
Assessment of Dried Serum Spots (DSS) and Volumetric-Absorptive Microsampling (VAMS) Techniques in Therapeutic Drug Monitoring of (Val)Ganciclovir—Comparative Study in Analytical and Clinical Practice

 ,
, 
Abstract
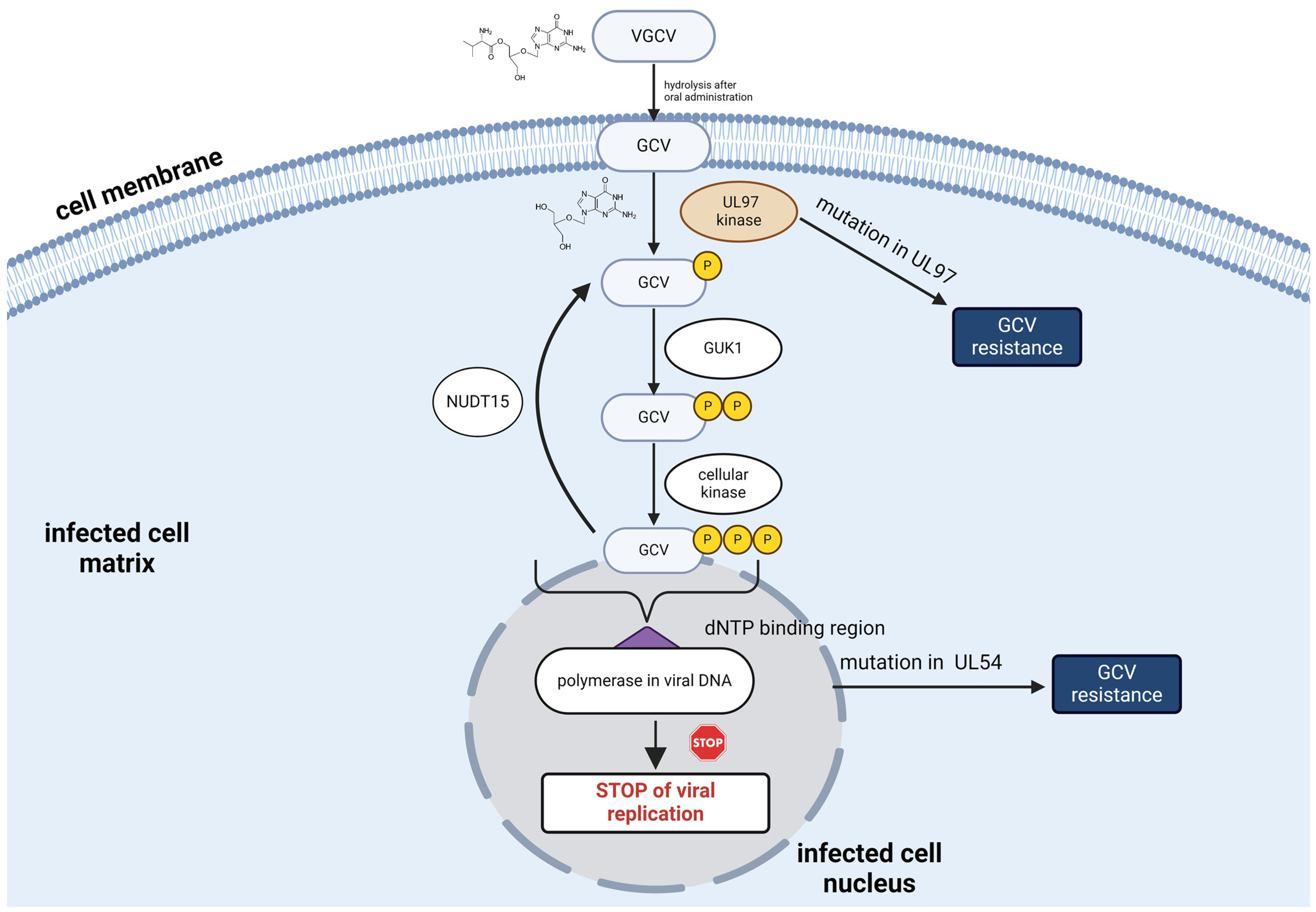
1. Introduction
2. Results
2.1. Method Development and Optimisation
2.2. Analytical Validation
- (1)
- serum-LC-MS/MS method:
- (a)
- IS: GCV-d5 − y = 0.032x + 0.004 (R2 = 0.993 ± 0.005),
- (b)
- IS: ACV − y = 0.024x + 0.001 (R2 = 0.995 ± 0.003).
