
Microbiota-Derived Extracellular Vesicle as Emerging Actors in Host Interactions

Abstract
:1. Introduction
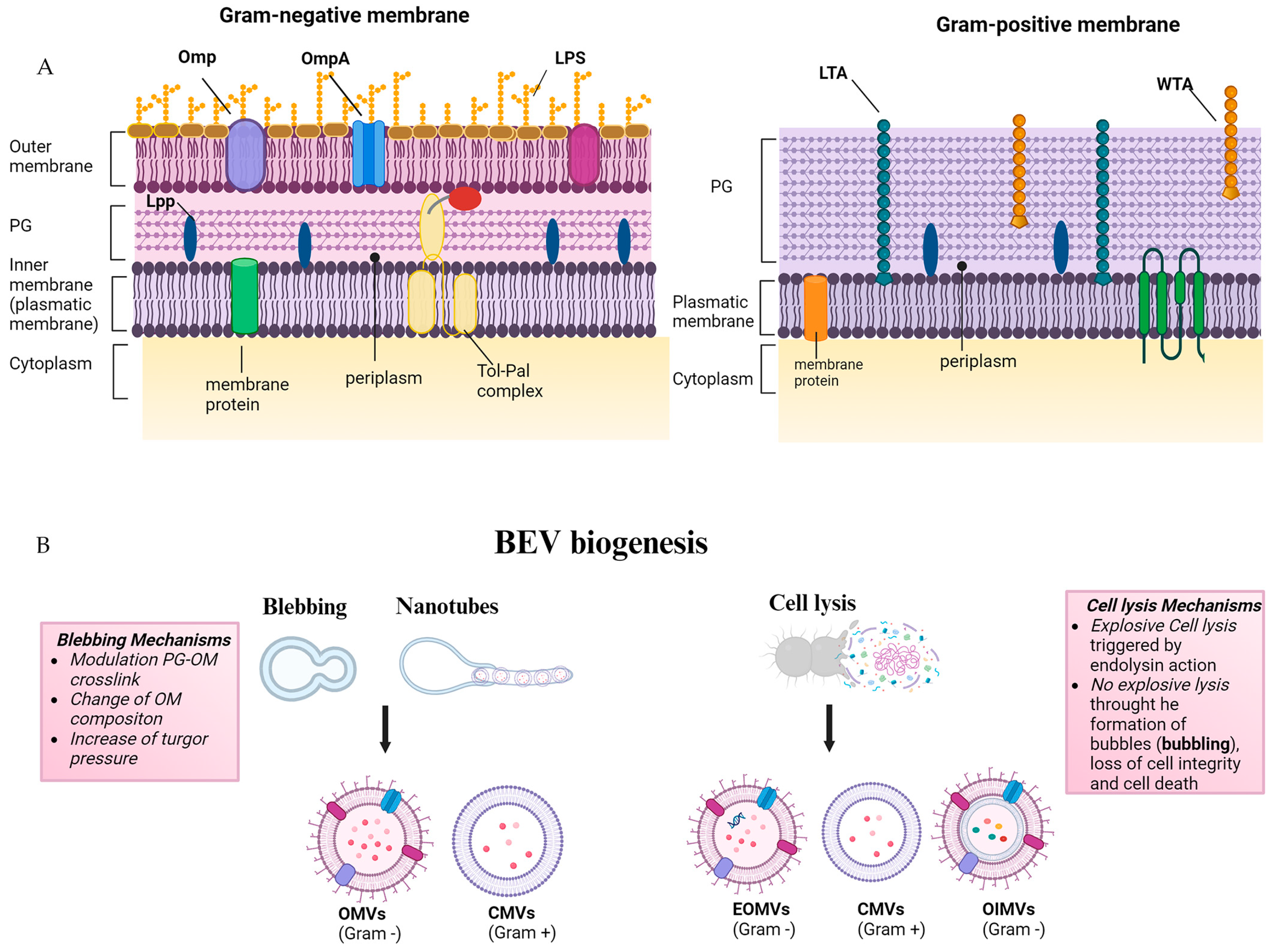
2. Origin and Composition of BEVs
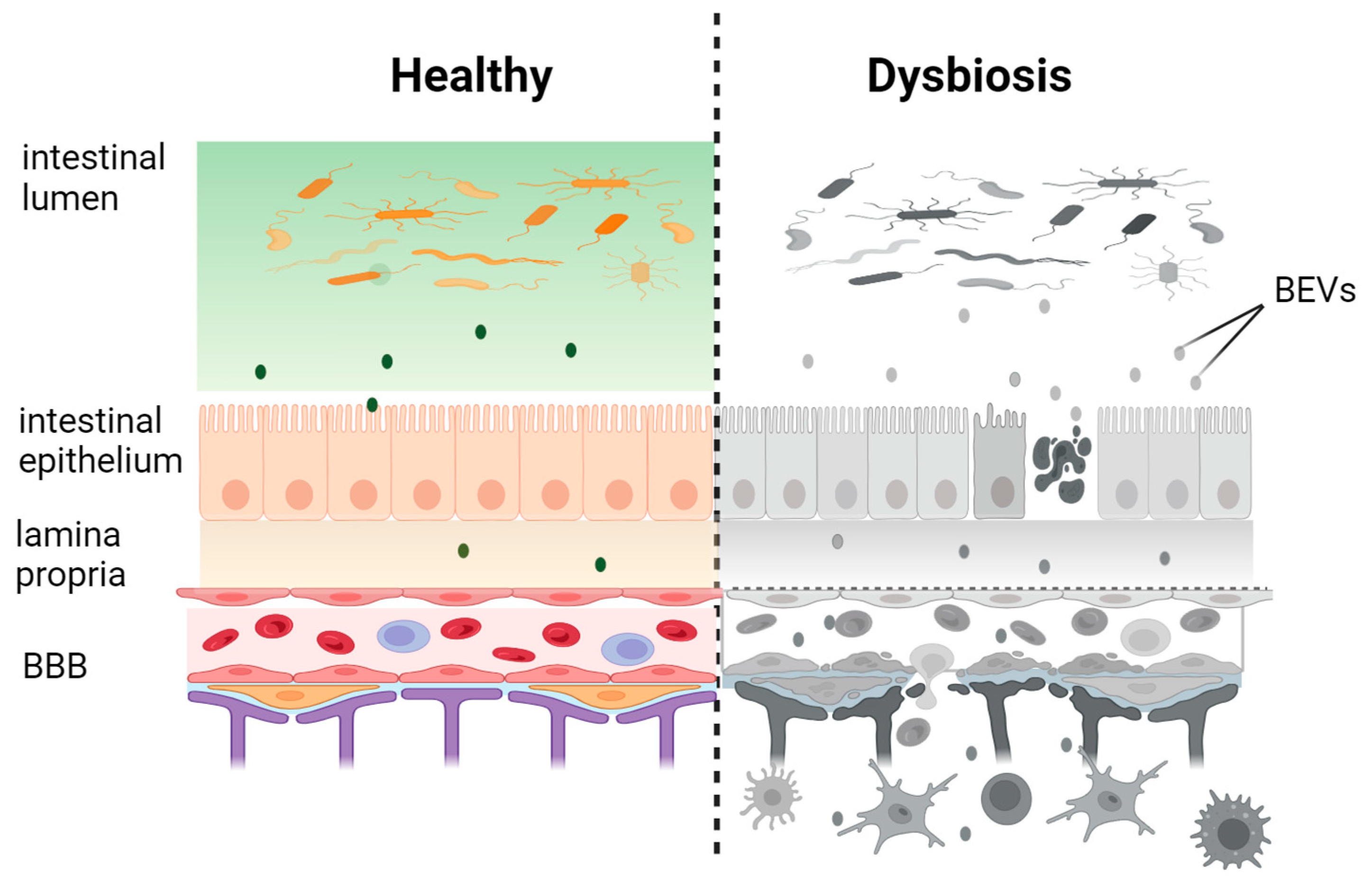
3. BEVs in Systemic Circulation
4. BEV Interaction with Epithelial Cells
4.1. BEV Interaction with Gastrointestinal Epithelium
4.2. BEV Interaction with Lung Epithelium
4.3. BEV Interaction with Skin Epithelium
4.4. BEV Interaction with Oral Epithelium
5. BEV Interaction with Immune System
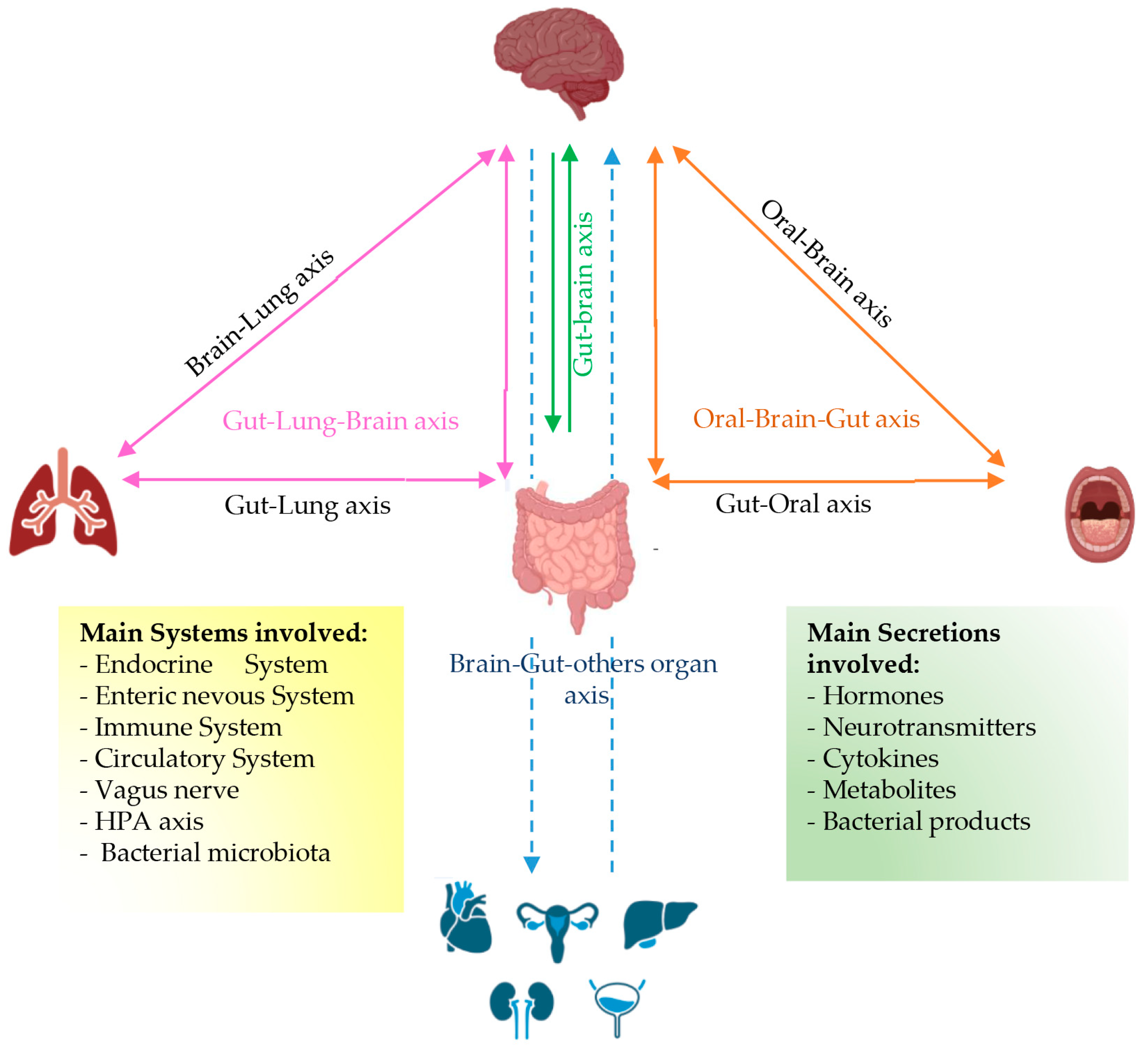
6. BEV Interaction with CNS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | References | Gram Stain |
|---|---|---|
| Aggregatibacter actinomycetemcomitans | [157,194,209,274,275] | Gram negative |
| Akkermansia muciniphila | [153,154,155] | Gram negative |
| Bacteroides fragilis | [127] | Gram negative |
| Bacteroides thetaiotaomicron | [88,128,231] | Gram negative |
| Bordetella pertussis | [200] | Gram negative |
| Campylobacter jejuni | [140] | Gram negative |
| E. coli Nissle 1917 | [126,151,152,213] | Gram negative |
| Enterobacter cloacae | [231] | Gram negative |
| Enterohemorrhagic E. coli | [91,193] | Gram negative |
| Enterotoxigenic E. coli | [109,123] | Gram negative |
| Escherichia coli | [82,126,138,156,163,205,213,219,226,267,268] | Gram negative |
| Fusobacterium nucleatum | [132] | Gram negative |
| Helicobacter pylori | [51,52,53,54,55,56,57,58,59,60,61,63,64,74,80,89,112,141,203,253,254] | Gram negative |
| Klebsiella pneuomoniae | [161] | Gram negative |
| Moraxella catarrhalis | [160] | Gram negative |
| Neisseria gonorrhoeae | [205,207] | Gram negative |
| Neisseria meningitidis | [218,220] | Gram negative |
| Paenalcaligenes hominis | [247] | Gram negative |
| Pediococcus pentosaceus | [206] | Gram positive |
| Porphyromonas gingivalis | [72,90,122,178,179,181,182,184,204,215,217,245,251,252] | Gram negative |
| Pseudomonas aeruginosa | [66,68,72,110,113,164,199,205] | Gram negative |
| Salmonella typhimurim | [72,191] | Gram negative |
| Staphylococcus aureus | [173,174,175,188,198,208] | Gram positive |
| Vibrio cholera | [114] | Gram negative |
7. Conclusions
Funding
Conflicts of Interest
References
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The human microbiota in health and disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- The Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Tierney, B.T.; Yang, Z.; Luber, J.M.; Beaudin, M.; Wibowo, M.C.; Baek, C.; Mehlenbacher, E.; Patel, C.J.; Kostic, A.D. The landscape of genetic content in the gut and oral human microbiome. Cell Host Microbe 2019, 26, 283–295.e8. [Google Scholar] [CrossRef] [PubMed]
- NIH HMP Working Group; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH human microbiome project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef]
- Lai, H.C.; Young, J.; Lin, C.-S.; Chang, C.J.; Lu, C.C.; Martel, J.; Ojcius, D.M.; Ko, Y.F.; Young, J.D.; Lai, H.C. Impact of the gut microbiota, prebiotics, and probiotics on human health and disease. Biomed. J. 2014, 37, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Virgin, H.W. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology 2014, 146, 1459–1469. [Google Scholar] [CrossRef]
- Palm, N.W.; de Zoete, M.R.; Flavell, R.A. Immune-microbiota interactions in health and disease. Clin. Immunol. 2015, 159, 122–127. [Google Scholar] [CrossRef]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol. 2018, 9, 417794. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut Reactions: How the Blood–Brain Barrier Connects the Microbiome and the Brain. Exp. Biol. Med. 2018, 243, 159–165. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Wang, Y.; Li, F.; Jia, J.; Song, X.; Qin, S.; Wang, R.; Jin, F.; Kitazato, K.; et al. The Gut-Microglia Connection: Implications for Central Nervous System Diseases. Front. Immunol. 2018, 9, 2325. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, I.; Izzo, L.; Tunesi, M.; Comar, M.; Albani, D.; Giordano, C. Organ-On-A-Chip In Vitro Models of the Brain and the Blood-Brain Barrier and Their Value to Study the Microbiota-Gut-Brain Axis in Neurodegeneration. Front. Bioeng. Biotechnol. 2020, 7, 435. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhu, H.; Feng, Y.; Guo, R.; Wan, D. The Impact of Gut Microbiota Disorders on the Blood–Brain Barrier. Infect. Drug Resist. 2020, 13, 3351–3363. [Google Scholar] [CrossRef]
- Macia, L.; Nanan, R.; Hosseini-Beheshti, E.; Grau, G.E. Host- and microbiota-derived extracellular vesicles, immune function, and disease development. Int. J. Mol. Sci. 2019, 21, 107. [Google Scholar] [CrossRef]
- Young, V.B. The role of the microbiome in human health and disease: An introduction for clinicians. BMJ 2017, 356, j831. [Google Scholar] [CrossRef]
- Takiishi, T.; Fenero, C.I.M.; Camara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [PubMed]
- Saffouri, G.B.; Shields-Cutler, R.R.; Chen, J.; Yang, Y.; Lekatz, H.R.; Hale, V.L.; Cho, J.M.; Battaglioli, E.J.; Bhattarai, Y.; Thompson, K.J.; et al. Small intestinal microbial dysbiosis underlies symptoms associated with functional gastrointestinal disorders. Nat. Commun. 2019, 10, 2012. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Zhu, S.; Jiang, Y.; Xu, K.; Cui, M.; Ye, W.; Zhao, G.; Jin, L.; Chen, X. The progress of gut microbiome research related to brain disorders. J. Neuroinflamm. 2020, 17, 25. [Google Scholar] [CrossRef]
- Jin, M.; Qian, Z.; Yin, J.; Xu, W.; Zhou, X. The role of intestinal microbiota in cardiovascular disease. J. Cell. Mol. Med. 2019, 23, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Deo, P.N.; Deshmukh, R. Oral microbiome: Unveiling the fundamentals. J. Oral Maxillofac. Pathol. 2019, 23, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Corrêa, J.D.; Silva, T.A. The Oral Microbiota Is Modified by Systemic Diseases. J. Dent. Res. 2019, 98, 148–156. [Google Scholar] [CrossRef]
- Baker, J.L.; Edlund, A. Exploiting the Oral Microbiome to Prevent Tooth Decay: Has Evolution Already Provided the Best Tools? Front. Microbiol. 2019, 9, 3323. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Chung, S.W.; Auh, Q.S.; Hong, S.J.; Lee, Y.A.; Jung, J.; Lee, G.J.; Park, H.J.; Shin, S.I.; Hong, J.Y. Progress in Oral Microbiome Related to Oral and Systemic Diseases: An Update. Diagnostics 2021, 11, 1283. [Google Scholar] [CrossRef] [PubMed]
- Sedghi, L.; DiMassa, V.; Harrington, A.; Lynch, S.V.; Kapila, Y.L. The oral microbiome: Role of key organisms and complex networks in oral health and disease. Periodontol. 2000 2021, 87, 107–131. [Google Scholar] [CrossRef]
