
Primary Progressive Multiple Sclerosis—A Key to Understanding and Managing Disease Progression
 , ,
, , 
Abstract
1. Introduction
2. Method—Literature Search
3. Terms and Definitions
4. Primary Progressive Multiple Sclerosis (PPMS)
4.1. Clinical Characteristics
4.2. Diagnostic Criteria
5. PPMS—Background for Progression
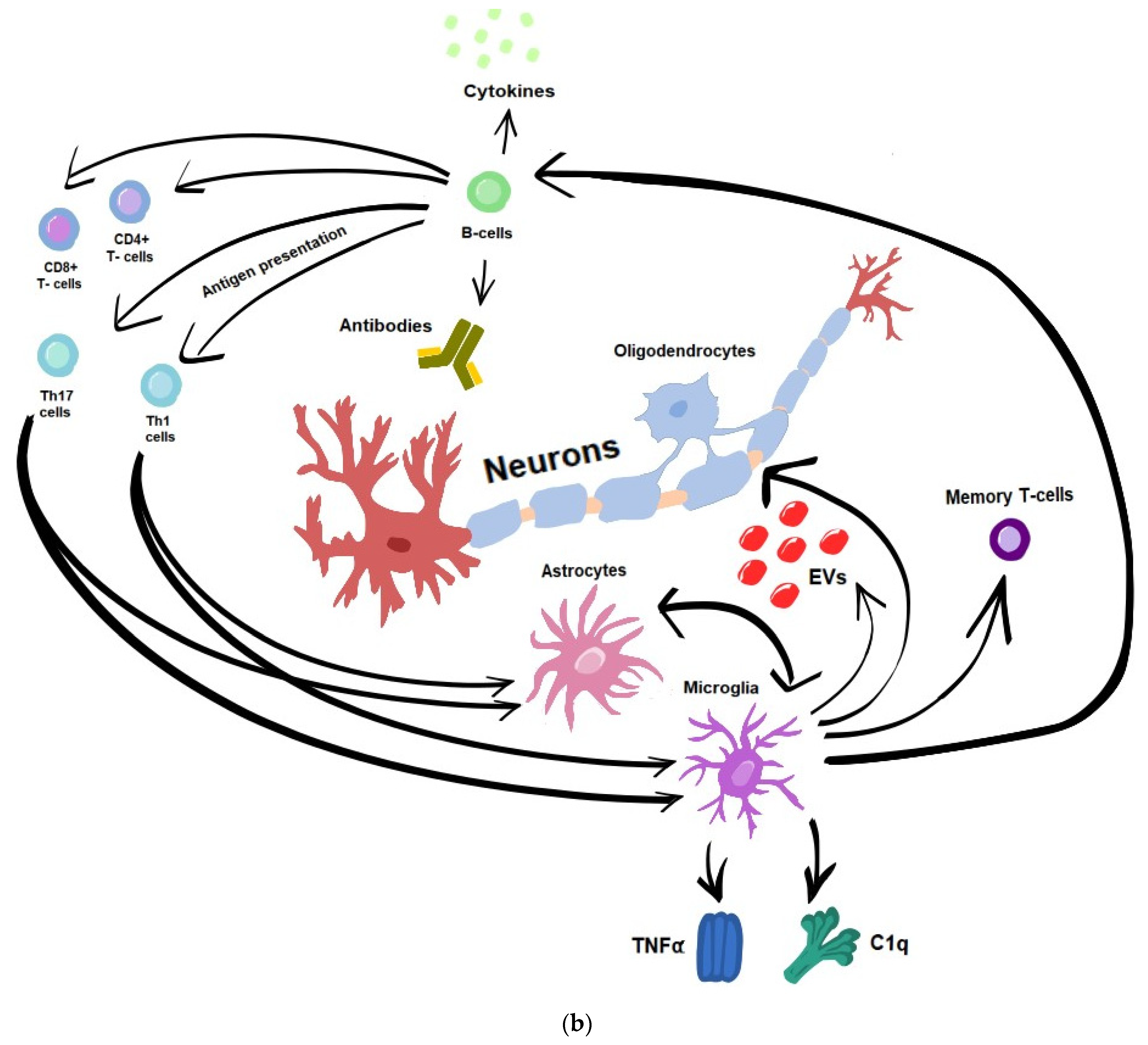
5.1. Immune-Mediated Inflammation and Neurodegeneration
5.1.1. T-Cells
5.1.2. B-Cells
5.1.3. Microglia
5.1.4. Astrocytes
5.2. Remyelination Failure
6. Monitoring and Predicting Progression in PPMS—Potential Biomarkers
6.1. Clinical Biomarkers
6.2. Biochemical Biomarkers
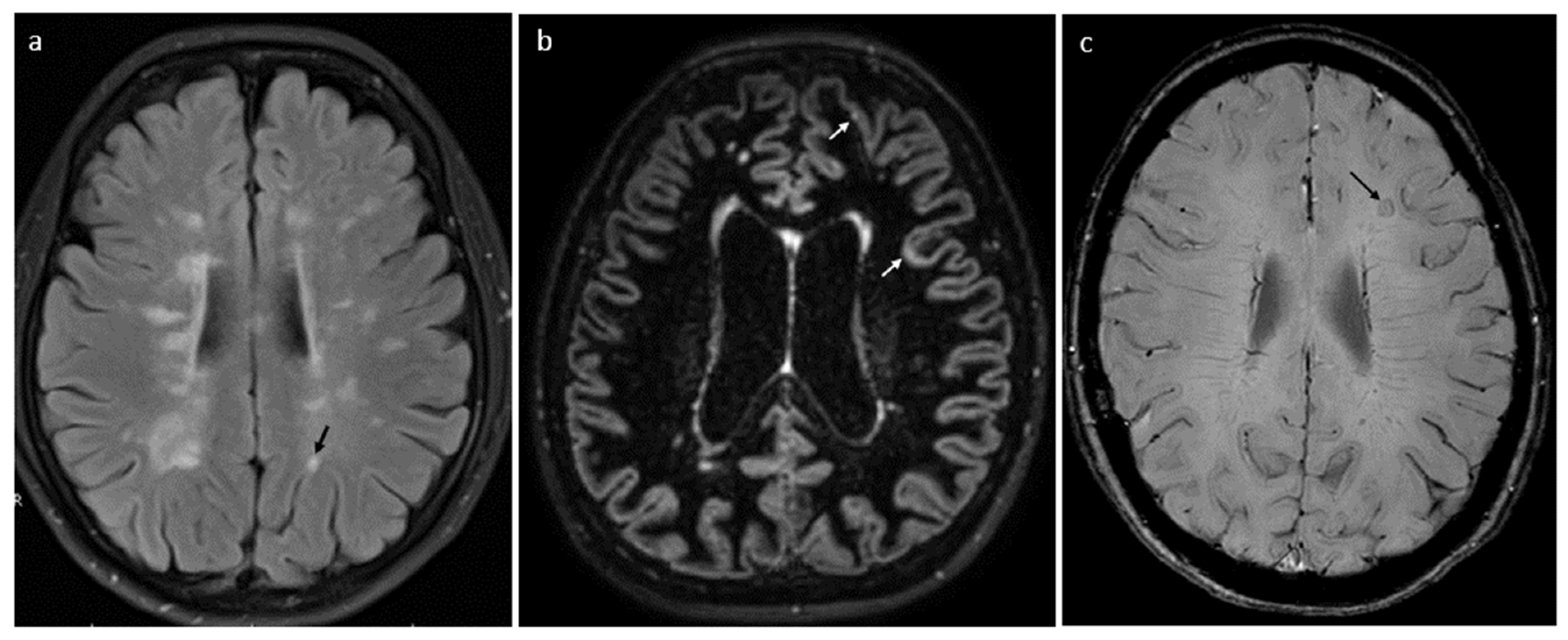
6.3. Radiological Biomarkers
7. PPMS—Therapeutic Options
8. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALCAM | Activated leukocyte cell-adhesion molecule |
| APC | Antigen-presenting cells |
| BICAMS | Brief International Cognitive Assessment for Multiple Sclerosis |
| BBB | Blood–brain barrier |
| BCMA | B-cell maturation antigen |
| BRB-N | Brief Repeatable Battery of Neuropsychological Tests |
| CHI3L1 | chitinase -3-like-1protein |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CVLT2 | California Verbal Learning |
| DMT | Disease-modifying therapies |
| EDSS | Expanded Disability Status Scale |
| EVs | Extracellular vesicles |
| GFAP | Glial fibrillary acidic protein |
| lncRNAs | Long non-coding RNAs |
| miRNAs | microRNAs |
| MS | Multiple sclerosis |
| MSFC | Multiple Sclerosis Functional Composite |
| NEDA | “No Evidence of Disease Activity” |
| NEP | “No Evidence of Progression” |
| NHPT | The nine-hole peg test |
| ncRNAs | Non-coding RNAs |
| OCB | Oligoclonal IgG bands |
| OCGB | Oligoclonal immunoglobulin G bands |
| OL | Oligodendrocytes |
| OS | Oxidative stress |
| PASAT | Paced Auditory Serial Addition Test |
| PIRA | Progression independent of relapses |
| PPMS | Primary progressive multiple sclerosis |
| PRL | Paramagnetic rim lesions |
| PROMs | Patient-reported outcome measures |
| RAW | Relapse-associated worsening |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RPMS | Relapsing–progressive multiple sclerosis |
| RRMS | Relapsing–remitting multiple sclerosis |
| SDMT | Symbol Digit Modality Test |
| SEL | Slowly expanding lesions |
| SPMS | Secondary progressive multiple sclerosis |
| TACI | Transmembrane activator and CAML interactor |
| TAS | Total Antioxidant Status |
References
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giovannoni, G.; Popescu, V.; Wuerfel, J.; Hellwig, K.; Iacobaeus, E.; Jensen, M.B.; García-Domínguez, J.M.; Sousa, L.; De Rossi, N.; Hupperts, R.; et al. Smouldering multiple sclerosis: The ‘real MS’. Ther. Adv. Neurol. Disord. 2022, 15, 17562864211066751. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a02893. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Cagol, A.; Lorscheider, J.; Tsagkas, C.; Benkert, P.; Yaldizli, Ö.; Kuhle, J.; Derfuss, T.; Sormani, M.P.; Thompson, A.; et al. Harmonizing Definitions for Progression Independent of Relapse Activity in Multiple Sclerosis: A Systematic Review. JAMA Neurol. 2023, 80, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kappos, L.; Wolinsky, J.S.; Giovannoni, G.; Arnold, D.L.; Wang, Q.; Bernasconi, C.; Model, F.; Koendgen, H.; Manfrini, M.; Belachew, S.; et al. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials. JAMA Neurol. 2020, 77, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Zanghì, A.; Galgani, S.; Bellantonio, P.; Zaffaroni, M.; Borriello, G.; Inglese, M.; Romano, S.; Conte, A.; Patti, F.; Trojano, M.; et al. Relapse-associated worsening in a real-life multiple sclerosis cohort: The role of age and pyramidal phenotype. Eur. J. Neurol. 2023, 30, 2736–2744. [Google Scholar] [CrossRef] [PubMed]
- Meca-Lallana, V.; Berenguer-Ruiz, L.; Carreres-Polo, J.; Eichau-Madueño, S.; Ferrer-Lozano, J.; Forero, L.; Higueras, Y.; Lara, N.T.; Vidal-Jordana, A.; Pérez-Miralles, F.C. Deciphering Multiple Sclerosis Progression. Front. Neurol. 2021, 12, 608491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rice, C.M.; Cottrell, D.; Wilkins, A.; Scolding, N.J. Primary progressive multiple sclerosis: Progress and challenges. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, D.A.; Kremenchutzky, M.; Rice, G.P.A.; Koopman, W.J.; Hader, W.; Baskerville, J.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study. 5. The clinical features and natural history of primary progressive multiple sclerosis. Brain 1999, 122 Pt 4, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Katsarogiannis, E.; Landtblom, A.-M.; Kristoffersson, A.; Wikström, J.; Semnic, R.; Berntsson, S.G. Absence of Oligoclonal Bands in Multiple Sclerosis: A Call for Differential Diagnosis. J. Clin. Med. 2023, 12, 4656. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; Masterman, T.; Casanova, B.; Gómez-Rial, J.; Espiño, M.; Sádaba, M.C.; González-Porqué, P.; Coret, F.; Álvarez-Cermeño, J.C. CSF oligoclonal band patterns reveal disease heterogeneity in multiple sclerosis. J. Neuroimmunol. 2009, 211, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Konen, F.F.; Hannich, M.J.; Schwenkenbecher, P.; Grothe, M.; Gag, K.; Jendretzky, K.F.; Gingele, S.; Sühs, K.-W.; Witte, T.; Skripuletz, T.; et al. Diagnostic Cerebrospinal Fluid Biomarker in Early and Late Onset Multiple Sclerosis. Biomedicines 2022, 10, 1629. [Google Scholar] [CrossRef]
