUMO and Ubiquitin Conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef]
- Munashingha, P.R.; Lee, C.H.; Kang, Y.H.; Shin, Y.K.; Nguyen, T.A.; Seo, Y.S. The Trans-Autostimulatory Activity of Rad27 Suppresses Dna2 Defects in Okazaki Fragment Processing. J. Biol. Chem. 2012, 287, 8675–8687. [Google Scholar] [CrossRef]
- Reagan, M.S.; Pittenger, C.; Siede, W.; Friedberg, E.C. Characterization of a Mutant Strain of Saccharomyces Cerevisiae with a Deletion of the RAD27 Gene, a Structural Homolog of the RAD2 Nucleotide Excision Repair Gene. J. Bacteriol. 1995, 177, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.R.; Gallo, D.; Leung, W.; Croissant, T.; Thu, Y.M.; Nguyen, H.D.; Starr, T.K.; Brown, G.W.; Bielinsky, A.K. Flap Endonuclease Overexpression Drives Genome Instability and DNA Damage Hypersensitivity in a PCNA-Dependent Manner. Nucleic Acids Res. 2018, 46, 5634–5650. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Groissman, M.; Liefshitz, B.; Katz, N.; Kuryachiy, M.; Kupiec, M. PCNA Unloading Is Crucial for the Bypass of DNA Lesions Using Homologous Recombination. Int. J. Mol. Sci. 2024, 25, 3359. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Keen, J.D.; Kricker, M.; Resnick, M.A.; Gordenin, D.A. Hypermutability of Homonucleotide Runs in Mismatch Repair and DNA Polymerase Proofreading Yeast Mutants. Mol. Cell. Biol. 1997, 17, 2859–2865. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, C.H.; Demin, A.A.; Munashingha, P.R.; Amangyeld, T.; Kwon, B.; Formosa, T.; Seo, Y.S. Rad52/Rad59-Dependent Recombination as a Means to Rectify Faulty Okazaki Fragment Processing. J. Biol. Chem. 2014, 289, 15064–15079. [Google Scholar] [CrossRef] [PubMed]
- Liefshitz, B.; Steinlauf, R.; Friedl, A.; Eckardt-Schupp, F.; Kupiec, M. Genetic Interactions between Mutants of the “error-Prone” Repair Group of Saccharomyces Cerevisiae and Their Effect on Recombination and Mutagenesis. Mutat. Res. DNA Repair 1998, 407, 135–145. [Google Scholar] [CrossRef]
- Lea, D.E.; Coulson, C.A. The Distribution of the Numbers of Mutants in Bacterial Populations. J. Genet. 1949, 49, 264–285. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Relevant Genotype | Source and Number in Lab Stock |
|---|---|---|
| E134 | MATα ade5-1 lys2::InsE-A14 his7-2 leu2-3,112 trp1-289 ura3-52 | [34] |
| E134 pol30-S152P | Same, POL30-S152P:LEU2 | This work 17,821 |
| E134 pol30-V180D | Same, pol30-V180D:LEU2 | This work 17,822 |
| E134 pol30-E143K | Same, pol30-E143K:LEU2 | This work 17,823 |
| E134 pol30-D150E | Same, pol30-D150E:LEU2 | This work 17,824 |
| E134 pol30-S152P mms2∆ | Same, pol30-S15SP:LEU2, mms2::KanMX | This work 17,825 |
| E134 pol30-S152P rev1∆ | Same, POL30-S152P:LEU2, rev1::URA3 | This work 17,826 |
| E134 pol30-S152P rev3∆ | Same, pol30-S152P:LEU2, rev3::KanMX | This work 18,081 |
| E134 pol30-S152P elg1∆ | Same, pol30-S152P:LEU2, elg1::HYG | This work 18,758 |