- Kapila, Y.L. Oral health’s inextricable connection to systemic health: Special populations bring to bear multimodal relationships and factors connecting periodontal disease to systemic diseases and conditions. Periodontol. 2000 2021, 87, 11–16. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive transmission of microbes along the gastrointestinal tract. eLife 2019, 8, e42693. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Shimomura, Y.; Higurashi, T.; Sugi, Y.; Arimoto, J.; Umezawa, S.; Uchiyama, S.; Matsumoto, M.; Nakajima, A. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut 2018, 68, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Hein, R.; Schmidt, T.; Kamada, N. The Bacterial Connection between the Oral Cavity and the Gut Diseases. J. Dent. Res. 2020, 99, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; Siegel, M.O.; Keiser, J.; Yakovleva, A.; Kim, A.; Davenport, L.; Devaney, J.; Hoffman, E.P.; Alsubail, R.; Crandall, K.A.; et al. Single-Molecule Long-Read 16S Sequencing to Characterize the Lung Microbiome from Mechanically Ventilated Patients with Suspected Pneumonia. J. Clin. Microbiol. 2014, 52, 3913–3921. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Beck, J.M.; Huffnagle, G.B.; Curtis, J. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann. Am. Thorac. Soc. 2015, 12, 821–830. [Google Scholar] [CrossRef] [PubMed]
- De Steenhuijsen Piters, W.A.A.; Binkowska, J.; Bogaert, D. Early life microbiota and respiratory tract infections. Cell Host Microbe 2020, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e8578. [Google Scholar] [CrossRef]
- Beck, J.M. ABCs of the lung microbiome. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 1), S3–S6. [Google Scholar] [CrossRef]
- Sulaiman, I.; Wu, B.G.; Li, Y.; Tsay, J.C.; Sauthoff, M.; Scott, A.S.; Ji, K.; Koralov, S.B.; Weiden, M.; Clemente, J.C.; et al. Functional lower airways genomic profiling of the microbiome to capture active microbial metabolism. Eur. Respir. J. 2021, 58, 2003434. [Google Scholar] [CrossRef]
- Vieira, W.A.; Pretorius, E. The impact of asthma on the gastrointestinal tract (GIT). J. Asthma Allergy 2010, 3, 123–130. [Google Scholar] [CrossRef]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; NISC Comparative Sequence Program; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Juodeikis, R.; Carding, S.R. Outer Membrane Vesicles: Biogenesis, Functions, and Issues. Microbiol. Mol. Biol. Rev. 2022, 86, e00032-22. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, Z.; Liu, X.; Tyler, B.M. Biogenesis and Biological Functions of Extracellular Vesicles in Cellular and Organismal Communication with Microbes. Front. Microbiol. 2022, 13, 817844. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Park, J.; Gho, Y.S. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin. Cell Dev. Biol. 2015, 40, 97–104. [Google Scholar] [CrossRef]
- Liu, Y.; Defourny, K.A.Y.; Smid, E.J.; Abee, T. Gram-Positive Bacterial Extracellular Vesicles and Their Impact on Health and Disease. Front. Microbiol. 2018, 9, 1502. [Google Scholar] [CrossRef]
- Lee, E.Y.; Choi, D.S.; Kim, K.P.; Gho, Y.S. Proteomics in gram-negative bacterial outer membrane vesicles. Mass Spectrom. Rev. 2008, 27, 535–555. [Google Scholar] [CrossRef]
- Zavan, L.; Bitto, N.J.; Johnston, E.L.; Greening, D.W.; Kaparakis-Liaskos, M. Helicobacter pylori Growth Stage Determines the Size, Protein Composition, and Preferential Cargo Packaging of Outer Membrane Vesicles. Proteomics 2019, 19, e1800209. [Google Scholar] [CrossRef] [PubMed]
- Turner, L.; Bitto, N.J.; Steer, D.L.; Lo, C.; D’Costa, K.; Ramm, G.; Shambrook, M.; Hill, A.F.; Ferrero, R.L.; Kaparakis-Liaskos, M. Helicobacter pylori Outer Membrane Vesicle Size Determines Their Mechanisms of Host Cell Entry and Protein Content. Front. Immunol. 2018, 9, 1466. [Google Scholar] [CrossRef] [PubMed]
- Fiocca, R.; Necchi, V.; Sommi, P.; Ricci, V.; Telford, J.; Cover, T.L.; Solcia, E. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J. Pathol. 1999, 188, 220–226. [Google Scholar] [CrossRef]
- Keenan, J.; Day, T.; Neal, S.; Cook, B.; Perez-Perez, G.; Allardyce, R.; Bagshaw, P. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol. Lett. 2000, 182, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Hynes, S.O.; Keenan, J.I.; Ferris, J.A.; Annuk, H.; Moran, A.P. Lewis Epitopes on Outer Membrane Vesicles of Relevance to Helicobacter pylori Pathogenesis. Helicobacter 2005, 10, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-I.; Choi, J.-P.; Seo, J.; Kim, B.J.; Rho, M.; Han, J.K.; Kim, J.G. Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Exp. Mol. Med. 2017, 49, e330. [Google Scholar] [CrossRef]
- Grande, R.; Di Marcantonio, M.C.; Robuffo, I.; Pompilio, A.; Celia, C.; Di Marzio, L.; Paolino, D.; Codagnone, M.; Muraro, R.; Stoodley, P.; et al. Helicobacter pylori ATCC 43629/NCTC 11639 Outer Membrane Vesicles (OMVs) from Biofilm and Planktonic Phase Associated with Extracellular DNA (eDNA). Front. Microbiol. 2015, 6, 1369. [Google Scholar] [CrossRef] [PubMed]
- Sommi, P.; Ricci, V.; Fiocca, R.; Necchi, V.; Romano, M.; Telford, J.L.; Solcia, E.; Ventura, U. Persistence of Helicobacter pylori VacA toxin and vacuolating potential in cultured gastric epithelial cells. Am. J. Physiol. Content 1998, 275, G681–G688. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Woo, T.; Kurata, S.; Zaman, C.; Hojo, F.; Hanawa, T.; Kato, S.; Kamiya, S. Analysis of outer membrane vesicle protein involved in biofilm formation of Helicobacter pylori. Anaerobe 2011, 17, 388–390. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Kurata, S.; Fukuda, M.; Kawakami, H.; Ochiai, K.; Hanawa, T.; Kamiya, S. Outer Membrane Vesicles of Helicobacter pylori TK1402 are Involved in Biofilm Formation. BMC Microbiol. 2009, 9, 197. [Google Scholar] [CrossRef]
- Jarzab, M.; Posselt, G.; Meisner-Kober, N.; Wessler, S. Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis? Microorganisms 2020, 8, 1328. [Google Scholar] [CrossRef] [PubMed]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.I.; Davis, K.A.; Beaugie, C.R.; McGovern, J.J.; Moran, A.P. Alterations in Helicobacter pylori outer membrane and outer membrane vesicle-associated lipopolysaccharides under iron-limiting growth conditions. Innate Immun. 2008, 14, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.I.; Allardyce, R.A. Iron influences the expression of Helicobacter pylori outer membrane vesicle-associated virulence factors. Eur. J. Gastroenterol. Hepatol. 2000, 12, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Takii, R.; Yamatake, K.; Kawakubo, T.; Tsukuba, T.; Yamamoto, K. A role for gingipains in cellular responses and bacterial survival in Porphyromonas gingivalis-infected cells. Front. Biosci. 2007, 12, 4800–4809. [Google Scholar] [CrossRef]
- Zavan, L.; Fang, H.; Johnston, E.L.; Whitchurch, C.; Greening, D.W.; Hill, A.F.; Kaparakis-Liaskos, M. The mechanism of Pseudomonas aeruginosa outer membrane vesicle biogenesis determines their protein composition. Proteomics 2023, 23, e2200464. [Google Scholar] [CrossRef]
- Chen, F.; Cui, G.; Wang, S.; Nair, M.K.M.; He, L.; Qi, X.; Han, X.; Zhang, H.; Zhang, J.R.; Su, J. Outer membrane vesicle-associated lipase FtlA enhances cellular invasion and virulence in Francisella tularensis LVS. Emerg. Microbes Infect. 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Kadurugamuwa, J.L.; Beveridge, T.J. Bacteriolytic effect of membrane vesicles from Pseudomonas aeruginosa on other bacteria including pathogens: Conceptually new antibiotics. J. Bacteriol. 1996, 178, 2767–2774. [Google Scholar] [CrossRef]