- Pannewitz-Makaj, K.; Wurster, U.; Jendretzky, K.F.; Gingele, S.; Sühs, K.-W.; Stangel, M.; Skripuletz, T.; Schwenkenbecher, P. Evidence of Oligoclonal Bands Does Not Exclude Non-Inflammatory Neurological Diseases. Diagnostics 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Karaaslan, Z. Hereditary Disorders Mimicking Progressive Multiple Sclerosis. Noro Psikiyatr. Arsivi 2019, 56, 1–2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Omerhoca, S.; Akkas, S.Y.; Icen, N.K. Multiple Sclerosis: Diagnosis and Differential Diagnosis. Noro Psikiyatr. Arsivi 2018, 55 (Suppl. S1), S1–S9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdelhak, A.; Weber, M.S.; Tumani, H. Primary Progressive Multiple Sclerosis: Putting Together the Puzzle. Front. Neurol. 2017, 8, 234. [Google Scholar] [CrossRef]
- Fernandes, M.G.F.; Mohammadnia, A.; Pernin, F.; Schmitz-Gielsdorf, L.E.; Hodgins, C.; Cui, Q.-L.; Yaqubi, M.; Blain, M.; Hall, J.; Dudley, R.; et al. Mechanisms of metabolic stress induced cell death of human oligodendrocytes: Relevance for progressive multiple sclerosis. Acta Neuropathol. Commun. 2023, 11, 108. [Google Scholar] [CrossRef]
- Vasić, M.; Topić, A.; Marković, B.; Milinković, N.; Dinčić, E. Oxidative stress-related risk of the multiple sclerosis development. J. Med. Biochem. 2023, 42, 1–8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Komori, M.; Blake, A.; Greenwood, M.; Lin, Y.C.; Kosa, P.; Ghazali, D.; Winokur, P.; Natrajan, M.; Wuest, S.C.; Romm, E.; et al. Cerebrospinal fluid markers reveal intrathecal inflammation in progressive multiple sclerosis. Ann. Neurol. 2015, 78, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Pukoli, D.; Vécsei, L. Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms. Int. J. Mol. Sci. 2023, 24, 12631. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Activated CD4+ and CD8+ T Cell Proportions in Multiple Sclerosis Patients. Inflammation 2016, 39, 2040–2044. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Kronisch, J.; Khorooshi, R.; Knier, B.; Toft-Hansen, H.; Gudi, V.; Floess, S.; Huehn, J.; Owens, T.; Korn, T.; et al. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflamm. 2017, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef]
- Comi, G.; Bar-Or, A.; Lassmann, H.; Uccelli, A.; Hartung, H.P.; Montalban, X.; Sørensen, P.S.; Hohlfeld, R.; Hauser, S.L. Expert Panel of the 27th Annual Meeting of the European Charcot Foundation. Role of B Cells in Multiple Sclerosis and Related Disorders. Ann. Neurol. 2021, 89, 13–23. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef]
- Thaler, F.S.; A Laurent, S.; Huber, M.; Mulazzani, M.; Dreyling, M.; Ködel, U.; Kümpfel, T.; Straube, A.; Meinl, E.; von Baumgarten, L. Soluble TACI and soluble BCMA as biomarkers in primary central nervous system lymphoma. Neuro Oncol. 2017, 19, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Bankoti, J.; Apeltsin, L.; Hauser, S.L.; Allen, S.; Albertolle, M.E.; Witkowska, H.E.; Von Büdingen, H.-C. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann. Neurol. 2014, 75, 266–276. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130 Pt 4, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Husseini, L.; Geladaris, A.; Weber, M.S. Toward identifying key mechanisms of progression in multiple sclerosis. Trends Neurosci. 2024, 47, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.-P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Healy, L.M.; Stratton, J.A.; Kuhlmann, T.; Antel, J. The role of glial cells in multiple sclerosis disease progression. Nat. Rev. Neurol. 2022, 18, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef]
- Robinson, R.R.; Dietz, A.K.; Maroof, A.M.; Asmis, R.; Forsthuber, T.G. The role of glial-neuronal metabolic cooperation in modulating progression of multiple sclerosis and neuropathic pain. Immunotherapy 2019, 11, 129–147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tobore, T.O. Oxidative/Nitroxidative Stress and Multiple Sclerosis. J. Mol. Neurosci. 2021, 71, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Yang, J.; Zhu, C.; Ding, Y.; Yang, S.; Xu, B.; He, D. Iron metabolism disorder and multiple sclerosis: A comprehensive analysis. Front. Immunol. 2024, 15, 1376838. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Mitochondrial Protein Nitration Primes Neurodegeneration in Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2006, 281, 31950–31962. [Google Scholar] [CrossRef] [PubMed]
- Vidaurre, O.G.; Haines, J.D.; Sand, I.K.; Adula, K.P.; Huynh, J.L.; McGraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain 2014, 137 Pt 8, 2271–2286, Erratum in: Brain 2015, 138 Pt 7, e367. [Google Scholar] [CrossRef] [PubMed]