- (2)
- DSS-LC-MS/MS method (ISACV):
- (3)
- s-VAMS-LC-MS/MS method (ISACV):
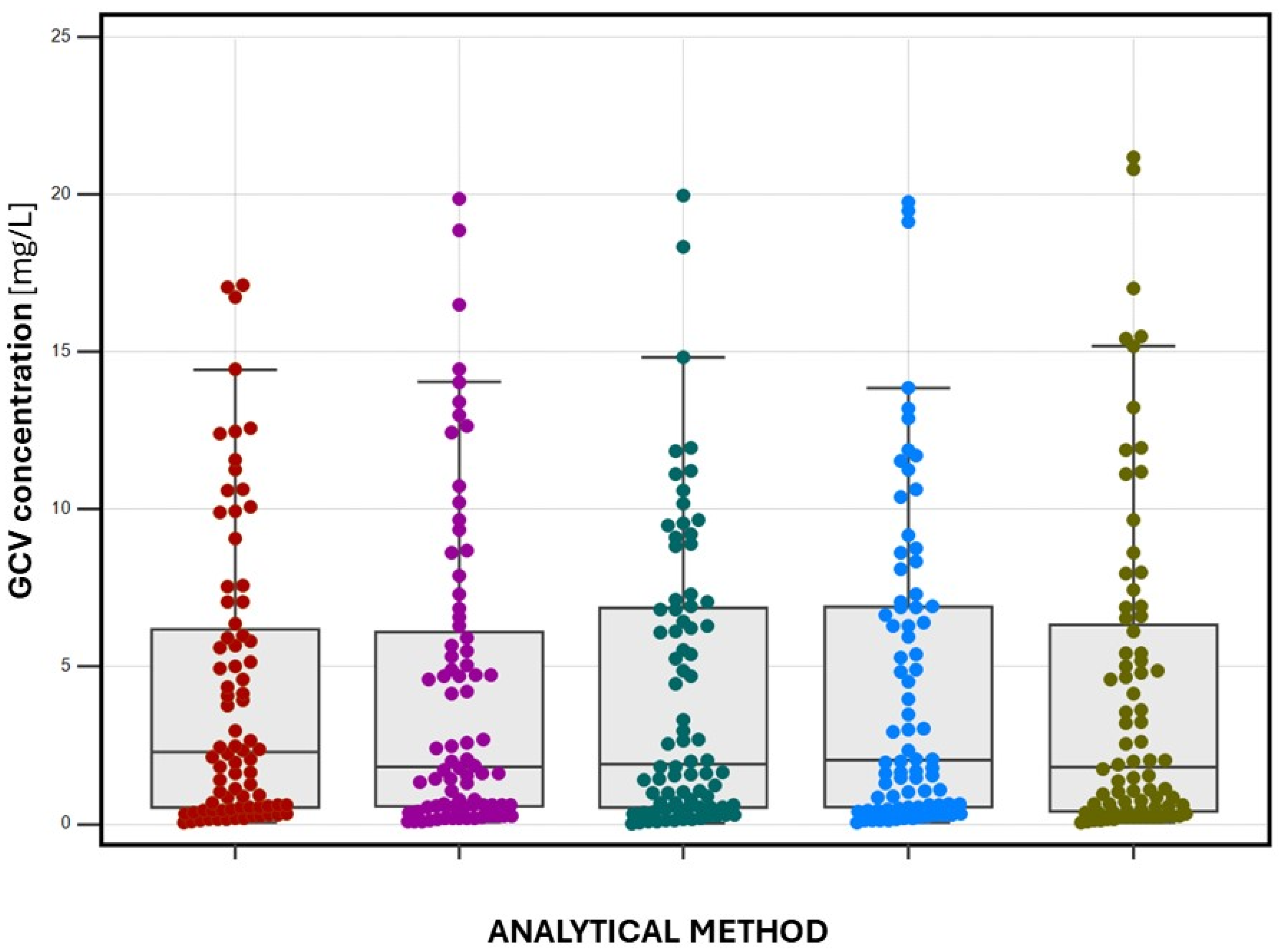
2.3. Patient Sample Determination
- (1)
- serum-LC-MS/MS (GCV-d5 as IS)—4.164 (0.054–19.854) mg/L,
- (2)
- serum-LC-MS/MS (ACV as IS)—4.185 (0.039–19.104) mg/L,
- (3)
- DSS-LC-MS/MS (ACV as IS)—4.167 (0.014–19.960) mg/L,
- (4)
- s-VAMS-LC-MS/MS (ACV as IS)—4.402 (0.039–19.760) mg/L,
- (5)
- HPLC/UV (ACV as IS)—4.003 (0.100–21.180) mg/L.