- Chattopadhyay, M.K.; Jagannadham, M.V. Vesicles-mediated resistance to antibiotics in bacteria. Front. Microbiol. 2015, 6, 758. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, L.; Miao, J.; Zhang, Z.; Ruan, J.; Xu, L.; Guo, H.; Zhang, M.; Qiao, W. Regulation of the formation and structure of biofilms by quorum sensing signal molecules packaged in outer membrane vesicles. Sci. Total Environ. 2022, 806, 151403. [Google Scholar] [CrossRef]
- Wang, W.; Chanda, W.; Zhong, M. The relationship between biofilm and outer membrane vesicles: A novel therapy overview. FEMS Microbiol. Lett. 2015, 362, fnv117. [Google Scholar] [CrossRef] [PubMed]
- Bitto, N.J.; Chapman, R.; Pidot, S.; Costin, A.; Lo, C.; Choi, J.; D’Cruze, T.; Reynolds, E.C.; Dashper, S.G.; Turnbull, L.; et al. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci. Rep. 2017, 7, 7072. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.R.; Sieber, K.B.; Robinson, K.M.; White, J.R.; Ganesan, A.; Nourbakhsh, S.; Dunning Hotopp, J.C. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput. Biol. 2013, 9, e1003107. [Google Scholar] [CrossRef] [PubMed]
- Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis 2008, 29, 2400–2405. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Stanton, B.A. Stanton. Transfer RNA-Derived Fragments, the Underappreciated Regulatory Small RNAs in Microbial Pathogenesis. Front. Microbiol. 2021, 12, 687632. [Google Scholar] [CrossRef]
- Dauros-Singorenko, P.; Blenkiron, C.; Phillips, A.; Swift, S. The functional RNA cargo of bacterial membrane vesicles. FEMS Microbiol. Lett. 2018, 365, fny023. [Google Scholar] [CrossRef]
- Fujishima, K.; Kanai, A. tRNA gene diversity in the three domains of life. Front. Genet. 2014, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wilson, B.; Kumar, P.; Dutta, A. Noncanonical roles of tRNAs: tRNA fragments and beyond. Annu. Rev. Genet. 2020, 54, 47–69. [Google Scholar] [CrossRef]
- Wang, Y.F.; Fu, J. Secretory and circulating bacterial small RNAs: A mini-review of the literature. ExRNA 2019, 1, 14. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Song, Z.; Li, R.; Ruan, H.; Liu, Q.; Huang, X. sncRNAs packaged by Helicobacter pylori outer membrane vesicles attenuate IL-8 secretion in human cells. Int. J. Med. Microbiol. 2020, 310, 151356. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, K.; Hampton, T.H.; Jarek, M.; Scharfe, M.; Gerber, S.A.; Mielcarz, D.W.; Demers, E.G.; Dolben, E.L.; Hammond, J.H.; Hogan, D.A.; et al. A novel mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 2016, 12, e1005672. [Google Scholar] [CrossRef] [PubMed]
- Celluzzi, A.; Masotti, A. How our other genome controls our epigenome. Trends Microbiol. 2016, 24, 777–787. [Google Scholar] [CrossRef] [PubMed]
- González Plaza, J.J. Small RNAs as Fundamental Players in the Transference of Information During Bacterial Infectious Diseases. Front. Mol. Biosci. 2020, 7, 101. [Google Scholar] [CrossRef]
- Stentz, R.; Carvalho, A.L.; Jones, E.J.; Carding, S.R. Fantastic voyage: The journey of intestinal microbiota-derived microvesicles through the body. Biochem. Soc. Trans. 2018, 46, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.-A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef]
- Schaack, B.; Hindré, T.; Quansah, N.; Hannani, D.; Mercier, C.; Laurin, D. Microbiota-Derived Extracellular Vesicles Detected in Human Blood from Healthy Donors. Int. J. Mol. Sci. 2022, 23, 13787. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Situ, B.; Huang, X.; Xue, Y.; He, X.; Li, Q.; Ou, D.; He, B.; Chen, J.; Huang, Y.; et al. Single-particle analysis of circulating bacterial extracellular vesicles reveals their biogenesis, changes in blood and links to intestinal barrier. J. Extracell. Vesicles 2023, 12, e12395. [Google Scholar] [CrossRef]
- Jones, E.J.; Booth, C.; Fonseca, S.; Parker, A.; Cross, K.; Miquel-Clopés, A.; Hautefort, I.; Mayer, U.; Wileman, T.; Stentz, R.; et al. The Uptake, Trafficking, and Biodistribution of Bacteroides thetaiotaomicron Generated Outer Membrane Vesicles. Front. Microbiol. 2020, 11, 57. [Google Scholar] [CrossRef]
- O’Donoghue, E.J.; Krachler, A.M. Mechanisms of outer membrane vesicle entry into host cells. Cell. Microbiol. 2016, 18, 1508–1517. [Google Scholar] [CrossRef]
- Furuta, N.; Tsuda, K.; Omori, H.; Yoshimori, T.; Yoshimura, F.; Amano, A. Porphyromonas gingivalis outer membrane vesicles enter human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. Infect. Immun. 2009, 77, 4187–4196. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Ruter, C.; Kunsmann, L.; Greune, L.; Bauwens, A.; Zhang, W.; Kuczius, T.; Kim, K.S.; Mellmann, A.; Schmidt, M.A.; et al. Enterohemorrhagic Escherichia coli hemolysin employs outer membrane vesicles to target mitochondria and cause endothelial and epithelial apoptosis. PLoS Pathog. 2013, 9, e1003797. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Abada, A.; Elazar, Z. Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 2014, 6, a018358. [Google Scholar] [CrossRef]
- Matlashov, M.E.; Shcherbakova, D.M.; Alvelid, J.; Baloban, M.; Pennacchietti, F.; Shemetov, A.A.; Testa, I.; Verkhusha, V.V. A Set of Monomeric Near-Infrared Fluorescent Proteins for Multicolor Imaging across Scales. Nat. Commun. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Caffaratti, C.; Blanquet, F.; Nicoud, O.; Josserand, V.; Laurin, D. Rapid Biodistribution of Fluorescent Outer-Membrane Vesicles from the Intestine to Distant Organs via the Blood in Mice. Int. J. Mol. Sci. 2024, 25, 1821. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, L.; Xiao, Z.; Li, M.; Wu, X.; Li, W.; Li, X.; Zhao, Q.; Wu, Y.; Zhang, H.; et al. Protective Role of γδ T Cells in Different Pathogen Infections and Its Potential Clinical Application. J. Immunol. Res. 2018, 2018, 5081634. [Google Scholar] [CrossRef] [PubMed]
- Girard, P.; Ponsard, B.; Charles, J.; Chaperot, L.; Aspord, C. Potent Bidirectional Cross-Talk Between Plasmacytoid Dendritic Cells and γδT Cells through BTN3A, Type I/II IFNs and Immune Checkpoints. Front. Immunol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Emery, D.C.; Shoemark, D.K.; Batstone, T.E.; Waterfall, C.M.; Coghill, J.A.; Cerajewska, T.L.; Davies, M.; West, N.X.; Allen, S.J. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front. Aging Neurosci. 2017, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Park, J.Y.; Choi, J.; Lee, Y.; Lee, J.E.; Lee, E.H.; Kwon, H.J.; Yang, J.; Jeong, B.R.; Kim, Y.K.; Han, P.L. Metagenome Analysis of Bodily Microbiota in a Mouse Model of Alzheimer Disease Using Bacteria-derived Membrane Vesicles in Blood. Exp. Neurobiol. 2017, 26, 369–379. [Google Scholar] [CrossRef]
- Lee, Y.; Park, J.Y.; Lee, E.H.; Yang, J.; Jeong, B.R.; Kim, Y.K.; Seoh, J.Y.; Lee, S.; Han, P.L.; Kim, E.J. Rapid assessment of microbiota changes in individuals with autism Spectrum disorder using Bacteria-derived membrane vesicles in urine. Exp. Neurobiol. 2017, 26, 307–317. [Google Scholar] [CrossRef]
- Tulkens, J.; De Wever, O.; Hendrix, A. Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nat. Protoc. 2020, 15, 40–67. [Google Scholar] [CrossRef]
- Natividad, J.M.; Petit, V.; Huang, X.; de Palma, G.; Jury, J.; Sanz, Y.; Philpott, D.; Garcia Rodenas, C.L.; McCoy, K.D.; Verdum, E.F. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−;Nod2−/− Mice. Inflamm. Bowel Dis. 2012, 18, 1434–1446. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [PubMed]