- Alcázar, A.; Regidor, I.; Masjuan, J.; Salinas, M.; Álvarez-Cermeño, J.C. Induction of apoptosis by cerebrospinal fluid from patients with primary-progressive multiple sclerosis in cultured neurons. Neurosci. Lett. 1998, 255, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Alcázar, A.; Regidor, I.; Masjuan, J.; Salinas, M.; Álvarez-Cermeño, J.C. Axonal damage induced by cerebrospinal fluid from patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2000, 104, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Cid, C.; Alvarez-Cermeño, J.C.; Regidor, I.; Plaza, J.; Salinas, M.; Alcázar, A. Caspase inhibitors protect against neuronal apoptosis induced by cerebrospinal fluid from multiple sclerosis patients. J. Neuroimmunol. 2003, 136, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Lin, J.; Kung, N.J.; Tse, A.L.; E Shimshak, S.J.; Roselle, A.K.; Cali, F.M.; Huang, J.; Beaty, J.M.; Shue, T.M.; et al. Cerebrospinal fluid immunoglobulins in primary progressive multiple sclerosis are pathogenic. Brain 2023, 146, 1979–1992. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kennedy, P.G.E.; George, W.; Yu, X. The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 7584. [Google Scholar] [CrossRef]
- Hollen, C.; Neilson, L.E.; Barajas RFJr Greenhouse, I.; Spain, R.I. Oxidative stress in multiple sclerosis-Emerging imaging techniques. Front. Neurol. 2023, 13, 1025659. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kallaur, A.P.; Reiche, E.M.V.; Oliveira, S.R.; Simão, A.N.C.; Pereira, W.L.C.J.; Alfieri, D.F.; Flauzino, T.; Proença, C.M.; Lozovoy, M.A.B.; Kaimen-Maciel, D.R.; et al. Immune-Inflammatory, and Oxidative Stress Biomarkers as Predictors for Disability and Disease Progression in Multiple Sclerosis. Mol. Neurobiol. 2017, 54, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.K.; Mahns, D.A.; Coorssen, J.R.; Shortland, P.J. The roles of microglia and astrocytes in phagocytosis and myelination: Insights from the cuprizone model of multiple sclerosis. Glia 2022, 70, 1215–1250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salles, D.; Samartini, R.S.; Alves, M.T.S.; Malinverni, A.C.M.; Stávale, J.N. Functions of astrocytes in multiple sclerosis: A review. Mult. Scler. Relat. Disord. 2022, 60, 103749. [Google Scholar] [CrossRef] [PubMed]
- Kerkering, J.; Muinjonov, B.; Rosiewicz, K.S.; Diecke, S.; Biese, C.; Schiweck, J.; Chien, C.; Zocholl, D.; Conrad, T.; Paul, F.; et al. iPSC-derived reactive astrocytes from patients with multiple sclerosis protect cocultured neurons in inflammatory conditions. J. Clin. Investig. 2023, 133, e164637. [Google Scholar] [CrossRef] [PubMed]
- Charabati, M.; Wheeler, M.A.; Weiner, H.L.; Quintana, F.J. Multiple sclerosis: Neuroimmune crosstalk and therapeutic targeting. Cell 2023, 186, 1309–1327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, X.; Jacob, C. Mechanisms of Demyelination and Remyelination Strategies for Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 6373. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Macchi, B.; Marino-Merlo, F.; Nocentini, U.; Pisani, V.; Cuzzocrea, S.; Grelli, S.; Mastino, A. Role of inflammation and apoptosis in multiple sclerosis: Comparative analysis between the periphery and the central nervous system. J. Neuroimmunol. 2015, 287, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Melchor, G.S.; Khan, T.; Reger, J.F.; Huang, J.K. Remyelination Pharmacotherapy Investigations Highlight Diverse Mechanisms Underlying Multiple Sclerosis Progression. ACS Pharmacol. Transl. Sci. 2019, 2, 372–386. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meca-Lallana, J.E.; Casanova, B.; Rodríguez-Antigüedad, A.; Eichau, S.; Izquierdo, G.; Durán, C.; Río, J.; Hernández, M.Á.; Calles, C.; Prieto-González, J.M.; et al. Consensus on early detection of disease progression in patients with multiple sclerosis. Front. Neurol. 2022, 13, 931014. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.W.; Mostert, J.P.; Wolinsky, J.S.; Lublin, F.D.; Uitdehaag, B.; Cutter, G.R. Comparison of the EDSS, Timed 25-Foot Walk, and the 9-Hole Peg Test as Clinical Trial Outcomes in Relapsing-Remitting Multiple Sclerosis. Neurology 2021, 97, e1560–e1570. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Niino, M.; Fukazawa, T.; Kira, J.I.; Okuno, T.; Mori, M.; Sanjo, N.; Ohashi, T.; Fukaura, H.; Fujimori, J.; Shimizu, Y.; et al. Validation of the Brief International Cognitive Assessment for Multiple Sclerosis in Japan. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317748972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meca-Lallana, V.; Gascón-Giménez, F.; Ginestal-López, R.C.; Higueras, Y.; Téllez-Lara, N.; Carreres-Polo, J.; Eichau-Madueño, S.; Romero-Imbroda, J.; Vidal-Jordana, Á.; Pérez-Miralles, F. Cognitive impairment in multiple sclerosis: Diagnosis and monitoring. Neurol. Sci. 2021, 42, 5183–5193. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Comi, G.; Bermel, R.; Bar-Or, A.; McGinley, M.; Arnold, D.; Henry, R.; Benedict, R.; Bhargava, P.; Butzkueven, H.; Chard, D.; et al. A multicenter, open label, single-arm, phase 3b study (CONSONANCE) to assess efficacy of ocrelizumab in patients with primary and secondary progressive multiple sclerosis: Year 1 interim analysis of cognition outcomes. In Proceedings of the AAN Annual Meeting, Seattle, WA, USA, 2–7 April 2022. [Google Scholar]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. ORATORIO Clinical Investigators. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Van Munster, C.E.; Uitdehaag, B.M. Outcome Measures in Clinical Trials for Multiple Sclerosis. CNS Drugs. 2017, 31, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hamade, M.; Wu, Q.; Wang, Q.; Axtell, R.; Giri, S.; Mao-Draayer, Y. Current and Future Biomarkers in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 5877. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barro, C.; Healy, B.C.; Liu, Y.; Saxena, S.; Paul, A.; Polgar-Turcsanyi, M.; Guttmann, C.R.G.; Bakshi, R.; Kropshofer, H.; Weiner, H.L.; et al. Serum GFAP and NfL Levels Differentiate Subsequent Progression and Disease Activity in Patients with Progressive Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 10, e200052. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, M.; Liu, N.; Xie, Q.; Li, X.; Sun, J.; Wang, H.; Wang, M. A candidate biomarker of glial fibrillary acidic protein in CSF and blood in differentiating multiple sclerosis and its subtypes: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2021, 51, 102870. [Google Scholar] [CrossRef] [PubMed]
- Ayrignac, X.; Le Bars, E.; Duflos, C.; Hirtz, C.; Maceski, A.M.; Carra-Dallière, C.; Charif, M.; Pinna, F.; Prin, P.; de Champfleur, N.M.; et al. Serum GFAP in multiple sclerosis: Correlation with disease type and MRI markers of disease severity. Sci. Rep. 2020, 10, 10923. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdelhak, A.; Huss, A.; Kassubek, J.; Tumani, H.; Otto, M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci. Rep. 2018, 8, 14798, Erratum in: Sci Rep. 2019, 9, 8433. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bar-Or, A.; Thanei, G.-A.; Harp, C.; Bernasconi, C.; Bonati, U.; Cross, A.H.; Fischer, S.; Gaetano, L.; Hauser, S.L.; Hendricks, R.; et al. Blood neurofilament light levels predict non-relapsing progression following anti-CD20 therapy in relapsing and primary progressive multiple sclerosis: Findings from the ocrelizumab randomised, double-blind phase 3 clinical trials. EBioMedicine 2023, 93, 104662. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Floro, S.; Carandini, T.; Pietroboni, A.M.; De Riz, M.A.; Scarpini, E.; Galimberti, D. Role of Chitinase 3-like 1 as a Biomarker in Multiple Sclerosis: A Systematic Review and Meta-analysis. Neurol Neuroimmunol. Neuroinflamm. 2022, 9, e1164. [Google Scholar] [CrossRef] [PubMed]
- Talaat, F.; Abdelatty, S.; Ragaie, C.; Dahshan, A. Chitinase-3-like 1-protein in CSF: A novel biomarker for progression in patients with multiple sclerosis. Neurol. Sci. 2023, 44, 3243–3252. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pérez-Miralles, F.; Prefasi, D.; García-Merino, A.; Gascón-Giménez, F.; Medrano, N.; Castillo-Villalba, J.; Cubas, L.; Alcalá, C.; Gil-Perotín, S.; Gómez-Ballesteros, R.; et al. CSF chitinase 3-like-1 association with disability of primary progressive MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e815. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nowak-Kiczmer, M.; Niedziela, N.; Zalejska-Fiolka, J.; Adamczyk-Sowa, M. Evaluation of antioxidant parameters of multiple sclerosis patients’ serum according to the disease course. Mult. Scler. Relat. Disord. 2023, 77, 104875. [Google Scholar] [CrossRef]
- Podbielska, M.; O’Keeffe, J.; Pokryszko-Dragan, A. New Insights into Multiple Sclerosis Mechanisms: Lipids on the Track to Control Inflammation and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 7319. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gutiérrez-Fernández, M.; de la Cuesta, F.; Tallón, A.; Cuesta, I.; Fernández-Fournier, M.; Laso-García, F.; Gómez-de Frutos, M.C.; Díez-Tejedor, E.; Otero-Ortega, L. Potential Roles of Extracellular Vesicles as Biomarkers and a Novel Treatment Approach in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 9011. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Irmer, B.; Chandrabalan, S.; Maas, L.; Bleckmann, A.; Menck, K. Extracellular Vesicles in Liquid Biopsies as Biomarkers for Solid Tumors. Cancers 2023, 15, 1307. [Google Scholar] [CrossRef]
- Prajjwal, P.; Shree, A.; Das, S.; Inban, P.; Ghosh, S.; Senthil, A.; Gurav, J.; Kundu, M.; Marsool, M.D.M.; Gadam, S.; et al. Vascular multiple sclerosis: Addressing the pathogenesis, genetics, pro-angiogenic factors, and vascular abnormalities, along with the role of vascular intervention. Ann. Med. Surg. 2023, 85, 4928–4938. [Google Scholar] [CrossRef] [PubMed]
- Pistono, C.; Osera, C.; Cuccia, M.; Bergamaschi, R. Roles of Extracellular Vesicles in Multiple Sclerosis: From Pathogenesis to Potential Tools as Biomarkers and Therapeutics. Sclerosis 2023, 1, 91–112. [Google Scholar] [CrossRef]
- Kaskow, B.J.; Baecher-Allan, C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a029025. [Google Scholar] [CrossRef] [PubMed]
