
Humanized L184Q Mutated Surfactant Protein C Gene Alters Alveolar Type 2 Epithelial Cell Fate

 ,
,
Abstract
:1. Introduction
2. Results
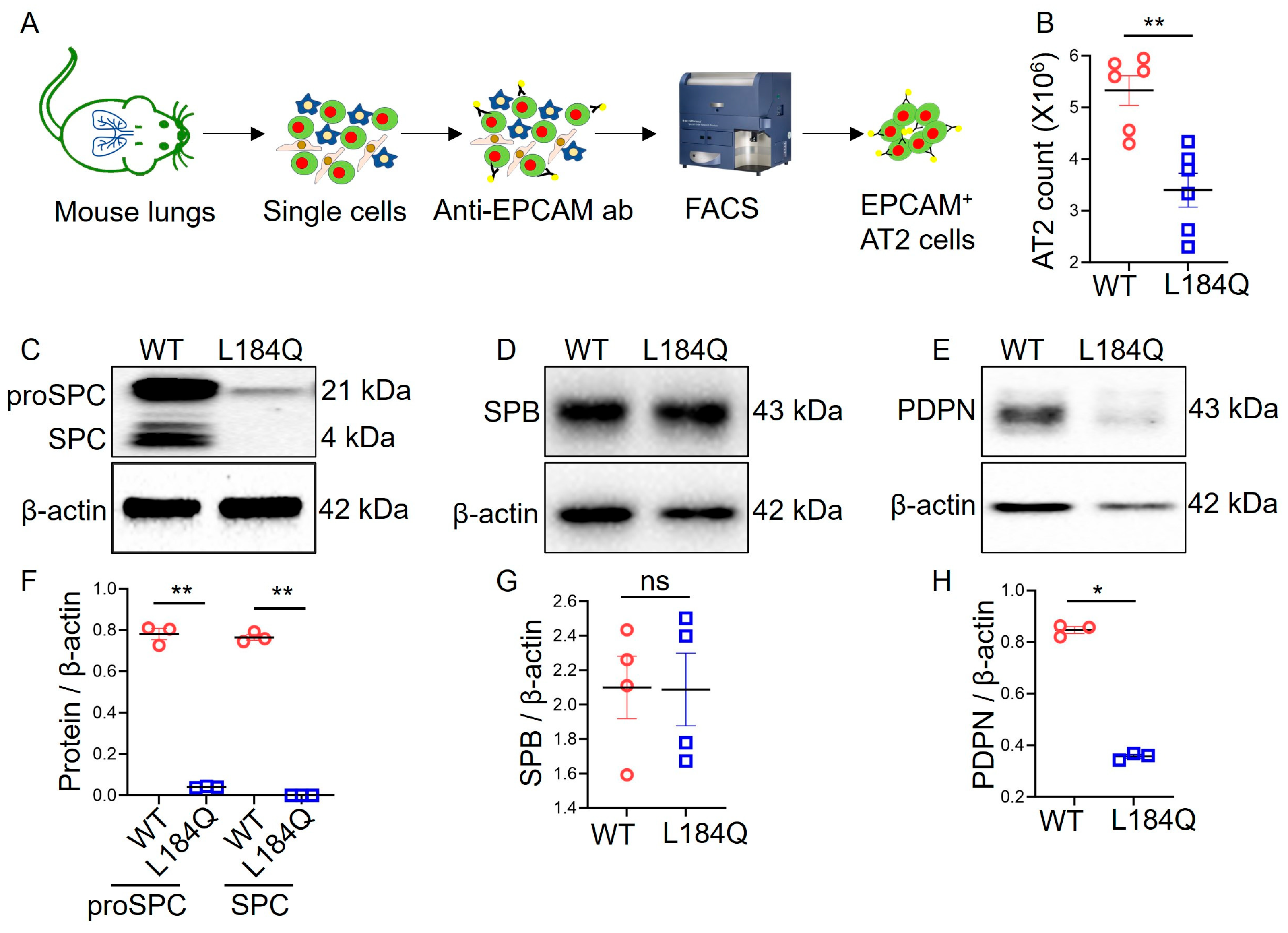
2.1. Sftpc L184Q Mutation Altered AT2 Yield and Marker Protein Expression In Vivo
2.2. SftpcL184Q Mutation Downregulated AT2 Lineage In Vitro
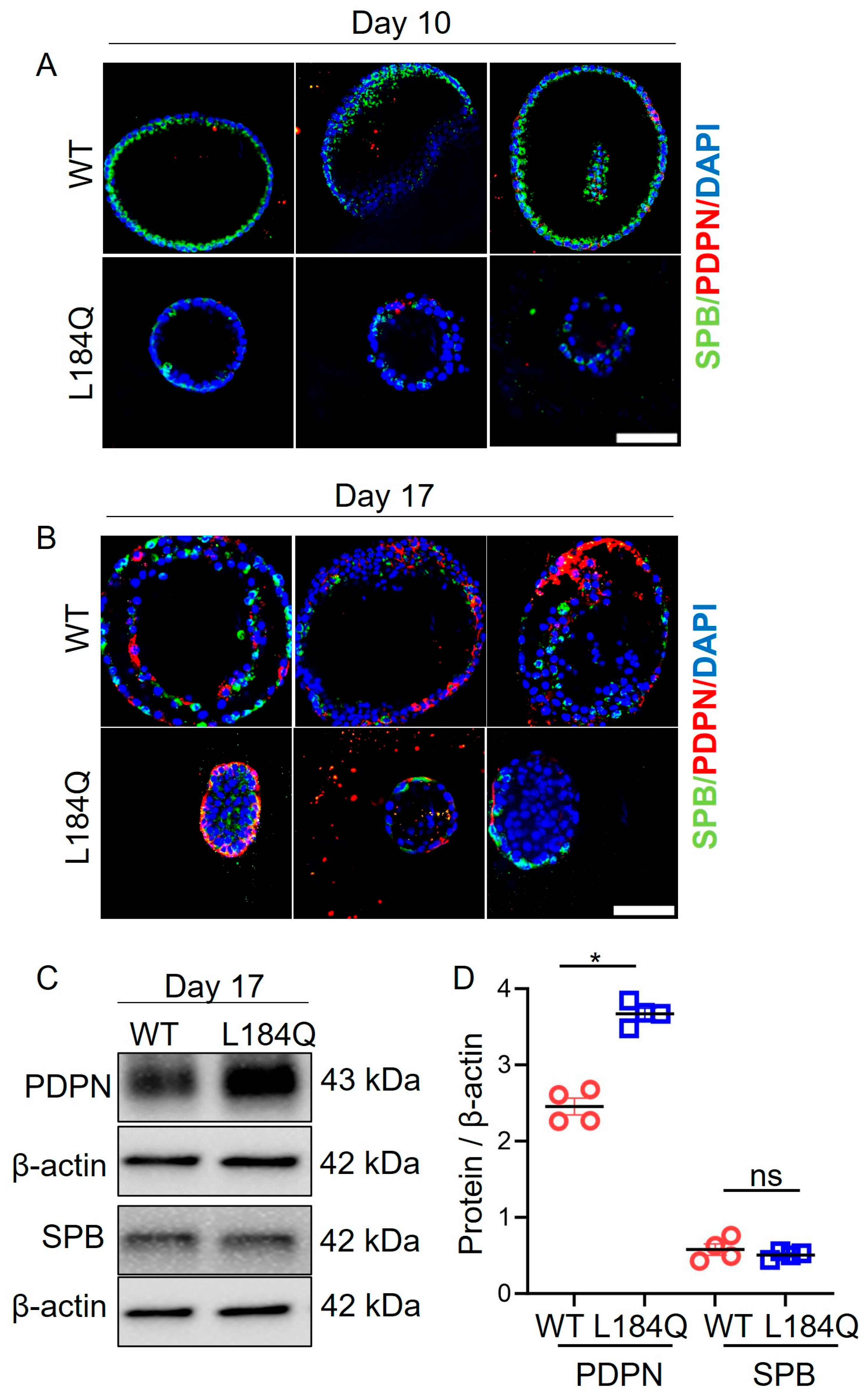
2.3. SftpcL184Q Disrupted Organoid Size and Structure
2.4. SftpcL184Q Halted Maturation of Transitional AT2 Cells into AT1 Cells
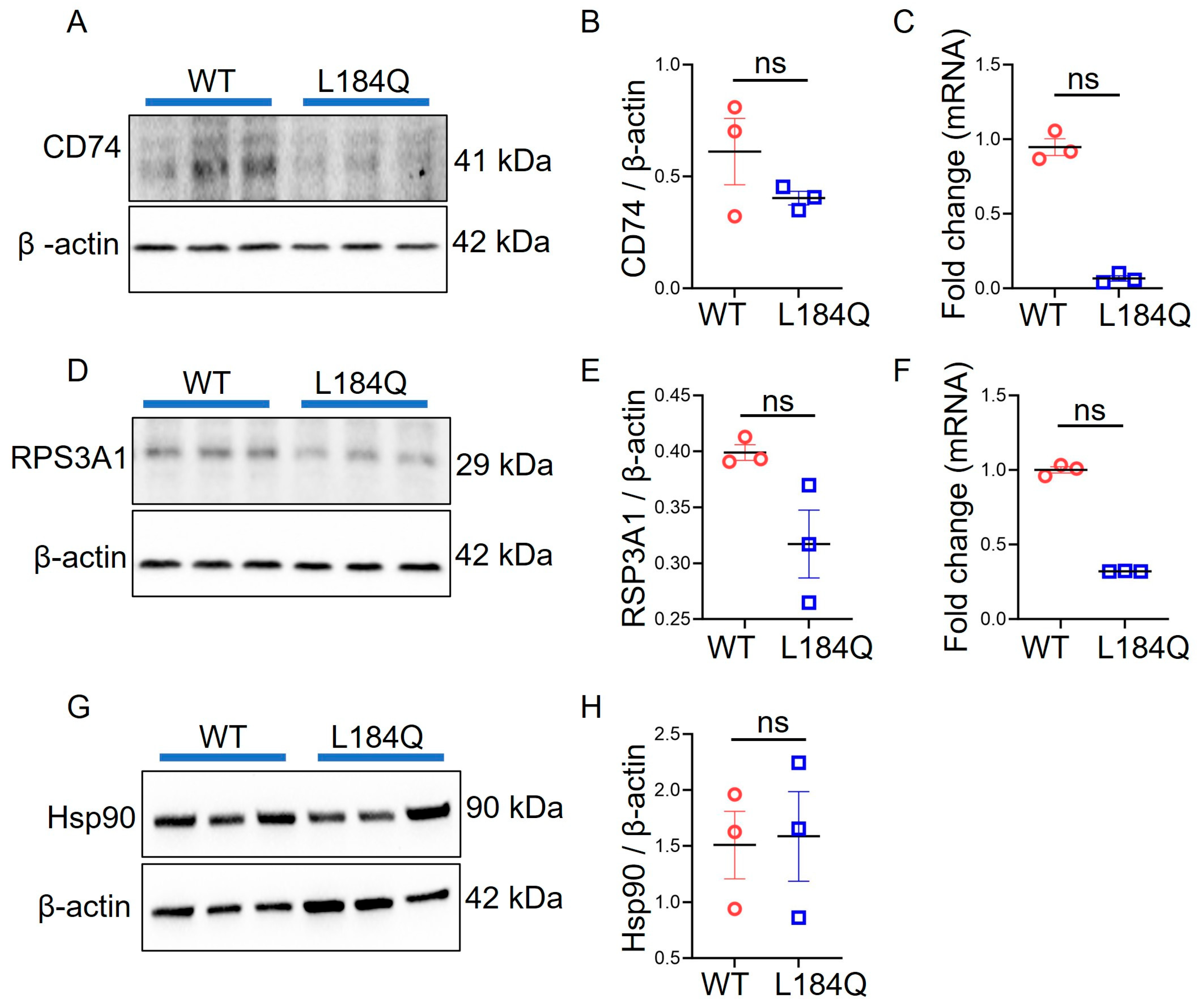
2.5. SftpcL184Q Did Not Alter CD74, HSP90, or RPS3A1 Expression in Organoids
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Fluorescence-Activated Cell Sorting (FACS) Mediated AT2 Cell Isolation
4.3. Feeder-Free Culture of Alveolar Organoids
4.4. Western Blotting
4.5. Immunofluorescence Staining
4.6. Flow Cytometry
4.7. EdU Labeling of Proliferating AT2 Cells
4.8. Quantitative Reverse Transcriptase PCR
4.9. Statistics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirastschijski, U.; Dembinski, R.; Maedler, K. Lung Surfactant for Pulmonary Barrier Restoration in Patients with COVID–19 Pneumonia. Front. Med. 2020, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Gorman, E.A.; O’Kane, C.M.; McAuley, D.F. Acute respiratory distress syndrome in adults: Diagnosis, outcomes, long–term sequelae, and management. Lancet 2022, 400, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J. Thoughts on the alveolar phase of COVID–19. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L115–L120. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.J.; Gattinoni, L.; Calfee, C.S. Acute respiratory distress syndrome. Lancet 2021, 398, 622–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ting, C.; Riemondy, K.A.; Douglas, M.; Foster, K.; Patel, N.; Kaku, N.; Linsalata, A.; Nemzek, J.; Varisco, B.M.; et al. Regulation of epithelial transitional states in murine and human pulmonary fibrosis. J. Clin. Investig. 2023, 133, e165612. [Google Scholar] [CrossRef] [PubMed]