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell. Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef]
- Devriese, S.; Van den Bossche, L.; Van Welden, S.; Holvoet, T.; Pinheiro, I.; Hindryckx, P.; De Vos, M.; Laukens, D. T84 monolayers are superior to Caco-2 as a model system of colonocytes. Histochem. Cell Biol. 2017, 148, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Caruana, J.C.; Walper, S.A. Bacterial membrane vesicles as mediators of microbe—Microbe and microbe—Host community interactions. Front. Microbiol. 2020, 11, 432. [Google Scholar] [CrossRef]
- Kesty, N.C.; Mason, K.M.; Reedy, M.; Miller, S.E.; Kuehn, M.J. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004, 23, 4538–4549. [Google Scholar] [CrossRef]
- Bomberger, J.M.; MacEachran, D.P.; Coutermarsh, B.A.; Ye, S.; O’Toole, G.A.; Stanton, B.A. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 2009, 5, e1000382. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Hampton, M.B.; Keenan, J.I. Helicobacter pylori outer membrane vesicles modulate proliferation and interleukin-8 production by gastric epithelial cells. Infect. Immun. 2003, 71, 5670–5675. [Google Scholar] [CrossRef]
- Bauman, S.J.; Kuehn, M.J. Purification of outer membrane vesicles from Pseudomonas aeruginosa and their activation of an IL-8 response. Microbes Infect. 2006, 8, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Chaudhuri, K. Vibrio cholerae O395 outer membrane vesicles modulate intestinal epithelial cells in a NOD1 protein-dependent manner and induce dendritic cell-mediated Th2/Th17 cell responses. J. Biol. Chem. 2013, 288, 4299–4309. [Google Scholar] [CrossRef]
- Qazi, B.S.; Tang, K.; Qazi, A. Recent Advances in Underlying Pathologies Provide Insight into Interleukin-8 Expression-Mediated Inflammation and Angiogenesis. Int. J. Inflamm. 2011, 2011, 908468. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Schertzer, J.W.; Whiteley, M. Bacterial outer membrane vesicles in trafficking, communication and the host-pathogen interaction. J. Mol. Microbiol. Biotechnol. 2013, 23, 118–130. [Google Scholar] [CrossRef]
- Kaparakis-Liaskos, M.; Ferrero, R.L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015, 15, 375–387. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef]
- Brewer, J.M.; Pollock, K.G.J.; Tetley, L.; Russell, D.G. Vesicle size influences the trafficking, processing, and presentation of antigens in lipid vesicles. J. Immunol. 2004, 173, 6143–6150. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Liu, S.; Zhang, S.; Pan, Y. The Role of Porphyromonas gingivalis Outer Membrane Vesicles in Periodontal Disease and Related Systemic Diseases. Front. Cell. Infect. Microbiol. 2021, 10, 585917. [Google Scholar] [CrossRef] [PubMed]
- Horstman, A.L.; Kuehn, M.J. Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J. Biol. Chem. 2000, 275, 12489–12496. [Google Scholar] [CrossRef]
- Suzuki, T. Regulation of intestinal epithelial permeability by tight junctions. Cell. Mol. Life Sci. 2013, 70, 631–659. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Fabrega, M.J.; Aguilera, L.; Gimenez, R.; Varela, E.; Cañas, M.A.; Antolín, M.; Badía, J.; Baldomà, L. Activation of immune and defense responses in the intestinal mucosa by outer membrane vesicles of commensal and probiotic Escherichia coli strains. Front. Microbiol. 2016, 7, 705. [Google Scholar] [CrossRef]
- Shen, Y.; Giardino Torchia, M.L.; Lawson, G.W.; Karp, C.L.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef]
- Hickey, C.A.; Kuhn, K.A.; Donermeyer, D.L.; Porter, N.T.; Jin, C.; Cameron, E.A.; Jung, H.; Kaiko, G.H.; Wegorzewska, M.; Malvin, N.P.; et al. Colitogenic Bacteroides thetaiotaomicron antigens access host immune cells in a sulfatase-dependent manner via outer membrane vesicles. Cell Host Microbe 2015, 17, 672–680. [Google Scholar] [CrossRef]
- Patten, D.A.; Hussein, E.; Davies, S.P.; Humphreys, P.N.; Collett, A. Commensal-derived OMVs elicit a mild proinflammatory response in intestinal epithelial cells. Microbiology 2017, 163, 702–711. [Google Scholar] [CrossRef]
- Horstman, A.L.; Kuehn, M.J. Bacterial surface association of heat-labile enterotoxin through lipopolysaccharide after secretion via the general secretory pathway. J. Biol. Chem. 2002, 277, 32538–32545. [Google Scholar] [CrossRef]
- Newton, K. Ripk1 and Ripk3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015, 25, 347–353. [Google Scholar] [CrossRef]
- Liu, L.; Liang, L.; Yang, C.; Zhou, Y.; Chen, Y. Extracellular vesicles of Fusobacterium nucleatum compromise intestinal barrier through targeting Ripk1-mediated cell death pathway. Gut Microbes 2021, 13, 1903826. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ni, J.; You, Y.; Feng, G.; Zhang, S.; Bao, W.; Hou, H.; Li, H.; Liu, L.; Zheng, M.; et al. Snx10-mediated Lps sensing causes intestinal barrier dysfunction via a Caspase-5-dependent signaling Cascade. EMBO J. 2021, 40, e108080. [Google Scholar] [CrossRef] [PubMed]
- Seksik, P.; Rigottier-Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Doré, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Tomita, M.; Ohkubo, R.; Hayashi, M. Lipopolysaccharide transport system across colonic epithelial cells in normal and infective rat. Drug Metab. Pharmacokinet. 2004, 19, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Awad, W.A.; Hess, C.; Hess, M. Enteric Pathogens and Their Toxin-Induced Disruption of the Intestinal Barrier through Alteration of Tight Junctions in Chickens. Toxins 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; McBain, A.J.; O’Neilla, C.A. Strain-dependent augmentation of tight-junction barrier function in human primary epidermal keratinocytes by Lactobacillus and Bifidobacterium Lysates. Appl. Environ. Microbiol. 2013, 79, 4887–4894. [Google Scholar] [CrossRef]
- Hecht, G. VII. Enteropathogenic Escherichia coli: Physiological alterations from an extracellular position. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1–G7. [Google Scholar] [CrossRef]
- Schoultz, I.; Keita, Å.V. The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells 2020, 9, 1909. [Google Scholar] [CrossRef]
- Elmi, A.; Nasher, F.; Jagatia, H.; Gundogdu, O.; Bajaj-Elliott, M.; Wren, B.; Wren, B.; Dorrell, N. Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occluding. Cell. Microbiol. 2016, 18, 561–572. [Google Scholar] [CrossRef]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.E.; Arnqvist, A.; Vikström, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell–cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362, fnv076. [Google Scholar] [CrossRef]
- Trebichavsky, I.; Splichal, I.; Rada, V.; Splichalova, A. Modulation of natural immunity in the gut by Escherichia coli Nissle 1917. Nutr. Rev. 2010, 68, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Wehkamp, J.; Altenhoefer, A.; Oelschlaeger, T.A.; Stange, E.F.; Fellermann, K. Induction of human β-defensin 2 by the probiotic Escherichia coli Nissle 1917 is mediated through flagellin. Infect. Immun. 2007, 75, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Ukena, S.N.; Singh, A.; Dringenberg, U.; Engelhardt, R.; Seidler, U.; Hansen, W.; Bleich, A.; Bruder, D.; Franzke, A.; Rogler, G.; et al. Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS ONE 2007, 2, e1308. [Google Scholar] [CrossRef] [PubMed]
