
Metal-Free, PPA-Mediated Fisher Indole Synthesis via Tandem Hydroamination–Cyclization Reaction between Simple Alkynes and Arylhydrazines

 , , , , , and
, , , , , and
Abstract
:1. Introduction
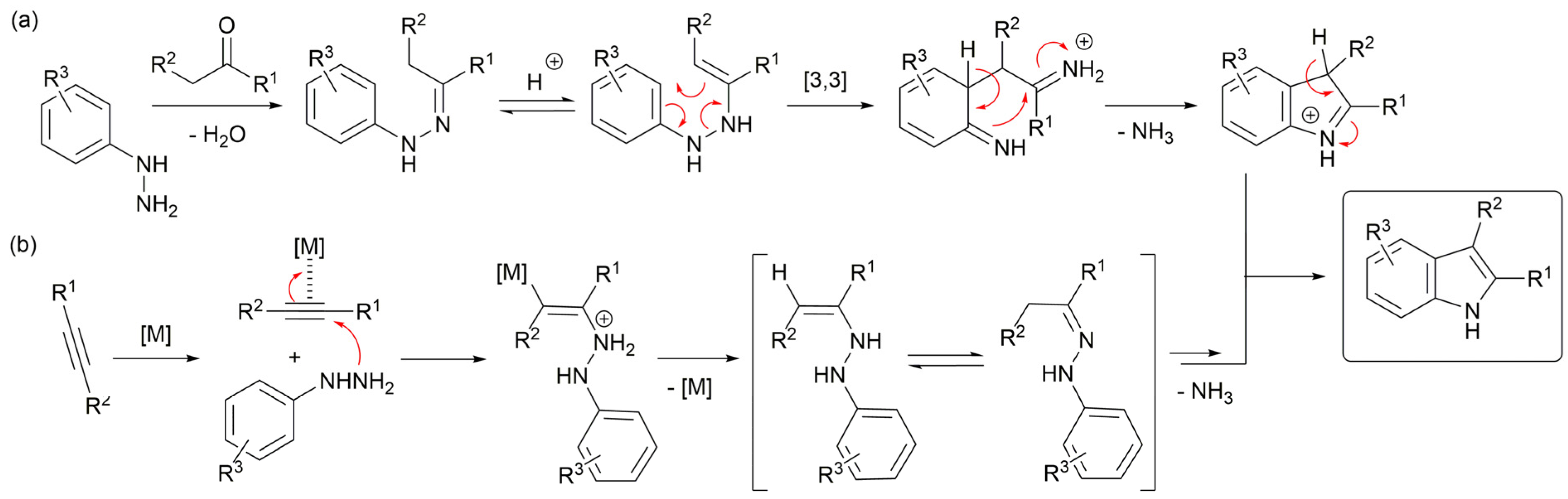
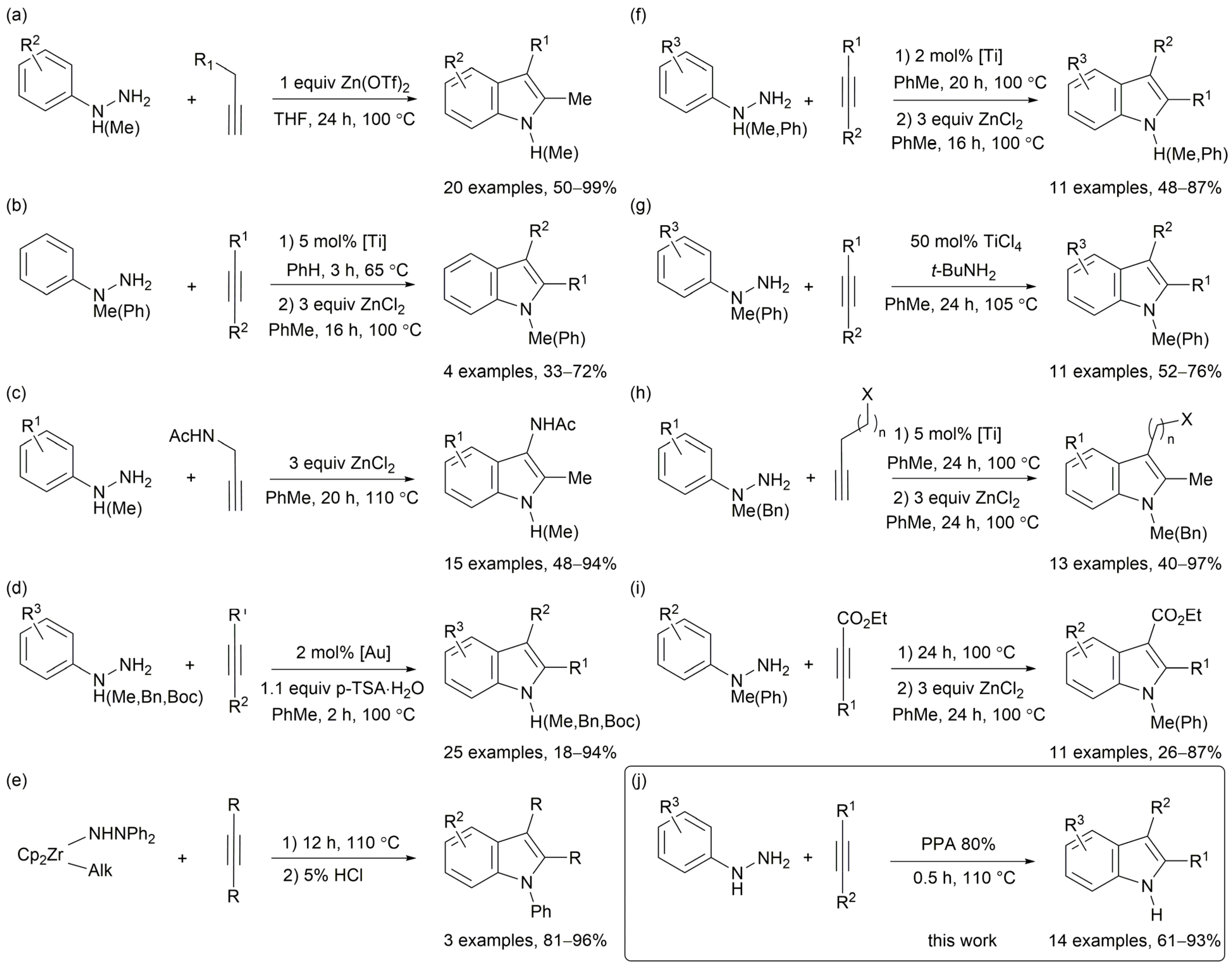
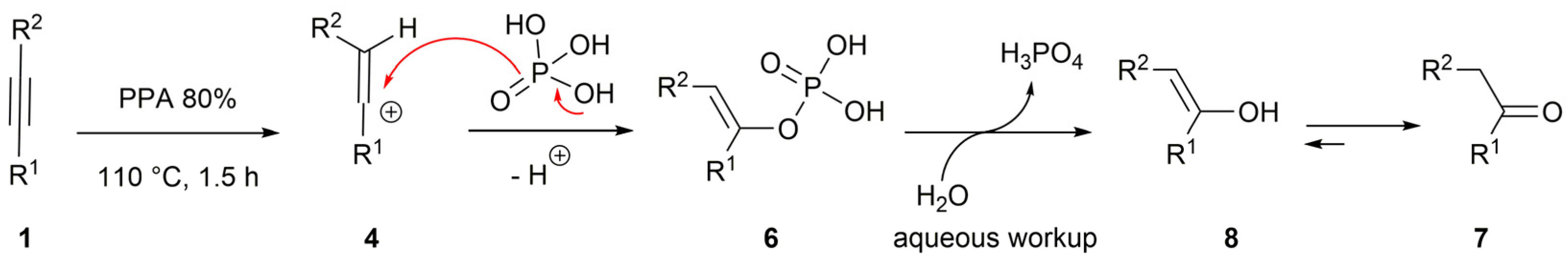
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Preparation of Indoles 3 (General Procedure)
3.3. Preparation of Acetophenones 7 (General Procedure)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaushik, N.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.; Verma, A.; Choi, E. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sharma, S.; Kalra, S.; Singh, G.; Monga, V.; Kumar, B. Medicinal Perspective of Indole Derivatives: Recent Developments and Structure-Activity Relationship Studies. Curr. Drug Targets 2020, 21, 864–891. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Han, C.; Mohammed, S.; Li, S.; Song, Y.; Sun, F.; Du, Y. Indole-Containing Pharmaceuticals: Targets, Pharmacological Activities, and SAR Studies. RSC Med. Chem. 2024, 15, 788–808. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery; Wiley: Hoboken, NJ, USA, 2016; ISBN 9780470512180. [Google Scholar]
- Das, S. Indole Frameworks via Transition-Metal-Free Annulation: A Current Perspective. New J. Chem. 2023, 47, 13729–13775. [Google Scholar] [CrossRef]
- Bugaenko, D.I.; Karchava, A.V.; Yurovskaya, M.A. Synthesis of Indoles: Recent Advances. Russ. Chem. Rev. 2019, 88, 99–159. [Google Scholar] [CrossRef]
- Youn, S.W.; Ko, T.Y. Metal-Catalyzed Synthesis of Substituted Indoles. Asian J. Org. Chem. 2018, 7, 1467–1487. [Google Scholar] [CrossRef]
- Ma, J.; Feng, R.; Dong, Z. Recent Advances in Indole Synthesis and the Related Alkylation. Asian J. Org. Chem. 2023, 12, e202300092. [Google Scholar] [CrossRef]
- Mathada, B.S.; Yernale, N.G. Current Advances in Transition Metal-Free Access to Indoles. A Review. Org. Prep. Proced. Int. 2023, 55, 299–316. [Google Scholar] [CrossRef]
- De, S. Synthesis of Some Heterocyclic Compounds Using Named Reactions. In Applied Organic Chemistry; Wiley: Hoboken, NJ, USA, 2021; pp. 469–506. [Google Scholar] [CrossRef]
- Inman, M.; Moody, C.J. Indole Synthesis—Something Old, Something New. Chem. Sci. 2013, 4, 29–41. [Google Scholar] [CrossRef]
- Humphrey, G.R.; Kuethe, J.T. Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef]