- Roig-Carles, D.; Willms, E.; Fontijn, R.D.; Martinez-Pacheco, S.; Mäger, I.; de Vries, H.E.; Hirst, M.; Sharrack, B.; Male, D.K.; Hawkes, C.A.; et al. Endothelial-Derived Extracellular Vesicles Induce Cerebrovascular Dysfunction in Inflammation. Pharmaceutics 2021, 13, 1525. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verderio, C.; Muzio, L.; Turola, E.; Bergami, A.; Novellino, L.; Ruffini, F.; Riganti, L.; Corradini, I.; Francolini, M.; Garzetti, L.; et al. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 2012, 72, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Gelibter, S.; Pisa, M.; Croese, T.; Finardi, A.; Mandelli, A.; Sangalli, F.; Colombo, B.; Martinelli, V.; Comi, G.; Filippi, M.; et al. Spinal Fluid Myeloid Microvesicles Predict Disease Course in Multiple Sclerosis. Ann. Neurol. 2021, 90, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Blonda, M.; Amoruso, A.; Grasso, R.; Di Francescantonio, V.; Avolio, C. Multiple Sclerosis Treatments Affect Monocyte-Derived Microvesicle Production. Front. Neurol. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, A.; Qurashi, A. Non-coding RNAs in the Pathogenesis of Multiple Sclerosis. Front. Genet. 2021, 12, 717922. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, N.; Zohar, K.; Linial, M. Challenging Cellular Homeostasis: Spatial and Temporal Regulation of miRNAs. Int. J. Mol. Sci. 2022, 23, 16152. [Google Scholar] [CrossRef]
- Dziadkowiak, E.; Baczyńska, D.; Wieczorek, M.; Olbromski, M.; Moreira, H.; Mrozowska, M.; Budrewicz, S.; Dzięgiel, P.; Barg, E.; Koszewicz, M. miR-31-5p as a Potential Circulating Biomarker and Tracer of Clinical Improvement for Chronic Inflammatory Demyelinating Polyneuropathy. Oxid. Med. Cell Longev. 2023, 2023, 2305163. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jalaiei, A.; Asadi, M.R.; Sabaie, H.; Dehghani, H.; Gharesouran, J.; Hussen, B.M.; Taheri, M.; Ghafouri-Fard, S.; Rezazadeh, M. Long Non-Coding RNAs, Novel Offenders or Guardians in Multiple Sclerosis: A Scoping Review. Front. Immunol. 2021, 12, 774002. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riva, P.; Ratti, A.; Venturin, M. The Long Non-Coding RNAs in Neurodegenerative Diseases: Novel Mechanisms of Pathogenesis. Curr. Alzheimer Res. 2016, 13, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.M.-S.; Gómez, I.; Quiroga-Varela, A.; Río, M.G.-D.; Cedeño, R.R.; Álvarez, G.; Buxó, M.; Miguela, A.; Villar, L.M.; Castillo-Villalba, J.; et al. miRNA Signature in CSF From Patients with Primary Progressive Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 10, e200069. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gupta, M.; Martens, K.; Metz, L.M.; de Koning, A.J.; Pfeffer, G. Long noncoding RNAs associated with phenotypic severity in multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 36, 101407. [Google Scholar] [CrossRef] [PubMed]
- Tiu, V.E.; Enache, I.; Panea, C.A.; Tiu, C.; Popescu, B.O. Predictive MRI Biomarkers in MS-A Critical Review. Medicina 2022, 58, 377. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Calvi, A.; Tur, C.; Chard, D.; Stutters, J.; Ciccarelli, O.; Cortese, R.; Battaglini, M.; Pietroboni, A.; De Riz, M.; Galimberti, D.; et al. Slowly expanding lesions relate to persisting black-holes and clinical outcomes in relapse-onset multiple sclerosis. Neuroimage Clin. 2022, 35, 103048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, D.H.; Lublin, F.D.; Sormani, M.P.; Kappos, L.; Yaldizli, Ö.; Freedman, M.S.; Cree, B.A.C.; Weiner, H.L.; Lubetzki, C.; Hartung, H.; et al. Brain atrophy and disability worsening in primary progressive multiple sclerosis: Insights from the INFORMS study. Ann. Clin. Transl. Neurol. 2018, 5, 346–356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Macaron, G.; Ontaneda, D. Diagnosis and Management of Progressive Multiple Sclerosis. Biomedicines 2019, 7, 56. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Andravizou, A.; Dardiotis, E.; Artemiadis, A.; Sokratous, M.; Siokas, V.; Tsouris, Z.; Aloizou, A.-M.; Nikolaidis, I.; Bakirtzis, C.; Tsivgoulis, G.; et al. Brain atrophy in multiple sclerosis: Mechanisms, clinical relevance and treatment options. Auto. Immun. Highlights 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Filippi, M.; Preziosa, P.; Langdon, D.; Lassmann, H.; Paul, F.; Rovira, À.M.D.; Schoonheim, M.M.; Solari, A.; Stankoff, B.; Rocca, M.A. Identifying Progression in Multiple Sclerosis: New Perspectives. Ann. Neurol. 2020, 88, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Colasanti, A.; Guo, Q.; Muhlert, N.; Giannetti, P.; Onega, M.; Newbould, R.D.; Ciccarelli, O.; Rison, S.; Thomas, C.; Nicholas, R.; et al. In Vivo Assessment of Brain White Matter Inflammation in Multiple Sclerosis with (18)F-PBR111 PET. J. Nucl. Med. 2014, 55, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Casserly, C.; Seyman, E.E.; Alcaide-Leon, P.; Guenette, M.; Lyons, C.; Sankar, S.; Svendrovski, A.; Baral, S.; Oh, J. Spinal Cord Atrophy in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J. Neuroimaging 2018, 28, 556–586. [Google Scholar] [CrossRef] [PubMed]
- Siger, M. Magnetic Resonance Imaging in Primary Progressive Multiple Sclerosis Patients: Review. Clin. Neuroradiol. 2022, 32, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Diao, W.; Tian, F.; Zhang, F.; He, L.; Long, X.; Zhou, F.; Jia, Z. Gray Matter Atrophy in the Cortico-Striatal-Thalamic Network and Sensorimotor Network in Relapsing-Remitting and Primary Progressive Multiple Sclerosis. Neuropsychol. Rev. 2021, 31, 703–720. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Grossman, R.I.; Babb, J.S.; He, J.; Mannon, L.J. Dirty-appearing white matter in multiple sclerosis: Volumetric MR imaging and magnetization transfer ratio histogram analysis. Am. J. Neuroradiol. 2003, 24, 1935–1940. [Google Scholar] [PubMed] [PubMed Central]