- Wambach, J.A.; Yang, P.; Wegner, D.J.; An, P.; Hackett, B.P.; Cole, F.S.; Hamvas, A. Surfactant protein–C promoter variants associated with neonatal respiratory distress syndrome reduce transcription. Pediatr. Res. 2010, 68, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Moorsel, C.H.M.V.; Vis, J.J.V.D.; Grutters, J.C. Genetic disorders of the surfactant system: Focus on adult disease. Eur. Respir. Rev. 2021, 30, 200085. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Mikawa, R.; Hagiwara, A.; Okuno, Y.; Awaya, T.; Yamamoto, Y.; Takahashi, S.; Yamaki, H.; Osawa, M.; Setoguchi, Y.; et al. Cryptotanshinone is a candidate therapeutic agent for interstitial lung disease associated with a BRICHOS–domain mutation of SFTPC. iScience 2023, 26, 107731. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.; van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.; van Es, H.W.; van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef]
- Sitaraman, S.; Martin, E.P.; Na, C.L.; Zhao, S.; Green, J.; Deshmukh, H.; Perl, A.T.; Bridges, J.P.; Xu, Y.; Weaver, T.E. Surfactant protein C mutation links postnatal type 2 cell dysfunction to adult disease. JCI Insight 2021, 6, e142501. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Hadisurya, M.; Ndetan, H.; Xi, N.M.; Adduri, S.; Konduru, N.V.; Samten, B.; Tao, W.A.; Singh, K.P.; Ji, H.L. Regenerative Signatures in Bronchioalveolar Lavage of Acute Respiratory Distress Syndrome. bioRxiv 2023. [Google Scholar] [CrossRef]
- Marsh, L.M.; Cakarova, L.; Kwapiszewska, G.; Wulffen, W.v.; Herold, S.; Seeger, W.; Lohmeyer, J. Surface expression of CD74 by type II alveolar epithelial cells: A potential mechanism for macrophage migration inhibitory factor–induced epithelial repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L442–L452. [Google Scholar] [CrossRef] [PubMed]