- Heravi, M.M.; Amiri, Z.; Kafshdarzadeh, K.; Zadsirjan, V. Synthesis of Indole Derivatives as Prevalent Moieties Present in Selected Alkaloids. RSC Adv. 2021, 11, 33540–33612. [Google Scholar] [CrossRef]
- Heravi, M.M.; Rohani, S.; Zadsirjan, V.; Zahedi, N. Fischer Indole Synthesis Applied to the Total Synthesis of Natural Products. RSC Adv. 2017, 7, 52852–52887. [Google Scholar] [CrossRef]
- Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L.J. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Catalysis and Regioselectivity in Hydrofunctionalization Reactions of Unsaturated Carbon Bonds. Part III. Russ. Chem. Rev. 2021, 90, 70–93. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Zeni, G. Recent Advances in the Synthesis of Indoles from Alkynes and Nitrogen Sources. Org. Chem. Front. 2020, 7, 155–210. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Zeni, G. Recent Developments in the Cyclization of Alkynes and Nitrogen Compounds for the Synthesis of Indole Derivatives. Asian J. Org. Chem. 2021, 10, 1282–1318. [Google Scholar] [CrossRef]
- Krüger (née Alex), K.; Tillack, A.; Beller, M. Catalytic Synthesis of Indoles from Alkynes. Adv. Synth. Catal. 2008, 350, 2153–2167. [Google Scholar] [CrossRef]
- Alex, K.; Tillack, A.; Schwarz, N.; Beller, M. Zinc-Promoted Hydrohydrazination of Terminal Alkynes: An Efficient Domino Synthesis of Indoles. Angew. Chemie Int. Ed. 2008, 47, 2304–2307. [Google Scholar] [CrossRef] [PubMed]
- Yim, J.C.H.; Bexrud, J.A.; Ayinla, R.O.; Leitch, D.C.; Schafer, L.L. Bis(Amidate)Bis(Amido) Titanium Complex: A Regioselective Intermolecular Alkyne Hydroamination Catalyst. J. Org. Chem. 2014, 79, 2015–2028. [Google Scholar] [CrossRef]
- Pews-Davtyan, A.; Tillack, A.; Schmöle, A.-C.; Ortinau, S.; Frech, M.J.; Rolfs, A.; Beller, M. A New Facile Synthesis of 3-Amidoindole Derivatives and Their Evaluation as Potential GSK-3β Inhibitors. Org. Biomol. Chem. 2010, 8, 1149. [Google Scholar] [CrossRef]
- Pews-Davtyan, A.; Beller, M. Efficient and Simple Zinc-Mediated Synthesis of 3-Amidoindoles. Org. Biomol. Chem. 2011, 9, 6331–6334. [Google Scholar] [CrossRef] [PubMed]
- Vedekhina, T.; Lukin, A.; Rogacheva, E.; Kraeva, L.; Krasavin, M. Zn(OTf)2-Catalyzed Arenehydrazination of Protected Propargylamines Leading to 3-Amidoindoles. Tetrahedron Lett. 2020, 61, 151430. [Google Scholar] [CrossRef]
- Patil, N.T.; Konala, A. Mechanistic Dichotomy with Alkynes in the Formal Hydrohydrazination/Fischer Indolization Tandem Reaction Catalyzed by a Ph3PAuNTf2/p-TSA Binary System. Eur. J. Org. Chem. 2010, 2010, 6831–6839. [Google Scholar] [CrossRef]