- Chisari, C.G.; Bianco, A.; Morra, V.B.; Calabrese, M.; Capone, F.; Cavalla, P.; Chiavazza, C.; Comi, C.; Danni, M.; Filippi, M.; et al. Effectiveness of Ocrelizumab in Primary Progressive Multiple Sclerosis: A Multicenter, Retrospective, Real-world Study (OPPORTUNITY). Neurotherapeutics 2023, 20, 1696–1706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Portaccio, E.; Fonderico, M.; Iaffaldano, P.; Pastò, L.; Razzolini, L.; Bellinvia, A.; De Luca, G.; Ragonese, P.; Patti, F.; Morra, V.B.; et al. Disease-Modifying Treatments and Time to Loss of Ambulatory Function in Patients with Primary Progressive Multiple Sclerosis. JAMA Neurol. 2022, 79, 869–878. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rose, D.R.; Cohen, J.A. Long-term ocrelizumab in progressive multiple sclerosis. Lancet Neurol. 2020, 19, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Buttmann, M.; Meuth, S.G.; Dirks, P.; Rouzic, E.M.-L.; Eggebrecht, J.C.; Hieke-Schulz, S.; Leemhuis, J.; Ziemssen, T. Safety, Adherence and Persistence in a Real-World Cohort of German MS Patients Newly Treated with Ocrelizumab: First Insights from the CONFIDENCE Study. Front. Neurol. 2022, 13, 863105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kappos, L.; Traboulsee, A.; Li, D.K.B.; Bar-Or, A.; Barkhof, F.; Montalban, X.; Leppert, D.; Baldinotti, A.; Schneble, H.-M.; Koendgen, H.; et al. Ocrelizumab exposure in relapsing-remitting multiple sclerosis: 10-year analysis of the phase 2 randomized clinical trial and its extension. J. Neurol. 2024, 271, 642–657. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weber, M.S.; Kappos, L.; Hauser, S.L.; Nicholas, J.A.; Schneble, H.M.; Wang, Q.; Giovannoni, G.; Filippi, M. The Patient Impact of 10 Years Ocrelizumab Treatment in Multiple Sclerosis: Long Term Data from Phase III OPERA and ORATORIO Studies—ECTRIMS 2023. In Proceedings of the 9th Joint ECTRIMS-ACTRIMS Meeting, Milan, Italy, 11–13 October 2023. [Google Scholar]
- Kremer, D.; Akkermann, R.; Küry, P.; Dutta, R. Current advancements in promoting remyelination in multiple sclerosis. Mult. Scler. 2019, 25, 7–14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amin, M.; Hersh, C.M. Updates and advances in multiple sclerosis neurotherapeutics. Neurodegener. Dis. Manag. 2023, 13, 47–70. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Allanach, J.R.; Farrell, J.W., 3rd; Mésidor, M.; Karimi-Abdolrezaee, S. Current status of neuroprotective and neuroregenerative strategies in multiple sclerosis: A systematic review. Mult. Scler. 2022, 28, 29–48. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krämer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martin, E.; Aigrot, M.S.; Grenningloh, R.; Stankoff, B.; Lubetzki, C.; Boschert, U.; Zalc, B. Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair. Brain Plast. 2020, 5, 123–133. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Saberi, D.; Geladaris, A.; Dybowski, S.; Weber, M.S. Bruton’s tyrosine kinase as a promising therapeutic target for multiple sclerosis. Expert Opin. Ther. Targets 2023, 27, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Geladaris, A.; Torke, S.; Weber, M.S. Bruton’s Tyrosine Kinase Inhibitors in Multiple Sclerosis: Pioneering the Path Towards Treatment of Progression? CNS Drugs 2022, 36, 1019–1030. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goodman, A.D.; Fedler, J.K.; Yankey, J.; Klingner, E.A.; Ecklund, D.J.; Goebel, C.V.; Bermel, R.A.; Chase, M.; Coffey, C.S.; Klawiter, E.C.; et al. Response to ibudilast treatment according to progressive multiple sclerosis disease phenotype. Ann. Clin. Transl. Neurol. 2021, 8, 111–118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ehrhardt, H.; Lambe, J.; Moussa, H.; Vasileiou, E.S.; Kalaitzidis, G.; Murphy, O.C.; Filippatou, A.G.; Pellegrini, N.; Douglas, M.; Davis, S.; et al. Effects of Ibudilast on Retinal Atrophy in Progressive Multiple Sclerosis Subtypes: Post Hoc Analyses of the SPRINT-MS Trial. Neurology 2023, 101, e1014–e1024. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakamura, K.; Thoomukuntla, B.; Bena, J.; Cohen, J.A.; Fox, R.J.; Ontaneda, D. Ibudilast reduces slowly enlarging lesions in progressive multiple sclerosis. Mult. Scler. 2024, 30, 369–380. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Espiritu, A.I.; Remalante-Rayco, P.P.M. High-dose biotin for multiple sclerosis: A systematic review and meta-analyses of randomized controlled trials. Mult. Scler. Relat. Disord. 2021, 55, 103159. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Fulton, D.; Douglas, M.R. Opicinumab: Is it a potential treatment for multiple sclerosis? Ann. Transl. Med. 2020, 8, 892. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Mellion, M.; Hupperts, R.; Edwards, K.R.; A Calabresi, P.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.L.; Fisher, E.; Rudick, R.; et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Gharagozloo, M.; Bannon, R.; Calabresi, P.A. Breaking the barriers to remyelination in multiple sclerosis. Curr. Opin. Pharmacol. 2022, 63, 102194. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, L.; Fung, E.; Bose, S.; Popp, A.; Böser, P.; Memmott, J.; Kutskova, Y.A.; Miller, R.; Tarcsa, E.; Klein, C.; et al. Elezanumab, a clinical stage human monoclonal antibody that selectively targets repulsive guidance molecule A to promote neuroregeneration and neuroprotection in neuronal injury and demyelination models. Neurobiol. Dis. 2021, 159, 105492. [Google Scholar] [CrossRef] [PubMed]