- (1)
- serum-LC-MS/MS (GCV-d5 as IS)—40% in the C0 range; 57.5% in the Cmax range,
- (2)
- serum-LC-MS/MS (ACV as IS)—42.5% in the C0 range, 65% in the Cmax range,
- (3)
- DSS-LC-MS/MS (ACV as IS)—42.5% in the C0 range, 70% in the Cmax range,
- (4)
- s-VAMS-LC-MS/MS (ACV as IS)—45% in the C0 range, 75% in the Cmax range,
- (5)
- HPLC/UV (ACV as IS)—45% in the C0 range, 77.5% in the Cmax range.
2.4. Cross-Validation and Methods Comparison
3. Discussion
4. Materials and Methods
4.1. Chemicals, Materials and Reagents
4.2. Stock Solutions
4.3. LC-MS/MS Analyses
- (1)
- 0.01–0.30 min 5% of B phase,
- (2)
- 0.31–1.20 min 75% of B phase,
- (3)
- 1.21–2.00 min 5% of B phase (re-equilibration).
4.4. Sampling Protocol
4.5. Sample Preparation Protocols
4.5.1. Plasma Sample Preparation
4.5.2. Dried Serum Spot (DSS) Sample Preparation
4.5.3. Dried Serum s-VAMS Sample Preparation
4.6. Method Validation
4.7. Stability Testing
- Autosampler stability (analysed at 24 h, 72 h, and 120 h after preparation),
- Working solution stability (analysed after one, three, and six months of storage at −20 °C),
- Freeze-thaw stability in the matrix (minimum of three freeze-thaw cycles, performed after a minimum of 12-h storage in the freezer),
- Bench-top (short-term) stability in the matrix (stored at ambient temperature, before and after extraction for 2 h and 4 h compared to a standard set of samples),
- Long-term stability in matrix during storage at room temperature (analysed after one, two, four, five, and seven weeks counted from the initial assay).
4.8. HPLC/UV Method for GCV Determination
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Märtson, A.G.; Edwina, A.E.; Kim, H.Y.; Knoester, M.; Touw, D.J.; Sturkenboom, M.G.G.; Alffenaar, J.C. Therapeutic Drug Monitoring of Ganciclovir: Where Are We. Ther. Drug Monit. 2022, 44, 138–147. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Cymevene: Summary of Product Characteristics, Labelling and Package Leaflet. 2023. Available online: https://www.ema.europa.eu/en/documents/referral/cymevene-article-30-referral-annex-iii_en.pdf (accessed on 7 August 2024).
- Electronic Medicines Compendium. Valcyte: Summary of product Characteristics, Labelling and Package Leaflet. 2023. Available online: https://www.medicines.org.uk/emc/product/14225/smpc (accessed on 7 August 2024).