- Walsh, P.J.; Carney, M.J.; Bergman, R.G. Generation, Dative Ligand Trapping, and N-N Bond Cleavage Reactions of the First Monomeric H1-Hydrazido Zirconocene Complex, Cp2Zr=NNPh2. A Zirconium-Mediated Synthesis of Indoles. J. Am. Chem. Soc. 1991, 113, 6343–6345. [Google Scholar] [CrossRef]
- Banerjee, S.; Barnea, E.; Odom, A.L. Titanium-Catalyzed Hydrohydrazination with Monosubstituted Hydrazines: Catalyst Design, Synthesis, and Reactivity. Organometallics 2008, 27, 1005–1014. [Google Scholar] [CrossRef]
- Cao, C.; Shi, Y.; Odom, A.L. Intermolecular Alkyne Hydroaminations Involving 1,1-Disubstituted Hydrazines. Org. Lett. 2002, 4, 2853–2856. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Born, R. TiCl4/t-BuNH2 as the Sole Catalyst for a Hydroamination-Based Fischer Indole Synthesis. Tetrahedron Lett. 2004, 45, 9541–9544. [Google Scholar] [CrossRef]
- Khedkar, V.; Tillack, A.; Michalik, M.; Beller, M. Convenient Synthesis of Tryptophols and Tryptophol Homologues by Hydroamination of Alkynes. Tetrahedron 2005, 61, 7622–7631. [Google Scholar] [CrossRef]
- Khedkar, V.; Tillack, A.; Michalik, M.; Beller, M. Efficient One-Pot Synthesis of Tryptamines and Tryptamine Homologues by Amination of Chloroalkynes. Tetrahedron Lett. 2004, 45, 3123–3126. [Google Scholar] [CrossRef]
- Schwarz, N.; Alex, K.; Sayyed, I.; Khedkar, V.; Tillack, A.; Beller, M. Titanium-Catalyzed Hydroamination of Propargyl Alcohol Derivatives: Synthesis of 3-Silyloxy-2-Methylindoles via Hydrohydrazination. Synlett 2007, 2007, 1091–1095. [Google Scholar] [CrossRef]
- Sayyed, I.A.; Alex, K.; Tillack, A.; Schwarz, N.; Michalik, D.; Beller, M. A Convenient and General Method for the Synthesis of Indole-2,3-dicarboxylates and 2-Arylindole-3-carboxylates. Eur. J. Org. Chem. 2007, 2007, 4525–4528. [Google Scholar] [CrossRef]
- Gehrmann, T.; Lloret Fillol, J.; Scholl, S.A.; Wadepohl, H.; Gade, L.H. Zirconium-Catalyzed Multistep Reaction of Hydrazines with Alkynes: A Non-Fischer-Type Pathway to Indoles. Angew. Chemie Int. Ed. 2011, 50, 5757–5761. [Google Scholar] [CrossRef]
- Zhao, D.; Shi, Z.; Glorius, F. Indole Synthesis by Rhodium(III)-Catalyzed Hydrazine-Directed C-H Activation: Redox-Neutral and Traceless by N-N Bond Cleavage. Angew. Chemie Int. Ed. 2013, 52, 12426–12429. [Google Scholar] [CrossRef]