- Havla, J.; Hohlfeld, R. Antibody Therapies for Progressive Multiple Sclerosis and for Promoting Repair. Neurotherapeutics 2022, 19, 774–784. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Antel, J.P.; Kennedy, T.E.; Kuhlmann, T. Seeking neuroprotection in multiple sclerosis: An ongoing challenge. J. Clin. Investig. 2023, 133, e168595. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Alam, S.S.; Kundu, S.; Ahmed, S.; Sultana, S.; Patar, A.; Hossan, T. Mesenchymal Stem Cell Therapy in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J. Clin. Med. 2023, 12, 6311. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gavasso, S.; Kråkenes, T.; Olsen, H.; Evjenth, E.C.; Ytterdal, M.; Haugsøen, J.B.; Kvistad, C.E. The Therapeutic Mechanisms of Mesenchymal Stem Cells in MS-A Review Focusing on Neuroprotective Properties. Int. J. Mol. Sci. 2024, 25, 1365. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S.; Arnold, D.L.; Brochet, B.; Hartung, H.P.; Montalban, X.; Naismith, R.T.; Manfrini, M.; Overell, J.; Koendgen, H.; Sauter, A.; et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: A post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2020, 19, 998–1009. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarkers | Relevance | References | ||
|---|---|---|---|---|
| Clinical (repeated assessment indicates worsening in particular aspects of disability) | Functional scales | EDSS | overall degree of disability (including several functional systems) | [63] |
| T25FW | mobility of gait | [63] | ||
| NHPT | manual dexterity | [63] | ||
| Neuropsychological tests | CVLT2 | verbal memory | [64] | |
| SDMT | attention and executive functions | [64,65] | ||
| PASAT | computing capacity, speed and flexibility of information processing | [65] | ||
| BICAMS BRB-NT | cognitive performance in multiple domains | [65] | ||
| PROMs | PROMIS 29 questionnaire, MS Quality of Life–54-MSQOL | patients’ perspective of their functioning in different spheres of life | [65] | |
| Biochemical–body fluid (serum, blood cells, CSF) | NfL | indicates neuronal/axonal injury | [69,70,71] | |
| GFAP | reflects astrocyte injury and reactive astrogliosis | [73,74] | ||
| CHI3L1 | reflects chronic inflammatory activity | [75,76,77,78] | ||
| Indices of oxidative stress | 8-iso-prostaglandin | predominance of pro-oxidative over anti-oxidative mechanisms | [37,52] | |
| platelet hemostatic function | [37,52] | |||
| advanced oxidation protein products and nitric oxide metabolites | [37,52] | |||
| Sphingolipids | ceramide derivatives | indices for microglia activation, signaling pathways linking inflammation with neurodegeneration | [79] | |
| EVs | markers of microglia and other CNS-resident cell activity (mediators propagating inflammation and preventing remyelination) | [83] | ||
| ncRNA | regulation of gene expression relevant for chronic inflammation and neuronal injury | [89,90,91,92,93,94,95] | ||
| Radiological (MRI) | SEL | constant and concentric volumetric expansion in T2, with concurrent reduction in T1-weighted sequence, revealed in subsequent 2–3 MRI scans | specific demyelinative lesions–sites of chronic inflammation | [96,97,98,99,100] |
| PRL | typical rim surrounding at least 75% of lesion, which reflects the layer of iron-laden microglia and macro-phages, accompanying demyelination and axonal transection (phase imaging, SWI or multi-gradient echo sequences) | specific demyelinative lesions—sites of chronic inflammation | [96,97,98,99,100] | |
| cortical lesions | visualization of cortical damage and ribbon-like subpial demyelination (ultra-high magnetic field resolution (>7 T) | chronic neuroinflammation | [96,102,103] | |
| measures of global and regional CNS atrophy | loss of cerebral structure volume | diffuse neurodegeneration with axonal loss | [100,101,102,103,104,105] | |
| “dirty-appearing white matter” | ill-defined hyperintense areas, located mainly around the lateral ventricles | diffuse neuronal damage with chronic inflammation | [104,106] | |
| Clinical Study | Description | References |
|---|---|---|