- Gatti, M.; Rinaldi, M.; Potena, L.; Salvaterra, E.; Morelli, M.C.; Giannella, M.; Viale, P.; Pea, F. Does therapeutic drug monitoring (TDM) of trough concentrations suffice for optimizing preemptive therapy with ganciclovir of cytomegalovirus infections in non-renal solid organ transplant recipients. Transpl. Infect. Dis. 2023, 25, e14107. [Google Scholar] [CrossRef] [PubMed]
- Märtson, A.G.; Edwina, A.E.; Burgerhof, J.G.M.; Berger, S.P.; de Joode, A.; Damman, K.; Verschuuren, E.A.M.; Blokzijl, H.; Bakker, M.; Span, L.F.; et al. Ganciclovir therapeutic drug monitoring in transplant recipients. J. Antimicrob. Chemother. 2021, 76, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Franck, B.; Autmizguine, J.; Marquet, P.; Ovetchkine, P.; Woillard, J.B. Pharmacokinetics, Pharmacodynamics, and Therapeutic Drug Monitoring of Valganciclovir and Ganciclovir in Transplantation. Clin. Pharmacol. Ther. 2022, 112, 233–276. [Google Scholar] [CrossRef]
- Li, Q.Y.; van den Anker, J.; Wu, Y.E.; Hao, G.X.; Zhao, W. Optimizing ganciclovir and valganciclovir dosing regimens in pediatric patients with cytomegalovirus infection: A spotlight on therapeutic drug monitoring. Expert Rev. Clin. Pharmacol. 2023, 16, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.D.; Ho, S.A.; Domazetovska, A.; Yong, M.K.; Rawlinson, W.D. Evidence supporting the use of therapeutic drug monitoring of ganciclovir in transplantation. Curr. Opin. Infect. Dis. 2023, 36, 505–513. [Google Scholar] [CrossRef] [PubMed]
- The Microsampling Blog. Neoteryx. How Stable Are Analytes in Dried Blood? 2023. Available online: https://www.neoteryx.com/microsampling-blog/how-stable-are-analytes-in-dried-blood (accessed on 7 August 2024).
- Capiau, S.; Veenhof, H.; Koster, R.A.; Bergqvist, Y.; Boettcher, M.; Halmingh, O.; Keevil, B.G.; Koch, B.; Linden, R.; Pistos, C.; et al. Official International Association for Therapeutic Drug Monitoring and Clinical Toxicology Guideline: Development and Validation of Dried Blood Spot-Based Methods for Therapeutic Drug Monitoring. Ther. Drug Monit. 2019, 41, 409–430. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Bioanalytical Method Validation. 2023. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (accessed on 7 August 2024).
- Food and Drug Administration Guidance for Industry: Bioanalytical Method Validation. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 7 August 2024).
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical method validation: New FDA guidance vs. EMA guideline. Better or worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Mansournia, M.A.; Waters, R.; Nazemipour, M.; Bland, M.; Altman, D.G. Bland-Altman methods for comparing methods of measurement and response to criticisms. Glob. Epidemiol. 2021, 3, 100045. [Google Scholar] [CrossRef] [PubMed]