- Li, D.Y.; Chen, H.J.; Liu, P.N. Rhodium-Catalyzed Oxidative Annulation of Hydrazines with Alkynes Using a Nitrobenzene Oxidant. Org. Lett. 2014, 16, 6176–6179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Wang, L.; Chen, K.; Song, C.; Zhu, J. Co(III)-Catalyzed, Internal and Terminal Alkyne-Compatible Synthesis of Indoles. Org. Lett. 2016, 18, 3806–3809. [Google Scholar] [CrossRef]
- Arutiunov, N.A.; Aksenov, A.V.; Aksenov, D.A.; Kurenkov, I.A.; Aksenova, I.V.; Zatsepilina, A.M.; Aksenov, N.A.; Kornienko, A. Convenient Synthesis of (Z)-3-(1-Aryl-2-Nitrovinyl)-Indoles. Tetrahedron Lett. 2023, 129, 154722. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Arutiunov, N.A.; Aksenov, A.V.; Kirilov, N.K.; Aksenova, I.V.; Aksenov, D.A.; Aleksandrova, E.V.; Rubin, M.; Kornienko, A. Synthesis of β-Carbolines with Electrocyclic Cyclization of 3-Nitrovinylindoles. Int. J. Mol. Sci. 2023, 24, 13107. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Yin, D. An Eco-Friendly Industrial Fischer Indole Cyclization Process. Org. Process Res. Dev. 2018, 22, 1115–1118. [Google Scholar] [CrossRef]
- Pohlki, F.; Doye, S. The Catalytic Hydroamination of Alkynes. Chem. Soc. Rev. 2003, 32, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Severin, R.; Doye, S. The Catalytic Hydroamination of Alkynes. Chem. Soc. Rev. 2007, 36, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Yim, J.C.-H.; Schafer, L.L. Efficient Anti-Markovnikov-Selective Catalysts for Intermolecular Alkyne Hydroamination: Recent Advances and Synthetic Applications. Eur. J. Org. Chem. 2014, 2014, 6825–6840. [Google Scholar] [CrossRef]
- Patil, N.T.; Singh, V. Alkyne Hydroamination Triggered Cyclizations: A Powerful Tool for the Construction of Biologically Important Structural Motifs. J. Organomet. Chem. 2011, 696, 419–432. [Google Scholar] [CrossRef]
- Escorihuela, J.; Lledós, A.; Ujaque, G. Anti-Markovnikov Intermolecular Hydroamination of Alkenes and Alkynes: A Mechanistic View. Chem. Rev. 2023, 123, 9139–9203. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Saunthwal, R.K.; Verma, A.K. Base-Mediated Hydroamination of Alkynes. Acc. Chem. Res. 2017, 50, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.S.S.; Zeni, G. Synthesis of Indoles from Alkynes and a Nitrogen Source under Metal-Free Conditions. Org. Biomol. Chem. 2020, 18, 4906–4915. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef] [PubMed]
- Coman, S.M.; Parvulescu, V.I. Nonprecious Metals Catalyzing Hydroamination and C–N Coupling Reactions. Org. Process Res. Dev. 2015, 19, 1327–1355. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Naájera, C.; Yus, M. Catalysis and Regioselectivity in Hydrofunctionalization Reactions of Unsaturated Carbon Bonds. Part II. Hydroamination. Russ. Chem. Rev. 2020, 89, 1074–1114. [Google Scholar] [CrossRef]