- Peronnet, E.; Venet, F.; Maucort–Boulch, D.; Friggeri, A.; Cour, M.; Argaud, L.; Allaouchiche, B.; Floccard, B.; Aubrun, F.; Rimmelé, T.; et al. Association between mRNA expression of CD74 and IL10 and risk of ICU–acquired infections: A multicenter cohort study. Intensive Care Med. 2017, 43, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wu, M.; Wang, F.; Jiang, L.; Tian, R.; Zhu, X.; He, S. CD74, a novel predictor for bronchopulmonary dysplasia in preterm infants. Medicine 2020, 99, e23477. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Sun, Y.; Wang, K.a.; Chen, Z.; Wang, X.; Chang, F.; Li, T.; Feng, P.; Xia, Z. Relationship between elevated soluble CD74 and severity of experimental and clinical ALI/ARDS. Sci. Rep. 2016, 6, 30067. [Google Scholar] [CrossRef]
- Westmeier, J.; Brochtrup, A.; Paniskaki, K.; Karakoese, Z.; Werner, T.; Sutter, K.; Dolff, S.; Limmer, A.; Mittermüller, D.; Liu, J.; et al. Macrophage migration inhibitory factor receptor CD74 expression is associated with expansion and differentiation of effector T cells in COVID–19 patients. Front. Immunol. 2023, 14, 1236374. [Google Scholar] [CrossRef]
- Ibañez, L.I.; Martinez, V.P.; Iglesias, A.A.; Bellomo, C.M.; Alonso, D.O.; Coelho, R.M.; Martinez Peralta, L.; Periolo, N. Decreased expression of surfactant Protein–C and CD74 in alveolar epithelial cells during influenza virus A(H1N1)pdm09 and H3N2 infection. Microb. Pathog. 2023, 176, 106017. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, X.; Liu, X.; Meng, W.; Guo, W.; Duan, C.; Liang, X.; Kang, L.; Lv, P.; Lin, Q.; et al. Tumor Necrosis Factor α–Dependent Lung Inflammation Promotes the Progression of Lung Adenocarcinoma Originating From Alveolar Type II Cells by Upregulating MIF–CD74. Lab. Invest. 2023, 103, 100034. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Schally, A.V.; Siejka, A. P53, GHRH, inflammation and cancer. EBioMedicine 2018, 37, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed]