- Pum, J. A practical guide to validation and verification of analytical methods in the clinical laboratory. Adv. Clin. Chem. 2019, 90, 215–281. [Google Scholar] [CrossRef] [PubMed]
- Pigliasco, F.; Cafaro, A.; Simeoli, R.; Barco, S.; Magnasco, A.; Faraci, M.; Tripodi, G.; Goffredo, B.M.; Cangemi, G. A UHPLC-MS/MS Method for Therapeutic Drug Monitoring of Aciclovir and Ganciclovir in Plasma and Dried Plasma Spots. Biomedicines 2021, 9, 1379. [Google Scholar] [CrossRef] [PubMed]
- Franzin, M.; Ruoso, R.; Del Savio, R.; Addobbati, R. Development and Validation of an HPLC-UV Method for the Quantification of Acyclovir and Ganciclovir in the Plasma of Pediatric Immunocompromised Patients. Int. J. Mol. Sci. 2024, 25, 2685. [Google Scholar] [CrossRef] [PubMed]
- Ärlemalm, A.; Helldén, A.; Karlsson, L.; Carlsson, B. Rapid determination of acyclovir, its main metabolite 9-carboxymethoxymethylguanine, ganciclovir, and penciclovir in human serum using LC-MS/MS. Biomed. Chromatogr. 2022, 36, e5315. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.R.; Li, X.N.; Chen, W.L.; Liu, G.Y.; Chu, N.N.; Yu, C. A sensitive assay for simultaneous determination of plasma concentrations of valganciclovir and its active metabolite ganciclovir by LC/MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 848, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.; Saxena, S.; Mishra, S.; Khuroo, A.; Monif, T. Determination of valganciclovir and ganciclovir in human plasma by liquid chromatography tandem mass spectrometric detection. Clin. Biochem. 2011, 44, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Märtson, A.G.; van Hateren, K.; van den Bosch, G.; van der Werf, T.; Touw, D.; Alffenaar, J.W. Determination of Ganciclovir and Acyclovir in Human Serum using Liquid Chromatography-Tandem Mass Spectrometry. J. Appl. Bioanal. 2018, 4, 175–186. [Google Scholar] [CrossRef]
- Asberg, A.; Hansen, C.N.; Reubsaet, L. Determination of ganciclovir in different matrices from solid organ transplanted patients treated with a wide range of concomitant drugs. J. Pharm. Biomed. Anal. 2007, 43, 1039–1044. [Google Scholar] [CrossRef]
- Peck Palmer, O.M.; Dasgupta, A. Review of the PreanalyticalPreanalytical Errors That Impact Therapeutic Drug Monitoring. Ther. Drug Monit. 2021, 43, 595–608. [Google Scholar] [CrossRef]
- Avataneo, V.; D’Avolio, A.; Cusato, J.; Cantù, M.; De Nicolò, A. LC-MS application for therapeutic drug monitoring in alternative matrices. J. Pharm. Biomed. Anal. 2019, 166, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, M.U.; Wouters, B.; Kindt, A.; Reiss, I.K.M.; Hankemeier, T. Blood microsampling technologies: Innovations and applications in 2022. Anal. Sci. Adv. 2023, 4, 154–180. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Parameter | LLOQ—0.10 mg/L | MQC1—0.75 mg/L | MQC2—7.50 mg/L | HQC—20.00 mg/L | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serum IS: GCV-d5 | Serum IS: ACV | DSS IS: ACV | s-VAMS IS: ACV | Serum IS: GCV-d5 | Serum IS: ACV | DSS IS: ACV | s-VAMS IS: ACV | Serum IS: GCV-d5 | Serum IS: ACV | DSS IS: ACV | s-VAMS IS: ACV | Serum IS: GCV-d5 | Serum IS: ACV | DSS IS: ACV | s-VAMS IS: ACV | |