- Bernoud, E.; Lepori, C.; Mellah, M.; Schulz, E.; Hannedouche, J. Recent Advances in Metal Free- and Late Transition Metal-Catalysed Hydroamination of Unactivated Alkenes. Catal. Sci. Technol. 2015, 5, 2017–2037. [Google Scholar] [CrossRef]
- Kang, H.-J.; Lee, J.-H.; Kim, D.-H.; Cho, C.-G. Imidazole-Selective Alkyne Hydroamination under Physiological Conditions. Org. Lett. 2020, 22, 7588–7593. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Grishin, I.Y.; Aksenov, N.A.; Malyuga, V.V.; Aksenov, D.A.; Nobi, M.A.; Rubin, M. Electrophilically Activated Nitroalkanes in Synthesis of 3,4-Dihydroquinozalines. Molecules 2021, 26, 4274. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.V.; Smirnov, A.N.; Aksenov, N.A.; Bijieva, A.S.; Aksenova, I.V.; Rubin, M. Benzimidazoles and Benzoxazoles via the Nucleophilic Addition of Anilines to Nitroalkanes. Org. Biomol. Chem. 2015, 13, 4289–4295. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, N.A.; Malyuga, V.V.; Abakarov, G.M.; Aksenov, D.A.; Voskressensky, L.G.; Aksenov, A.V. Synthesis of 3,4-Dihydroisoquinolines Using Nitroalkanes in Polyphosphoric Acid. Russ. Chem. Bull. 2019, 68, 1047–1051. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Khamraev, V.; Aksenov, N.A.; Kirilov, N.K.; Domenyuk, D.A.; Zelensky, V.A.; Rubin, M. Electrophilic Activation of Nitroalkanes in Efficient Synthesis of 1,3,4-Oxadiazoles. RSC Adv. 2019, 9, 6636–6642. [Google Scholar] [CrossRef] [PubMed]
- Popp, F.D.; McEwen, W.E. Polyphosphoric Acids as a Reagent in Organic Chemistry. Chem. Rev. 1958, 58, 321–401. [Google Scholar] [CrossRef]
- Dodd, J.H. Polyphosphoric Acid. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2001; pp. 183–202. ISBN 0471936235. [Google Scholar]
- Pinchukova, N.A.; Chebanov, V.A.; Gorobets, N.Y.; Gudzenko, L.V.; Ostras, K.S.; Shishkin, O.V.; Hulshof, L.A.; Voloshko, A.Y. Beneficial Energy-Efficiencies in the Microwave-Assisted Vacuum Preparation of Polyphosphoric Acid. Chem. Eng. Process. Process Intensif. 2011, 50, 1193–1197. [Google Scholar] [CrossRef]