- Sibinska, Z.; Tian, X.; Korfei, M.; Kojonazarov, B.; Kolb, J.S.; Klepetko, W.; Kosanovic, D.; Wygrecka, M.; Ghofrani, H.A.; Weissmann, N.; et al. Amplified canonical transforming growth factor–β signalling via heat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1501941. [Google Scholar] [CrossRef]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Štorkánová, H.; Oreská, S.; Špiritović, M.; Heřmánková, B.; Bubová, K.; Komarc, M.; Pavelka, K.; Vencovský, J.; Distler, J.H.W.; Šenolt, L.; et al. Plasma Hsp90 levels in patients with systemic sclerosis and relation to lung and skin involvement: A cross–sectional and longitudinal study. Sci. Rep. 2021, 11, 1. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, L.D.; Zhang, F.; Liang, Q.Z.; Jiao, Y.; Shi, F.S.; He, B.; Xu, P.; Huang, Y.W. Heat shock protein 90 facilitates SARS–CoV–2 structural protein–mediated virion assembly and promotes virus–induced pyroptosis. J. Biol. Chem. 2023, 299, 104668. [Google Scholar] [CrossRef] [PubMed]
- Solopov, P.; Biancatelli, R.; Marinova, M.; Dimitropoulou, C.; Catravas, J.D. The HSP90 Inhibitor, AUY–922, Ameliorates the Development of Nitrogen Mustard–Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 4740. [Google Scholar] [CrossRef] [PubMed]
- Antonov, A.; Snead, C.; Gorshkov, B.; Antonova, G.N.; Verin, A.D.; Catravas, J.D. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 551–559. [Google Scholar] [CrossRef]
- De Graeve, S.; Marinelli, S.; Stolz, F.; Hendrix, J.; Vandamme, J.; Engelborghs, Y.; Van Dijck, P.; Thevelein, J.M. Mammalian ribosomal and chaperone protein RPS3A counteracts α–synuclein aggregation and toxicity in a yeast model system. Biochem. J. 2013, 455, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Wang, J.; Liu, Y.; Qiu, L.; Li, J.Z.; Hu, H.G.; Hu, Z.L.; Zhang, W.; Lu, B.; Zhang, J.P. Esculentoside A specifically binds to ribosomal protein S3a and impairs LPS–induced signaling in macrophages. Int. Immunopharmacol. 2018, 54, 254–260. [Google Scholar] [CrossRef]
- Lim, K.H.; Kim, K.H.; Choi, S.I.; Park, E.S.; Park, S.H.; Ryu, K.; Park, Y.K.; Kwon, S.Y.; Yang, S.I.; Lee, H.C.; et al. RPS3a over–expressed in HBV–associated hepatocellular carcinoma enhances the HBx–induced NF–κB signaling via its novel chaperoning function. PLoS ONE 2011, 6, e22258. [Google Scholar] [CrossRef]