- Zyrek, A.; Cichon, C.; Helms, S.; Enders, C.; Sonnenborn, U.; Schmidt, M.A. Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCzeta redistribution resulting in tight junction and epithelial barrier repair. Cell. Microbiol. 2007, 9, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Hering, N.A.; Richter, J.F.; Fromm, A.; Wieser, A.; Hartmann, S.; Günzel, D.; Bücker, R.; Fromm, M.; Schulzke, J.D.; Troeger, H. TcpC protein from E. coli Nissle improves epithelial barrier function involving PKCζ and ERK1/2 signaling in HT-29/B6 cells. Mucosal Immunol. 2014, 7, 369–378. [Google Scholar] [CrossRef]
- Grabig, A.; Paclik, D.; Guzy, C.; Dankof, A.; Baumgart, D.C.; Erckenbrecht, J.; Raupach, B.; Sonnenborn, U.; Eckert, J.; Schumann, R.R.; et al. Escherichia coli strain Nissle 1917 ameliorates experimental colitis via toll-like receptor 2- and toll-like receptor 4-dependent pathways. Infect. Immun. 2006, 74, 4075–4082. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Mesa, N.; Utrilla, P.; Comalada, M.; Zorrilla, P.; Garrido-Mesa, J.; Zarzuelo, A.; Rodríguez-Cabezas, M.E.; Gálvez, J. The association of minocycline and the probiotic Escherichia coli Nissle 1917 results in an additive beneficial effect in a DSS model of reactivated colitis in mice. Biochem. Pharmacol. 2011, 82, 1891–1900. [Google Scholar] [CrossRef]
- Olier, M.; Marcq, I.; Salvador-Cartier, C.; Secher, T.; Dobrindt, U.; Boury, M.; Bacquié, V.; Pénary, M.; Gaultier, E.; Nougayrède, J.P.; et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 2012, 3, 501–509. [Google Scholar] [CrossRef]
- Souza, É.L.; Elian, S.D.; Paula, L.M.; Garcia, C.C.; Vieira, A.T.; Teixeira, M.M.; Arantes, R.M.; Nicoli, J.R.; Martins, F.S. Escherichia coli strain Nissle 1917 ameliorates experimental colitis by modulating intestinal permeability, the inflammatory response and clinical signs in a faecal transplantation model. J. Med. Microbiol. 2016, 65, 201–210. [Google Scholar] [CrossRef]
- Cañas, M.A.; Giménez, R.; Fábrega, M.J.; Toloza, L.; Baldomà, L.; Badia, J. Outer membrane vesicles from the probiotic Escherichia coli Nissle 1917 and the commensal ECOR12 enter intestinal epithelial cells via clathrin-dependent endocytosis and elicit differential effects on DNA damage. PLoS ONE 2016, 11, e0160374. [Google Scholar] [CrossRef]
- Alvarez, C.S.; Badia, J.; Bosch, M.; Giménez, R.; Baldomà, L. Outer membrane vesicles and soluble factors released by probiotic Escherichia coli Nissle 1917 and commensal Ecor63 enhance barrier function by regulating expression of tight junction proteins in intestinal epithelial cells. Front. Microbiol. 2016, 7, 1981. [Google Scholar] [CrossRef] [PubMed]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Vigsnæs, L.K.; Brynskov, J.; Steenholdt, C.; Wilcks, A.; Licht, T.R. Gram-negative bacteria account for main differences between faecal microbiota from patients with ulcerative colitis and healthy controls. Benef. Microbes 2012, 3, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietilä, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef] [PubMed]
- Cañas, M.A.; Fabrega, M.J.; Gimenez, R.; Badia, J.; Baldoma, L. Outer membrane vesicles from probiotic and commensal Escherichia coli activate NOD1-mediated immune responses in intestinal epithelial cells. Front. Microbiol. 2018, 9, 498. [Google Scholar] [CrossRef] [PubMed]
- Thay, B.; Damm, A.; Kufer, T.A.; Wai, S.N.; Oscarsson, J. Aggregatibacter actinomycetemcomitans outer membrane vesicles are internalized in human host cells and trigger NOD1- and NOD2-dependent NF-κB activation. Infect. Immun. 2014, 82, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.T.; Mimuro, H.; Kufer, T.A.; Lo, C.; Wheeler, R.; Turner, L.J.; Thomas, B.J.; Malosse, C.; Gantier, M.P.; Casillas, L.N.; et al. The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signalling. Cell Host Microbe 2014, 15, 623–635. [Google Scholar] [CrossRef]
- Ryu, S.; Ni, K.; Wang, C.; Sivanantham, A.; Carnino, J.M.; Ji, H.L.; Jin, Y. Bacterial Outer Membrane Vesicles Promote Lung Inflammatory Responses and Macrophage Activation via Multi-Signaling Pathways. Biomedicines 2023, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Schaar, V.; de Vries, S.P.; Perez Vidakovics, M.L.; Bootsma, H.J.; Larsson, L.; Hermans, P.W.; Bjartell, A.; Mörgelin, M.; Riesbeck, K. Multicomponent Moraxella catarrhalis outer membrane vesicles induce an inflammatory response and are internalized by human epithelial cells. Cell. Microbiol. 2011, 13, 432–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, E.J.; Lee, J.H.; Jun, S.H.; Choi, C.W.; Kim, S.I.; Kang, S.S.; Hyun, S. Klebsiella pneumonia secretes outer membrane vesicles that induce the innate immune response. FEMS Microbiol. Lett 2012, 331, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Tsai, Y.F.; Pan, Y.L.; Hwang, T.L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2021, 44, 439–446. [Google Scholar] [CrossRef]
- Kim, J.H.; Yoon, Y.J.; Lee, J.; Choi, E.J.; Yi, N.; Park, K.S.; Park, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli up-regulate expression of endothelial cell adhesion molecules in vitro and in vivo. PLoS ONE 2013, 8, e59276. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Lee, J.; Jang, S.C.; Kim, S.R.; Jang, M.H.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Pulmonary inflammation induced by bacteria-free outer membrane vesicles from Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2013, 49, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.T.; Abrams, M.; Tlougan, B.; Rademaker, A.; Paller, A.S. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics 2009, 123, e808–e814. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Choi, E.J.; Lee, W.H.; Choi, S.J.; Roh, T.Y.; Park, J.; Jee, Y.K.; Zhu, Z.; Koh, Y.Y.; Gho, Y.S.; et al. Extracellular vesicles, especially derived from gram-negative bacteria, in indoor dust induce neutrophilic pulmonary inflammation associated with both Th1 and Th17 cell responses. Clin. Exp. Allergy 2013, 43, 443–454. [Google Scholar] [CrossRef]
- Asher, M.I.; Montefort, S.; Björkstén, B.; Lai, C.K.; Strachan, D.P.; Weiland, S.K.; Williams, H.; ISAAC Phase Three Study Group. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 2006, 368, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Oh, S.Y.; Jeon, S.G.; Park, H.W.; Lee, S.Y.; Chun, E.Y.; Bang, B.; Lee, H.S.; Oh, M.H.; Kim, Y.S.; et al. Airway exposure levels of lipopolysaccharide determine type 1 versus type 2 experimental asthma. J. Immunol. 2007, 178, 5375–5382. [Google Scholar] [CrossRef]
- Pastacaldi, C.; Lewis, P.; Howarth, P. Staphylococci and staphylococcal superantigens in asthma and rhinitis: A systematic review and meta-analysis. Allergy 2011, 66, 549–555. [Google Scholar] [CrossRef]
- Bisgaard, H.; Hermansen, M.N.; Buchvald, F.; Loland, L.; Halkjaer, L.B.; Bønnelykke, K.; Brasholt, M.; Heltberg, A.; Vissing, N.H.; Thorsen, S.V.; et al. Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 2007, 357, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, A.B. Skin anatomy, physiology, and pathophysiology. Nurs. Clin. N. Am. 1999, 34, 777–797. [Google Scholar] [CrossRef]