- Guy, A.; Guetté, J.-P.; Lang, G. Utilization of Polyphosphoric Acid in the Presence of a Co-Solvent. Synthesis 1980, 1980, 222–223. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Prajapati, N.P.; Patel, H.D. Silica-Supported Polyphosphoric Acid (PPA-SiO2): An Efficient and Reusable Heterogeneous Catalyst for Ecofriendly Organic Synthesis. Synth. Commun. 2016, 46, 197–219. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, A.V.; Kornienko, A.; De Carvalho, A.; Mathieu, V.; Aksenov, D.A.; Ovcharov, S.N.; Griaznov, G.D.; Rubin, M. A Nitroalkane-Based Approach to One-Pot Three-Component Synthesis of Isocryptolepine and Its Analogs with Potent Anti-Cancer Activities. RSC Adv. 2018, 8, 36980–36986. [Google Scholar] [CrossRef]
- Sil, S.; Krishnapriya, A.U.; Mandal, P.; Kuniyil, R.; Mandal, S.K. Cross-Coupling Between Aryl Halides and Aryl Alkynes Catalyzed by an Odd Alternant Hydrocarbon. Chem. Eur. J. 2024, 30, e202400895. [Google Scholar] [CrossRef]
- Liang, S.; Hammond, G.B.; Xu, B. Efficient Hydration of Alkynes through Acid-Assisted Brønsted Acid Catalysis. Chem. Commun. 2015, 51, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Hagemeyer, H.J.; Hull, D.C. Reactions of Isopropenyl Acetate. Ind. Eng. Chem. 1949, 41, 2920–2924. [Google Scholar] [CrossRef]
- Hintermann, L.; Labonne, A. Catalytic Hydration of Alkynes and Its Application in Synthesis. Synthesis 2007, 2007, 1121–1150. [Google Scholar] [CrossRef]
- Salvio, R.; Bassetti, M. Sustainable Hydration of Alkynes Promoted by First Row Transition Metal Complexes. Background, Highlights, and Perspectives. Inorganica Chim. Acta 2021, 522, 120288. [Google Scholar] [CrossRef]
- Sedikides, A.T.; Lennox, A.J.J. Silver-Catalyzed (Z)-β-Fluoro-Vinyl Iodonium Salts from Alkynes: Efficient and Selective Syntheses of Z-Monofluoroalkenes. J. Am. Chem. Soc. 2024, 146, 15672–15680. [Google Scholar] [CrossRef] [PubMed]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis–Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Gholinejad, M.; Esmailoghli, H.; Khosravi, F.; Sansano, J.M. Ionic Liquid Modified Carbon Nanotube Supported Palladium Nanoparticles for Efficient Sonogashira-Hagihara Reaction. J. Organomet. Chem. 2022, 963, 122295. [Google Scholar] [CrossRef]
- Hering, T.; Hari, D.P.; König, B. Visible-Light-Mediated α-Arylation of Enol Acetates Using Aryl Diazonium Salts. J. Org. Chem. 2012, 77, 10347–10352. [Google Scholar] [CrossRef]
- Yu, X.; Park, E.-J.; Kondratyuk, T.P.; Pezzuto, J.M.; Sun, D. Synthesis of 2-Arylindole Derivatives and Evaluation as Nitric Oxide Synthase and NFκB Inhibitors. Org. Biomol. Chem. 2012, 10, 8835. [Google Scholar] [CrossRef]