| E134 pol30-S152P rev1∆, rev3∆ | Same, pol30-S152P:LEU2, rev1::URA3, rev3::KanMX | This work 19,204 |
| E134 POL30-V180D mms2∆ | Same, pol30-V180D:LEU2, mms2::KanMX | This work 17,827 |
| E134 POL30-V180D rev1∆ | Same, pol3030-V180D:LEU2, rev1::URA3 | This work 17,831 |
| E134 POL30-V180D rev3∆ | Same, pol30-V1D80:LEU2, rev3::KanMX | This work 18,082 |
| E134 pol30-V180D elg1∆ | Same, pol30-V180D:LEU2, elg1::HygMX | This work 18,065 |
| E134 POL30-V180D rev1∆, rev3∆ | Same, pol3030-V180D:LEU2, rev1::URA3, rev3::KanMX | This work 19,205 |
| E134 POL30-E143K mms2∆ | Same, pol3030-E143K:LEU2, mms2::KanMX | This work 18,367 |
| E134 pol30-E143K rev1∆ | Same, pol30-E143K:LEU2, rev1::URA3 | This work 17,829 |
| E134 pol30-E143K rev3∆ | Same, pol30-E143K:LEU2, rev3::KanMX | This work 18,368 |
| E134 pol30-E143K elg1∆ | Same, pol30-E143K:LEU2, Elg1::HygMX | This work 18,052 |
| E134 POL30-E143K rev1∆, rev3∆ | Same, pol30-E143K:LEU2, rev1::URA3, rev3::KanMX | This work 18,366 |
| E134 pol30-D150E mms2∆ | Same, POL30-D150E:LEU2, mms2::KanMX | This work 18,912 |
| E134 pol30-D150E rev1∆ | Same, pol30-D150E:LEU2, rev1::URA3 | This work 17,828 |
| E134 pol30-D150E rev3∆ | Same, pol30-D150E:LEU2, rev3::KanMX | This work 18,083 |
| E134 pol30-D150E elg1∆ | Same, pol30-D150E:LEU2, elg1::HygMX | This work 17,830 |
| E134 pol30-D150E rev1∆, rev3∆ | Same, pol30-D150E:LEU2, rev1::URA3, rev3::KanMX | This work 19,203 |
| E134 pol30-S152P,K164R | Same, pol30-S152P, K164R:LEU2 | This work 19,684 |
| E134 pol30-V180D,K164R | Same, pol30-V180D, K164R:LEU2 | This work 19,311 |
| E134 pol30-E143K,K164R | Same, pol30-E143K, K164R:LEU2 | This work 19,525 |
| E134 pol30-D150E,K164R | Same, pol30-D150E, K164R, K127R:LEU2 | This work 19,604 |
| E134 pol30-S152P,K164R,K127R | Same, pol30-S152P, K164R, K127R:LEU2 | This work 19,685 |
| E134 pol30-V180D,K164R,K127R | Same, pol30-V180D, K164R, K127R:LEU2 | This work 19,678 |
| E134 pol30-E143K,K164R,K127R | Same, pol30-E143K, K164R, K127R:LEU2 | This work 19,527 |
| E134 pol30-D150E,K164R,K127R | Same, pol30-D150E, K164R, K127R:LEU2 | This work 19,604 |
| E134 pol30-K164R,K127R | Same, pol30-K164R:LEU2 | This work 19,809 |
| E134 pol30-K164R | Same, pol30-K164R: LEU2 | This work 19,526 |
| pRS325-rad27-DA | rad27-DA with ADH1 promoter in pRS325 (LEU2 Marker, 2micron) | [35] 3559 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbel-Groissman, M.; Liefshitz, B.; Kupiec, M. Effects of PCNA Stability on the Formation of Mutations. Int. J. Mol. Sci. 2024, 25, 8646. https://doi.org/10.3390/ijms25168646
Arbel-Groissman M, Liefshitz B, Kupiec M. Effects of PCNA Stability on the Formation of Mutations. International Journal of Molecular Sciences. 2024; 25(16):8646. https://doi.org/10.3390/ijms25168646
Chicago/Turabian StyleArbel-Groissman, Matan, Batia Liefshitz, and Martin Kupiec. 2024. "Effects of PCNA Stability on the Formation of Mutations" International Journal of Molecular Sciences 25, no. 16: 8646. https://doi.org/10.3390/ijms25168646