- Breuer, K.; Haussler, S.; Kapp, A.; Werfel, T. Staphylococcus aureus: Colonizing features and influence of an antibacterial treatment in adults with atopic dermatitis. Br. J. Dermatol. 2002, 147, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P.; et al. Gram-positive bacteria produce membrane vesicles: Proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef]
- Gurung, M.; Moon, D.C.; Choi, C.W.; Lee, J.H.; Bae, Y.C.; Kim, J.; Lee, Y.C.; Seol, S.Y.; Cho, D.T.; Kim, S.I.; et al. Staphylococcus aureus produces membrane-derived vesicles that induce host cell death. PLoS ONE 2011, 6, e27958. [Google Scholar] [CrossRef]
- Jun, S.H.; Lee, J.H.; Kim, S.I.; Choi, C.W.; Park, T.I.; Jung, H.R.; Cho, J.W.; Kim, S.H.; Lee, J.C. Staphylococcus aureus-derived membrane vesicles exacerbate skin inflammation in atopic dermatitis. Clin. Exp. Allergy 2017, 47, 85–96. [Google Scholar] [CrossRef]
- Seo, M.D.; Kang, T.J.; Lee, C.H.; Lee, A.Y.; Noh, M. HaCaT Keratinocytes and Primary Epidermal Keratinocytes Have Different Transcriptional Profiles of Cornified Envelope-Associated Genes to T Helper Cell Cytokines. Biomol. Ther. 2012, 20, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Amano, A. Disruption of epithelial barrier and impairment of cellular function by Porphyromonas gingivalis. Front. Biosci. 2007, 12, 3965–3974. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Takashiba, S.; Kosono, S.; Yoshida, M.; Watanabe, H.; Ohnishi, M.; Senpuku, H. Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain-mediated detachment of cultured oral epithelial cells and immune responses. Microbes Infect. 2014, 16, 6–16. [Google Scholar] [CrossRef]
- Veith, P.D.; Chen, Y.Y.; Gorasia, D.G.; Chen, D.; Glew, M.D.; O’Brien-Simpson, N.M.; Cecil, J.D.; Holden, J.A.; Reynolds, E.C. Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J. Proteome Res. 2014, 13, 2420–2432. [Google Scholar] [CrossRef]
- Amano, A.; Nakagawa, I.; Okahashi, N.; Hamada, N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J. Periodontal Res. 2004, 39, 136–142. [Google Scholar] [CrossRef]
- Furuta, N.; Takeuchi, H.; Amano, A. Entry of Porphyromonas gingivalis outer membrane vesicles into epithelial cells causes cellular functional impairment. Infect. Immun. 2009, 77, 4761–4770. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Inaba, H.; Kato, T.; Tagashira, M.; Honma, D.; Kanda, T.; Ohtake, Y.; Amano, A. Inflammatory responses of gingival epithelial cells stimulated with Porphyromonas gingivalis vesicles are inhibited by hop-associated polyphenols. J. Periodontol. 2008, 79, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.N.; Barr, C.E.; Seshamma, T.; Reidy, J.; Pomerantz, R.J.; Bagasra, O. Infection of oral mucosal cells by human immunodeficiency virus type 1 in seropositive persons. J. Infect. Dis. 1995, 171, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.H.; Ho, M.H.; Liu, B.; Hildreth, J.; Dash, C.; Goodwin, J.S.; Balasubramaniam, M.; Chen, C.H.; Xie, H. Role of Porphyromonas gingivalis outer membrane vesicles in oral mucosal transmission of HIV. Sci. Rep. 2018, 8, 8812. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Belogortseva, N.I.; Wu, J.; Lai, W.H.; Chen, C.H. Inhibition of human immunodeficiency virus type 1 entry by a binding domain of Porphyromonas gingivalis gingipain. Antimicrob. Agents Chemother. 2006, 50, 3070–3074. [Google Scholar] [CrossRef]
- Pascal, M.; Perez-Gordo, M.K.; Caballero, T.; Escribese, M.M.; Natividad Lopez Longo, M.; Luengo, O.; Manso, L.; Matheu, V.; Seoane, E.; Zamorano, M.; et al. Microbiome and allergic diseases. Front. Immunol. 2018, 9, 1584. [Google Scholar] [CrossRef]
- De Luca, F.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bitto, N.J.; Cheng, L.; Johnston, E.L.; Pathirana, R.; Phan, T.K.; Poon, I.K.H.; O’Brien-Simpson, N.M.; Hill, A.F.; Stinear, T.P.; Kaparakis-Liaskos, M. Staphylococcus aureus membrane vesicles contain immunostimulatory DNA, RNA and peptidoglycan that activate innate immune receptors and induce autophagy. J. Extracell. Vesicles 2021, 10, e12080. [Google Scholar] [CrossRef] [PubMed]
- Lelouard, H.; Fallet, M.; De Bovis, B.; Meresse, S.; Gorvel, J.P. Peyer’s patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 2012, 142, 592–601.e3. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gallausiaux, C.; Malabirade, A.; Habier, J.; Wilmes, P. Fusobacterium nucleatum extracellular vesicles modulate gut epithelial cell innate immunity Via FomA and TLR2. Front. Immunol. 2020, 11, 583644. [Google Scholar] [CrossRef]
- Yang, J.; Hwang, I.; Lee, E.; Shin, S.J.; Lee, E.J.; Rhee, J.H.; Yu, J.W. Bacterial Outer Membrane Vesicle-Mediated Cytosolic Delivery of Flagellin Triggers Host NLRC4 Canonical Inflammasome Signaling. Front. Immunol. 2020, 11, 581165. [Google Scholar] [CrossRef]
- Skerniškytė, J.; Karazijaitė, E.; Lučiūnaitė, A.; Sužiedėlienė, E. OmpA proteindeficient Acinetobacter baumannii outer membrane vesicles trigger reduced inflammatory response. Pathogens 2021, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Marejková, M.; Bauwens, A.; Kunsmann-Prokscha, L.; Mellmann, A.; Karch, H. Enterohemorrhagic Escherichia coli O157 outer membrane vesicles induce interleukin 8 production in human intestinal epithelial cells by signaling Via toll-like receptors TLR4 and TLR5 and activation of the nuclear factor NF-κB. Int. J. Med. Microbiol. 2018, 308, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.Y.; Choi, S.Y.; Lee, J.H.; Hong, S.H.; Lee, H.J. Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL-6 promotion by RNA cargo. Front. Mol. Biosci. 2020, 7, 596366. [Google Scholar] [CrossRef]
- Balmer, M.L.; Schürch, C.M.; Saito, Y.; Geuking, M.B.; Li, H.; Cuenca, M.; Kovtonyuk, L.V.; McCoy, K.D.; Hapfelmeier, S.; Ochsenbein, A.F.; et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J. Immunol. 2014, 193, 5273–5283. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, A.; Yáñez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef]
- Athman, J.J.; Wang, Y.; McDonald, D.J.; Boom, W.H.; Harding, C.V.; Wearsch, P.A. Bacterial membrane vesicles mediate the release of Mycobacterium tuberculosis lipoglycans and lipoproteins from infected macrophages. J. Immunol. 2015, 195, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Eagen, W.J.; Lee, J.C. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 3174–3184. [Google Scholar] [CrossRef]
- Bitto, N.J.; Baker, P.J.; Dowling, J.K.; Wray-McCann, G.; De Paoli, A.; Tran, L.S.; Leung, P.L.; Stacey, K.J.; Mansell, A.; Masters, S.L.; et al. Membrane vesicles from Pseudomonas aeruginosa activate the noncanonical inflammasome through caspase-5 in human monocytes. Immunol. Cell Biol. 2018, 96, 1120–1130. [Google Scholar] [CrossRef]
- Elizagaray, M.L.; Gomes, M.T.R.; Guimaraes, E.S.; Rumbo, M.; Hozbor, D.F.; Oliveira, S.C.; Moreno, G. Canonical and Non-canonical Inflammasome Activation by Outer Membrane Vesicles Derived from Bordetella pertussis. Front. Immunol. 2020, 11, 1879. [Google Scholar] [CrossRef] [PubMed]