- Kashuba, E.; Yurchenko, M.; Szirak, K.; Stahl, J.; Klein, G.; Szekely, L. Epstein–Barr virus–encoded EBNA–5 binds to Epstein–Barr virus–induced Fte1/S3a protein. Exp. Cell Res. 2005, 303, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Naora, H.; Naora, H. Involvement of ribosomal proteins in regulating cell growth and apoptosis: Translational modulation or recruitment for extraribosomal activity? Immunol. Cell Biol. 1999, 77, 197–205. [Google Scholar] [CrossRef]
- Tang, Y.; He, Y.; Li, C.; Mu, W.; Zou, Y.; Liu, C.; Qian, S.; Zhang, F.; Pan, J.; Wang, Y.; et al. RPS3A positively regulates the mitochondrial function of human periaortic adipose tissue and is associated with coronary artery diseases. Cell Discov. 2018, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Schröder, B. The multifaceted roles of the invariant chain CD74—More than just a chaperone. Biochim. Biophys. Acta 2016, 1863, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.P.; Wert, S.E.; Nogee, L.M.; Weaver, T.E. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J. Biol. Chem. 2003, 278, 52739–52746. [Google Scholar] [CrossRef] [PubMed]
- Venosa, A.; Katzen, J.; Tomer, Y.; Kopp, M.; Jamil, S.; Russo, S.J.; Mulugeta, S.; Beers, M.F. Epithelial Expression of an Interstitial Lung Disease–Associated Mutation in Surfactant Protein–C Modulates Recruitment and Activation of Key Myeloid Cell Populations in Mice. J. Immunol. 2019, 202, 2760–2771. [Google Scholar] [CrossRef] [PubMed]
- Woischnik, M.; Sparr, C.; Kern, S.; Thurm, T.; Hector, A.; Hartl, D.; Liebisch, G.; Mulugeta, S.; Beers, M.F.; Schmitz, G.; et al. A non–BRICHOS surfactant protein c mutation disrupts epithelial cell function and intercellular signaling. BMC Cell Biol. 2010, 11, 88. [Google Scholar] [CrossRef]