| Within-run (intra-day) accuracy and precision [n = 10] | ||||||||||||||||
| CGCV [mg/L] | 0.11 ± 0.02 | 0.10 ± 0.01 | 0.12 ± 0.02 | 0.52 ± 0.04 | 0.77 ± 0.05 | 0.76 ± 0.03 | 0.72 ± 0.04 | 0.76 ± 0.03 | 7.13 ± 0.46 | 7.45 ± 0.14 | 7.38 ± 0.19 | 7.50 ± 0.24 | 20.20 ± 0.82 | 19.99 ± 0.10 | 20.17 ± 0.49 | 20.12 ± 0.27 |
| Accuracy [%] | 107.76 | 104.41 | 103.61 | 103.96 | 101.50 | 105.23 | 96.66 | 103.12 | 100.59 | 97.83 | 98.46 | 99.86 | 99.80 | 99.15 | 99.74 | 100.18 |
| Precision [%] | 16.26 | 10.77 | 17.87 | 14.33 | 5.92 | 3.99 | 5.58 | 4.18 | 5.49 | 2.41 | 2.60 | 3.24 | 4.08 | 0.51 | 2.42 | 1.33 |
| Between-run (inter-day) accuracy and precision [n = 10] | ||||||||||||||||
| CGCV [mg/L] | 0.11 ± 0.01 | 0.10 ± 0.01 | 0.12 ± 0.02 | 0.11 ± 0.02 | 0.75 ± 0.04 | 0.77 ± 0.05 | 0.77 ± 0.05 | 0.77 ± 0.05 | 7.67 ± 0.24 | 7.63 ± 0.18 | 7.49 ± 0.14 | 7.46 ± 0.26 | 20.67 ± 0.41 | 20.13 ± 0.24 | 20.22 ± 0.19 | 20.18 ± 0.20 |
| Accuracy [%] | 102.29 | 104.41 | 103.61 | 105.59 | 99.19 | 101.50 | 101.50 | 101.50 | 100.20 | 100.24 | 100.01 | 99.48 | 100.55 | 99.82 | 100.00 | 100.46 |
| Precision [%] | 11.80 | 10.77 | 17.87 | 13.64 | 5.33 | 5.92 | 5.92 | 5.92 | 3.08 | 2.36 | 1.92 | 3.51 | 1.98 | 1.21 | 0.92 | 0.99 |
| GCV Concentration [mg/L] | Calculated Concentration [mg/L] and Stability at 4 °C [%] | |||
|---|---|---|---|---|
| Initial Day (T = 0) | After 24 h | After 72 h | After 120 h | |
| IS: GCV-d5 [n = 6]—LC-MS/MS method in serum | ||||
| LLOQ (0.10) | 0.12 ± 0.04; 100.00 | 0.11 ± 0.05; 93.67 | 0.12 ± 0.06; 95.96 | 0.10 ± 0.08; 91.33 |
| MQC1 (0.75) | 0.76 ± 0.08; 100.00 | 0.74 ± 0.05; 97.86 | 0.74 ± 0.07; 97.96 | 0.72 ± 0.12; 93.38 |
| MQC2 (7.50) | 7.54 ± 0.23; 100.00 | 7.55 ± 0.20; 100.09 | 7.49 ± 0.32; 97.87 | 7.48 ± 0.29; 96.94 |
| HQC (20.00) | 20.09 ± 1.01; 100.00 | 20.13 ± 1.21; 101.99 | 20.06 ± 1.19; 98.90 | 19.37 ± 1.65; 94.86 |
| IS: ACV [n = 6]—LC-MS/MS method in serum | ||||
| LLOQ (0.10) | 0.11 ± 0.03; 100.00 | 0.09 ± 0.04; 89.81 | 0.09 ± 0.03; 90.11 | 0.09 ± 0.05; 89.87 |
| MQC1 (0.75) | 0.76 ± 0.11; 100.00 | 0.72 ± 0.13; 94.76 | 0.73 ± 0.14; 94.23 | 0.74 ± 0.19; 95.02 |
| MQC2 (7.50) | 7.52 ± 0.19; 100.00 | 7.54 ± 0.23; 101.57 | 7.57 ± 0.25; 103.21 | 7.34 ± 0.33; 96.23 |
| HQC (20.00) | 20.23 ± 1.34; 100.00 | 20.59 ± 1.45; 101.26 | 19.98 ± 1.61; 98.87 | 18.59 ± 2.16; 93.43 |
| IS: ACV [n = 6]—LC-MS/MS method in DSS | ||||
| LLOQ (0.10) | 0.14 ± 0.06; 100.00 | 0.12 ± 0.07; 89.71 | 0.10 ± 0.03; 81.43 | 0.09 ± 0.01; 74.69 |
| MQC1 (0.75) | 0.77 ± 0.12; 100.00 | 0.75 ± 0.18; 98.77 | 0.70 ± 0.17; 91.87 | 0.67 ± 0.18; 87.29 |