- Cecil, J.D.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Holden, J.A.; Singleton, W.; Perez-Gonzalez, A.; Mansell, A.; Reynolds, E.C. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front. Immunol. 2017, 8, 1017. [Google Scholar] [CrossRef]
- Ding, P.-H.; Yang, M.-X.; Wang, N.-N.; Jin, L.J.; Dong, Y.; Cai, X.; Chen, L.L. Porphyromonas gingivalis-Induced NLRP3 Inflammasome Activation and Its Downstream Interleukin-1β Release Depend on Caspase-4. Front. Microbiol. 2020, 11, 1881. [Google Scholar] [CrossRef]
- Winter, J.; Letley, D.; Rhead, J.; Atherton, J.; Robinson, K. Helicobacter pylori membrane vesicles stimulate innate pro- and anti-inflammatory responses and induce apoptosis in Jurkat T cells. Infect. Immun. 2014, 82, 1372–1381. [Google Scholar] [CrossRef]
- Duncan, L.; Yoshioka, M.; Chandad, F.; Grenier, D. Loss of lipopolysaccharide receptor CD14 from the surface of human macrophage-like cells mediated by Porphyromonas gingivalis outer membrane vesicles. Microb. Pathog. 2004, 36, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Deo, P.; Chow, S.H.; Han, M.-L.; Speir, M.; Huang, C.; Schittenhelm, R.B.; Dhital, S.; Emery, J.; Li, J.; Kile, B.T.; et al. Mitochondrial dysfunction caused by outer membrane vesicles from gram-negative bacteria activates intrinsic apoptosis and inflammation. Nat. Microbiol. 2020, 5, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Alpdundar, B.E.; Bayyurt, K.B.; Yazar, V.; Aykut, G.; Guler, U.; Salih, B.; Yilmaz, N.S.; Ayanoglu, I.C.; Polat, M.M.; Akcali, K.C.; et al. Human gut commensal membrane vesicles modulate inflammation by generating M2-like macrophages and myeloid-derived suppressor cells. J. Immunol. 2020, 205, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Deo, P.; Chow, S.H.; Hay, L.D.; Kleifeld, O.; Costin, A.; Elgass, K.D.; Jiang, J.-H.; Ramm, G.; Gabriel, K.; Dougan, G.; et al. Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLoS Pathog. 2018, 14, e1006945. [Google Scholar] [CrossRef]
- Rodriguez, B.V.; Kuehn, M.J. Staphylococcus aureus secretes immunomodulatory RNA and DNA via membrane vesicles. Sci. Rep. 2020, 10, 18293. [Google Scholar] [CrossRef]
- Han, E.C.; Choi, S.-Y.; Lee, Y.; Park, J.-W.; Hong, S.-H.; Lee, H.-J. Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood–brain barrier in mice. FASEB J. 2019, 33, 13412–13422. [Google Scholar] [CrossRef]
- Randolph, G.J. Dendritic cells: The first step. J. Exp. Med. 2021, 218, e20202077. [Google Scholar] [CrossRef]
- Alaniz, R.C.; Deatherage, B.L.; Lara, J.C.; Cookson, B.T. Membrane vesicles are immunogenic facsimiles of Salmonella typhimurium that potently activate dendritic cells, prime B and T cell responses, and stimulate protective immunity in vivo. J. Immunol. 2007, 179, 7692–7701. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef]
- Diaz-Garrido, N.; Fábrega, M.-J.; Vera, R.; Giménez, R.; Badia, J.; Baldomà, L. Membrane vesicles from the probiotic Nissle 1917 and gut resident Escherichia coli strains distinctly modulate human dendritic cells and subsequent T cell responses. J. Funct. Foods 2019, 61, 103495. [Google Scholar] [CrossRef]
- Avouac, J.; Gossec, L.; Dougados, M. Diagnostic and predictive value of anti-cyclic citrullinated protein antibodies in rheumatoid arthritis: A systematic literature review. Ann. Rheum. Dis. 2006, 65, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Gabarrini, G.; Heida, R.; van Ieperen, N.; Curtis, M.A.; van Winkelhoff, A.J.; van Dijl, J.M. Dropping anchor: Attachment of peptidylarginine deiminase via A-LPS to secreted outer membrane vesicles of Porphyromonas gingivalis. Sci. Rep. 2018, 8, 8949. [Google Scholar] [CrossRef]
- Gomez-Banuelos, E.; Mukherjee, A.; Darrah, E.; Andrade, F. Rheumatoid arthritis-associated mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. J. Clin. Med. 2019, 8, 1309. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Hirota, K.; Yoshida, K.; Weng, Y.; He, Y.; Shiotsu, N.; Ikegame, M.; Uchida-Fukuhara, Y.; Tanai, A.; Guo, J. Outer membrane vesicles of Porphyromonas gingivalis: Novel communication tool and strategy. Jpn. Dent. Sci. Rev. 2021, 57, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Mirlashari, M.R.; Høiby, E.A.; Holst, J.; Lyberg, T. Outer membrane vesicles from Neisseria meningitidis: Effects on cytokine production in human whole blood. Cytokine 2001, 13, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Choi, K.H.; Kim, Y.S.; Hong, B.S.; Kim, O.Y.; Kim, J.H.; Yoon, C.M.; Koh, G.Y.; Kim, Y.K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS ONE 2010, 5, e11334. [Google Scholar] [CrossRef] [PubMed]
- Namork, E.; Brandtzaeg, P. Fatal meningococcal septicaemia with “blebbing” meningococcus. Lancet 2002, 360, 1741. [Google Scholar] [CrossRef]
- Blackwell, T.S.; Christman, J.W. Sepsis and cytokines: Current status. Br. J. Anaesth. 1996, 77, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, N.C.; Guo, R.F.; Ward, P.A. Novel strategies for the treatment of sepsis. Nat. Med. 2003, 9, 517–524. [Google Scholar] [CrossRef]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.J.; Kubes, P. Intravascular immunity: The host-pathogen encounter in blood vessels. Nat. Rev. Immunol. 2009, 9, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Liu, Y.; Qiu, X.; Tang, Y.; Wang, H.; Xiao, X.; Chen, F.; Lu, B. Bacteria-released outer membrane vesicles promote disseminated intravascular coagulation. Thromb. Res. 2019, 178, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 1997, 112, 321S–329S. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef]
- Karst, S.M. The influence of commensal bacteria on infection with enteric viruses. Nat. Rev. Microbiol. 2016, 14, 197–204. [Google Scholar] [CrossRef]
- Bhar, S.; Zhao, G.; Bartel, J.D.; Sterchele, H.; Del Mazo, A.; Emerson, L.E.; Edelmann, M.J.; Jones, M.K. Bacterial extracellular vesicles control murine norovirus infection through modulation of antiviral immune responses. Front. Immunol. 2022, 13, 909949. [Google Scholar] [CrossRef]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [PubMed]
- Bragazzi Cunha, J.; Wobus, C.E. Select membrane proteins modulate MNV-1 infection of macrophages and dendritic cells in a cell type-specific manner. Virus Res. 2016, 222, 64–70. [Google Scholar] [CrossRef]
- Mosby, C.A.; Bhar, S.; Phillips, M.B.; Edelmann, M.J.; Jones, M.K. Interaction with mammalian enteric viruses alters outer membrane vesicle production and content by commensal bacteria. J. Extracell. Vesicles 2022, 11, e12172. [Google Scholar] [CrossRef]
- Intili, G.; Paladino, L.; Rappa, F.; Alberti, G.; Plicato, A.; Calabrò, F.; Fucarino, A.; Cappello, F.; Bucchieri, F.; Tomasello, G.; et al. From Dysbiosis to Neurodegenerative Diseases through Different Communication Pathways: An Overview. Biology 2023, 12, 195. [Google Scholar] [CrossRef] [PubMed]