- Principi, L.; Ferrini, E.; Ciccimarra, R.; Pagani, L.; Chinello, C.; Previtali, P.; Smith, A.; Villetti, G.; Zoboli, M.; Ravanetti, F.; et al. Proteomic Fingerprint of Lung Fibrosis Progression and Response to Therapy in Bleomycin–Induced Mouse Model. Int. J. Mol. Sci. 2023, 24, 4410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Ali, G.; Chang, J.; Komatsu, S.; Tsukasaki, Y.; Nie, H.G.; Chang, Y.; Zhang, M.; Liu, Y.; Jain, K.; et al. Proliferative regulation of alveolar epithelial type 2 progenitor cells by human Scnn1d gene. Theranostics 2019, 9, 8155–8170. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.G.; Zhao, R.; Liu, Y.; Guo, X.; Yi, G.; Ji, H.L. Wnt5a/β–catenin axis is involved in the downregulation of AT2 lineage by PAI–1. Am. J. Physiol. Lung Cell Mol. Physiol. 2022, 323, L515–L524. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda–Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open–source platform for biological–image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| CD74 | GCTGGATGAAGCAGTGGCTCTT | GATGTGGCTGACTTCTTCCTGG |
| RPS3A1 | GGTACGATGTGAAAGCTCCAGC | GGCTCACTTCAAACACACGACC |
| GAPDH | CATCACTGCCACCCAGAAGACTG | ATGCCAGTGAGCTTCCCGTTCAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, K.G.; Liu, Y.; Zhao, R.; Muire, P.J.; Zhang, J.; Zang, Q.S.; Ji, H.-L. Humanized L184Q Mutated Surfactant Protein C Gene Alters Alveolar Type 2 Epithelial Cell Fate. Int. J. Mol. Sci. 2024, 25, 8723. https://doi.org/10.3390/ijms25168723
Jain KG, Liu Y, Zhao R, Muire PJ, Zhang J, Zang QS, Ji H-L. Humanized L184Q Mutated Surfactant Protein C Gene Alters Alveolar Type 2 Epithelial Cell Fate. International Journal of Molecular Sciences. 2024; 25(16):8723. https://doi.org/10.3390/ijms25168723
Chicago/Turabian StyleJain, Krishan G., Yang Liu, Runzhen Zhao, Preeti J. Muire, Jiwang Zhang, Qun Sophia Zang, and Hong-Long Ji. 2024. "Humanized L184Q Mutated Surfactant Protein C Gene Alters Alveolar Type 2 Epithelial Cell Fate" International Journal of Molecular Sciences 25, no. 16: 8723. https://doi.org/10.3390/ijms25168723