- Lai, R.-Y.; Surekha, K.; Hayashi, A.; Ozawa, F.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Intra- and Intermolecular Hydroamination of Alkynes Catalyzed by Ortho-Metalated Iridium Complexes. Organometallics 2007, 26, 1062–1068. [Google Scholar] [CrossRef]
- Bhunia, S.K.; Polley, A.; Natarajan, R.; Jana, R. Through-Space 1,4-Palladium Migration and 1,2-Aryl Shift: Direct Access to Dibenzo[a,c]Carbazoles through a Triple C-H Functionalization Cascade. Chem. Eur. J. 2015, 21, 16786–16791. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Annaka, K.; Fujita, A.; Sato, A.; Konakahara, T. InBr3-Promoted Divergent Approach to Polysubstituted Indoles and Quinolines from 2-Ethynylanilines: Switch from an Intramolecular Cyclization to an Intermolecular Dimerization by a Type of Terminal Substituent Group. J. Org. Chem. 2008, 73, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Lou, Y.; Wang, C.; Qi, L.; Fang, T.; Zhang, F.; Xu, H.; Zhou, L.; Li, W.; Zhang, G.; et al. Chiral Brønsted Acid from Chiral Phosphoric Acid Boron Complex and Water: Asymmetric Reduction of Indoles. Angew. Chem. Int. Ed. 2020, 59, 3294–3299. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Leslie, B.E.; Driver, T.G. Dirhodium(II)-Catalyzed Intramolecular C-H Amination of Aryl Azides. Angew. Chem. Int. Ed. 2008, 47, 5056–5059. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xia, Y.; Jin, Y.; Lu, M. Green and Reusable Homogeneous Oxidative System with Ceric Ammonium Nitrate/[Imim-PEG 1000-TEMPO] for Efficient Aerobic Oxidation of Alcohols and One-pot Synthesis of Benzimidazoles from Alcohols under Ambient Conditions. Appl. Organomet. Chem. 2015, 29, 109–112. [Google Scholar] [CrossRef]
- Jung, J.; Kim, J.; Park, G.; You, Y.; Cho, E.J. Selective Debromination and A-Hydroxylation of α-Bromo Ketones Using Hantzsch Esters as Photoreductants. Adv. Synth. Catal. 2016, 358, 74–80. [Google Scholar] [CrossRef]
- Chai, H.; Wang, L.; Liu, T.; Yu, Z. A Versatile Ru(II)-NNP Complex Catalyst for the Synthesis of Multisubstituted Pyrroles and Pyridines. Organometallics 2017, 36, 4936–4942. [Google Scholar] [CrossRef]
- Li, X.; Che, X.; Chen, G.-H.; Zhang, J.; Yan, J.-L.; Zhang, Y.-F.; Zhang, L.-S.; Hsu, C.-P.; Gao, Y.Q.; Shi, Z.-J. Direct Oxidation of Aliphatic C–H Bonds in Amino-Containing Molecules under Transition-Metal-Free Conditions. Org. Lett. 2016, 18, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleh, B.; El-Apasery, M.A.; Elnagdi, M.H. Synthesis of New Azolyl Azoles and Azinyl Azoles. J. Heterocycl. Chem. 2005, 42, 483–486. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Y.; Pan, X.; Wang, F.; Huang, S. Oxidation of Tertiary Aromatic Alcohols to Ketones in Water. Adv. Synth. Catal. 2018, 360, 3607–3612. [Google Scholar] [CrossRef]