- Ait-Belgnaoui, A.; Durand, H.; Cartier, C.; Chaumaz, G.; Eutamene, H.; Ferrier, L.; Houdeau, E.; Fioramonti, J.; Bueno, L.; Theodorou, V. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 2012, 37, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Ning, M.X.; Chen, D.K.; Ma, W.T. Interactions between the gut microbiota and the host innate immune response against pathogens. Front. Immunol. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Varesi, A.; Campagnoli, L.I.M.; Fahmideh, F.; Pierella, E.; Romeo, M.; Ricevuti, G.; Nicoletta, M.; Chirumbolo, S.; Pascale, A. The Interplay between Gut Microbiota and Parkinson’s Disease: Implications on Diagnosis and Treatment. Int. J. Mol. Sci. 2022, 23, 12289. [Google Scholar] [CrossRef]
- Azzoni, R.; Marsland, B.J. The lung-brain axis: A new frontier in host-microbe interactions. Immunity 2022, 55, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Hosang, L.; Canals, R.C.; van der Flier, F.J.; Hollensteiner, J.; Daniel, R.; Flügel, A.; Odoardi, F. The lung microbiome regulates brain autoimmunity. Nature 2022, 603, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Blank, T.; Prinz, M. Fine-tuning of type I IFN-signaling in microglia—Implications for homeostasis, CNS autoimmunity and interferonopathies. Curr. Opin. Neurobiol. 2016, 36, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Nakao, R.; Ito, A.; Uematsu, H.; Senpuku, H. Immunoreactive antigens recognized in serum samples from mice intranasally immunized with Porphyromonas gingivalis outer membrane vesicles. Pathog. Dis. 2015, 73, ftu006. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The transport mechanism of extracellular vesicles at the blood-brain barrier. Curr. Pharm. Des. 2017, 23, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Kim, J.K.; Han, S.K.; Lee, D.Y.; Lee, H.J.; Yim, S.V.; Kim, D.H. The extracellular vesicle of gut microbial Paenalcaligenes hominis is a risk factor for vagus nerve-mediated cognitive impairment. Microbiome 2020, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Peng, W.; Mai, Y.; Li, K.; Wei, W.; Hu, L.; Zhu, S.; Zhou, H.; Jie, W.; Wei, Z.; et al. Outer membrane vesicles enhance tau phosphorylation and contribute to cognitive impairment. J. Cell. Physiol. 2020, 235, 4843–4855. [Google Scholar] [CrossRef]
- Yaghoubfar, R.; Behrouzi, A.; Ashrafian, F.; Shahryari, A.; Moradi, H.R.; Choopani, S.; Hadifar, S.; Vaziri, F.; Nojoumi, S.A.; Fateh, A.; et al. Modulation of serotonin signaling/metabolism by Akkermansia muciniphila and its extracellular vesicles through the gut-brain axis in mice. Sci. Rep. 2020, 10, 22119. [Google Scholar] [CrossRef]
- Gong, T.; Chen, Q.; Mao, H.; Zhang, Y.; Ren, H.; Xu, M.; Chen, H.; Yang, D. Outer membrane vesicles of Porphyromonas gingivalis trigger NLRP3 inflammasome and induce neuroinflammation, tau phosphorylation, and memory dysfunction in mice. Front. Cell. Infect. Microbiol. 2022, 12, 925435. [Google Scholar] [CrossRef]
- Ma, X.; Shin, Y.J.; Yoo, J.W.; Park, H.S.; Kim, D.H. Extracellular vesicles derived from Porphyromonas gingivalis induce vagus nerve-mediated cognitive impairment. J. Adv. Res. 2023, 54, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.; Lobos-González, L.; Guerrero, S.; Kogan, M.J.; Shao, B.; Heinecke, J.W.; Quest, A.F.G.; Leyton, L.; Valenzuela-Valderrama, M. Helicobacter pylori outer membrane vesicles induce astrocyte reactivity through nuclear factor-κappa B activation and cause neuronal damage in vivo in a murine model. J. Neuroinflamm. 2023, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Cools, L.; Van Imschoot, G.; Van Wonterghem, E.; Pauwels, M.J.; Vlaeminck, I.; De Witte, C.; El Andaloussi, S.; Wierda, K.; De Groef, L.; et al. Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. J. Extracell. Vesicles 2023, 12, e12306. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3β (GSK3β) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurons at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Jang, S.E.; Lim, S.M.; Jeong, J.J.; Jang, H.M.; Lee, H.J.; Han, M.J.; Kim, D.H. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018, 11, 369–379. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, K.A.; Ahn, Y.T.; Jeong, J.J.; Huh, C.S.; Kim, D.H. Comparative analysis of gut microbiota in elderly people of urbanized towns and longevity villages. BMC Microbiol. 2015, 15, 49. [Google Scholar] [CrossRef]
- Doulberis, M.; Kotronis, G.; Thomann, R.; Polyzos, S.A.; Boziki, M.; Gialamprinou, D.; Deretzi, G.; Katsinelos, P.; Kountouras, J. Review: Impact of Helicobacter pylori on Alzheimer’s disease: What do we know so far? Helicobacter 2018, 23, e12454. [Google Scholar] [CrossRef]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The impact of systemic inflammation on Alzheimer’s disease pathology. Front. Immunol. 2022, 12, 796867. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 2018, 100, 1337–1353.e5. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, A.; Upadhyaya, B.B.; Fritz, J.V.; Heintz-Buschart, A.; Desai, M.S.; Yusuf, D.; Huang, D.; Baumuratov, A.; Wang, K.; Galas, D.; et al. The extracellular RNA complement of Escherichia coli. Microbiologyopen 2015, 4, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hong, S.H. Analysis of microRNA-size, small RNAs in Streptococcus mutans by deep sequencing. FEMS Microbiol. Lett. 2012, 326, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Choi, J.W.; Lee, Y.; Hong, S.H.; Lee, H.J. Identification of microRNA-Size, small RNAs in Escherichia coli. Curr. Microbiol. 2013, 67, 609–613. [Google Scholar] [CrossRef]
- Blenkiron, C.; Simonov, D.; Muthukaruppan, A.; Tsai, P.; Dauros, P.; Green, S.; Hong, J.; Print, C.G.; Swift, S.; Phillips, A.R. Uropathogenic Escherichia coli releases extracellular vesicles that are associated with RNA. PLoS ONE 2016, 11, e0160440. [Google Scholar] [CrossRef]
- Lee, H.J. Microbial extracellular RNAs and their roles in human diseases. Exp. Biol. Med. 2020, 245, 845–850. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Pirraglia, E.; Dasanayake, A.P.; Norman, R.G.; Boylan, R.J.; Nehorayoff, A.; Glodzik, L.; Brys, M.; de Leon, M.J. TNF-α and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J. Neuroimmunol. 2009, 216, 92–97. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J. Neuropathol. Exp. Neurol. 2010, 69, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef]
- Ha, J.Y.; Seok, J.; Kim, S.J.; Jung, H.J.; Ryu, K.Y.; Nakamura, M.; Jang, I.S.; Hong, S.H.; Lee, Y.; Lee, H.J. Periodontitis promotes bacterial extracellular vesicle-induced neuroinflammation in the brain and trigeminal ganglion. PLoS Pathog. 2023, 19, e1011743. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N. Retrograde signaling via axonal transport through signaling endosomes. J. Pharmacol. Sci. 2019, 141, 91–96. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margutti, P.; D’Ambrosio, A.; Zamboni, S. Microbiota-Derived Extracellular Vesicle as Emerging Actors in Host Interactions. Int. J. Mol. Sci. 2024, 25, 8722. https://doi.org/10.3390/ijms25168722
Margutti P, D’Ambrosio A, Zamboni S. Microbiota-Derived Extracellular Vesicle as Emerging Actors in Host Interactions. International Journal of Molecular Sciences. 2024; 25(16):8722. https://doi.org/10.3390/ijms25168722
Chicago/Turabian StyleMargutti, Paola, Antonella D’Ambrosio, and Silvia Zamboni. 2024. "Microbiota-Derived Extracellular Vesicle as Emerging Actors in Host Interactions" International Journal of Molecular Sciences 25, no. 16: 8722. https://doi.org/10.3390/ijms25168722