| 2012–2016 | The objective of this clinical trial was to evaluate the safety of a single intravenous infusion of autologous bone marrow-derived mesenchymal stem cells (MSCs) in multiple sclerosis (MS) with progressive disease. | ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2013–2017 | This study reported that the treatment effect of ibudilast, in comparison with placebo, was associated with slower rate of brain atrophy in patients with PPMS. | [120] ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2014–2019 | The hypothesis of this project was based on the results of animal model studies in which quetiapine fumarate was shown to have remyelinating and neuroprotective effects in models of inflammatory and non-inflammatory demyelination. | [130,131] ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2016–2016 | This study confirms the efficacy of opicinumab with regard to clinical outcomes; thus, the remyelination potential of the drug (as well as its putative impact on progressive MS) remains unclear. | [115,124,125] ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2017–2026 | The aim of this study is to assess the safety and efficacy of Glatiramer Acetate (GA) Depot in slowing the progression of disability in patients with PPMS. | ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2020–2025 | The efficacy of SAR442168 (Tolebrutinib) compared to placebo in delaying disability progression in PPMS. SAR442168, a CNS-penetrant Bruton’s tyrosine kinase (BTK) inhibitor, has the potential for a dual mechanism of action: modulation and consequent inhibition of antigen-induced B-cell activation responsible for inflammation, and modulation of macrophages and dysfunctional microglial cells linked to neuroinflammation in the brain and spinal cord. | ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2021–2025 | An assessment of the safety of intrathecal administration of DUOC-01 cells to adults with PPMS. DUOC-01 is a population of cells isolated from donated human umbilical cord blood mononuclear cells. DUOC-01 cells are derived from CB CD14+ monocytes. | ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2022–2029 | Observation of patients who completed the CONSONANCE trial after a 4-year study period to assess the effect of ocrelizumab on disability such as the need to use an assistive device or a wheelchair. | ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2022–2025 | This study evaluates the efficacy and safety of oral masitinib, which acts as a selective tyrosine kinase inhibitor targeting immune cells (mast cells and microglia). The study involves the treatment of patients with primary progressive or secondary progressive forms of multiple sclerosis without relapsing–remitting disease. | ClinicalTrials.gov (accessed 3 August 2024) |
| ||
| 2024–2026 | Remyelination is a promising therapeutic strategy in PPMS—remote ischemic conditioning (RIC) at a dose of four cycles per day prevents gait deterioration in patients with PPMS. | ClinicalTrials.gov (accessed 3 August 2024) |
|
| Milestone | Description | References |
|---|---|---|
| 1996—standardized definition of PPMS | Definition of clinical course of MS, based on an international survey (USA National Multiple Sclerosis Society—NMSS). PPMS—separate subtype of disease with continuous progression from onset of clinical manifestation | [9,10,133] |
| 2013—modified definitions of the clinical course in MS | PPMS treatment remains a separate clinical course because of the absence of exacerbations prior to clinical progression; PPMS can be additionally modified by the temporary presence of activity or progression. | [5] |
| 2017—recent revision of McDonald diagnostic criteria | The recognition of PPMS requires the confirmation of continued clinical progression, independent of relapse activity, observed retrospectively or prospectively for at least one year, with concomitance of at least two of the following categories: ≥one lesion detected in MRI of the brain (in typical regions), ≥two lesions detected in MRI of the spinal cord, the presence of OCB in CSF. | [16] |
| 2017–18—the approval of ocrelizumab (OCR) as the first disease-modifying therapy for PPMS | OCR-humanized monoclonal antibody targeted at CD20 antigen on B-cells. Beneficial effects of OCR on clinical and radiological measures of progression and activity in PPMS confirmed by ORATORIO clinical trial. Approved by FDA (2017) and EMA (2018) for use in the active type of PPMS. | [66,67,134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sempik, I.; Dziadkowiak, E.; Moreira, H.; Zimny, A.; Pokryszko-Dragan, A. Primary Progressive Multiple Sclerosis—A Key to Understanding and Managing Disease Progression. Int. J. Mol. Sci. 2024, 25, 8751. https://doi.org/10.3390/ijms25168751
Sempik I, Dziadkowiak E, Moreira H, Zimny A, Pokryszko-Dragan A. Primary Progressive Multiple Sclerosis—A Key to Understanding and Managing Disease Progression. International Journal of Molecular Sciences. 2024; 25(16):8751. https://doi.org/10.3390/ijms25168751
Chicago/Turabian StyleSempik, Izabela, Edyta Dziadkowiak, Helena Moreira, Anna Zimny, and Anna Pokryszko-Dragan. 2024. "Primary Progressive Multiple Sclerosis—A Key to Understanding and Managing Disease Progression" International Journal of Molecular Sciences 25, no. 16: 8751. https://doi.org/10.3390/ijms25168751
APA StyleSempik, I., Dziadkowiak, E., Moreira, H., Zimny, A., & Pokryszko-Dragan, A. (2024). Primary Progressive Multiple Sclerosis—A Key to Understanding and Managing Disease Progression. International Journal of Molecular Sciences, 25(16), 8751. https://doi.org/10.3390/ijms25168751