- Pathoor, R.; Bahulayan, D. MCR-Click Synthesis, Molecular Docking and Cytotoxicity Evaluation of a New Series of Indole–Triazole–Coumarin Hybrid Peptidomimetics. New J. Chem. 2018, 42, 6810–6816. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| # | Acid, g | Temperature, °C | Solvent | Time, h | Yield, a % |
| 1 | PPA 80%, 2 | 90 | 0.5 | 88 | |
| 2 | PPA 80%, 2 | 100 | 0.5 | 90 | |
| 3 | PPA 80%, 2 | 110 | 0.5 | 93 | |
| 4 | PPA 80%, 1 | 110 | 1 | 85 | |
| 5 | PPA 80%, 4 | 110 | 0.5 | 92 | |
| 6 | PPA 87%, 2 | 100 | 1 | 50 | |
| 7 | H3PO4, 1 mL | 100 | 3 | 65 | |
| 8 | H3PO4, 0.5 mL | reflux | EtOH (1 mL) | 3 | 33 |
| 9 | PPA 80%, 2 | reflux | Toluene (1 mL) | 1.5 | 91 |
 | ||||
|---|---|---|---|---|
| # | Acid, g | Temperature, °C | Solvent | Yield a, % |
| 1 | PPA 80%, 2 | 90 | 88 | |
| 2 | PPA 80%, 2 | 100 | 90 | |
| 3 | PPA 80%, 2 | 110 | 93 | |
| 4 | PPA 80%, 1 | 110 | 85 | |
| 5 | PPA 80%, 4 | 110 | 92 | |
| 6 | PPA 87%, 2 | 100 | 50 | |
| 7 | H3PO4, 2 | 100 | 65 | |
| 8 | H3PO4, 0.5 | reflux | EtOH (1 mL) | 33 |
| 9 | PPA 80%, 2 | reflux | Toluene (1 mL) | 91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aksenov, A.V.; Makieva, D.C.; Arestov, R.A.; Arutiunov, N.A.; Aksenov, D.A.; Aksenov, N.A.; Leontiev, A.V.; Aksenova, I.V. Metal-Free, PPA-Mediated Fisher Indole Synthesis via Tandem Hydroamination–Cyclization Reaction between Simple Alkynes and Arylhydrazines. Int. J. Mol. Sci. 2024, 25, 8750. https://doi.org/10.3390/ijms25168750
Aksenov AV, Makieva DC, Arestov RA, Arutiunov NA, Aksenov DA, Aksenov NA, Leontiev AV, Aksenova IV. Metal-Free, PPA-Mediated Fisher Indole Synthesis via Tandem Hydroamination–Cyclization Reaction between Simple Alkynes and Arylhydrazines. International Journal of Molecular Sciences. 2024; 25(16):8750. https://doi.org/10.3390/ijms25168750
Chicago/Turabian StyleAksenov, Alexander V., Dinara C. Makieva, Rodion A. Arestov, Nikolai A. Arutiunov, Dmitrii A. Aksenov, Nicolai A. Aksenov, Alexander V. Leontiev, and Inna V. Aksenova. 2024. "Metal-Free, PPA-Mediated Fisher Indole Synthesis via Tandem Hydroamination–Cyclization Reaction between Simple Alkynes and Arylhydrazines" International Journal of Molecular Sciences 25, no. 16: 8750. https://doi.org/10.3390/ijms25168750