| MQC2 (7.50) | 7.58 ± 0.36; 100.00 | 7.54 ± 0.31; 98.25 | 7.42 ± 0.38; 96.88 | 7.09 ± 0.41; 93.62 |
| HQC (20.00) | 20.34 ± 1.98; 100.00 | 20.19 ± 1.69; 97.95 | 19.22 ± 1.93; 94.19 | 18.93 ± 2.31; 90.38 |
| IS: ACV [n = 6]—LC-MS/MS method in s-VAMS | ||||
| LLOQ (0.10) | 0.11 ± 0.02; 100.00 | 0.09 ± 0.03; 89.99 | 0.10 ± 0.03; 90.79 | 0.09 ± 0.02; 87.37 |
| MQC1 (0.75) | 0.74 ± 0.13; 100.00 | 0.73 ± 0.14; 98.34 | 0.76 ± 0.20; 101.97 | 0.69 ± 0.18; 97.49 |
| MQC2 (7.50) | 7.58 ± 0.29; 100.00 | 7.59 ± 0.33; 98.76 | 7.44 ± 0.66; 98.15 | 7.26 ± 0.73; 96.07 |
| HQC (20.00) | 20.16 ± 0.99; 100.00 | 20.49 ± 1.71; 102.48 | 20.06 ± 1.28; 99.05 | 19.09 ± 1.44; 96.99 |
| GCV Concentration [mg/L] | Calculated Concentration [mg/L] and Stability at RT [%] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serum (IS: GCV-d5) [n = 6] | Serum (IS: ACV) [n = 6] | DSS (IS: ACV) [n = 6] | s-VAMS(IS: ACV) [n = 6] | |||||||||
| 0 h | −2 h | +2 h | 0 h | −2 h | +2 h | 0 h | −2 h | +2 h | 0 h | −2 h | +2 h | |
| MQC1 (0.75) | 0.74 ± 0.08 100.00% | 0.71 ± 0.07 95.59% | 0.73 ± 0.09 98.33% | 0.72 ± 0.09 100.00% | 0.73 ± 0.06 101.23% | 0.76 ± 0.08 102.37% | 0.74 ± 0.02 100.00% | 0.74 ± 0.04 99.13% | 0.71 ± 0.06 96.91% | 0.74 ± 0.08 100.00% | 0.76 ± 0.09 101.70% | 0.75 ± 0.03 99.64% |
| MQC2 (7.50) | 7.79 ± 0.46 100.00% | 7.58 ± 0.48 97.10% | 7.31 ± 0.37 95.93% | 7.65 ± 0.54 100.00% | 7.55 ± 0.41 96.20% | 7.71 ± 0.39 99.81% | 7.61 ± 0.21 100.00% | 7.58 ± 0.48 97.28% | 7.42 ± 0.29 93.46% | 7.46 ± 0.18 100.00% | 7.51 ± 0.23 100.98% | 7.33 ± 0.31 97.11% |
| Parameter | LLOQ—0.10 mg/L | MQC1—0.75 mg/L | MQC2—7.50 mg/L | HQC—20.00 mg/L | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GCV | IS | F | GCV | IS | F | GCV | IS | F | GCV | IS | F | |
| serum-LC-MS/MS [n = 6] IS: GCV-d5 | ||||||||||||
| ME [%] | −12.25 ± 2.21 | −16.24 ± 2.73 | −0.92 ± 0.67 | −17.89 ± 4.24 | −14.31 ± 3.06 | −1.29 ± 0.24 | −13.21 ± 3.89 | −12.88 ± 2.98 | 1.09 ± 0.19 | −10.43 ± 5.89 | −12.36 ± 3.41 | −0.89 ± 0.19 |
| PE [%] | 68.93 ± 2.87 | 68.10 ± 3.46 | 99.32 ± 5.21 | 70.09 ± 7.21 | 72.22 ± 5.01 | 100.09 ± 7.21 | 64.23 ± 4.87 | 66.93 ± 5.87 | 97.34 ± 2.07 | 77.31 ± 4.68 | 73.03 ± 2.99 | 100.33 ± 5.07 |
| AR [%] | 74.26 ± 1.56 | 78.55 ± 2.24 | 98.79 ± 7.75 | 69.57 ± 2.34 | 78.23 ± 3.14 | 96.23 ± 4.01 | 69.89 ± 3.76 | 73.58 ± 4.94 | 98.31 ± 2.17 | 70.63 ± 5.24 | 63.10 ± 3.39 | 98.76 ± 6.40 |
| serum-LC-MS/MS [n = 6] IS:ACV | ||||||||||||
| ME [%] | −13.98 ± 3.54 | −15.27 ± 6.34 | 0.89 ± 0.56 | −12.55 ± 4.31 | −9.99 ± 2.14 | 1.04 ± 0.78 | −13.39 ± 6.98 | −16.15 ± 11.23 | −0.76 ± 0.34 | −9.74 ± 3.26 | −13.13 ± 6.12 | 1.12 ± 0.87 |
| PE [%] | 70.42 ± 4.47 | 67.67 ± 5.41 | 102.62 ± 10.51 | 65.34 ± 7.23 | 66.56 ± 9.95 | 93.15 ± 7.99 | 67.22 ± 8.14 | 71.72 ± 6.69 | 94.56 ± 3.67 | 78.22 ± 5.67 | 73.75 ± 4.24 | 100.91 ± 11.99 |
| AR [%] | 66.20 ± 6.25 | 59.33 ± 7.22 | 103.33 ± 4.68 | 68.11 ± 4.43 | 66.15 ± 10.79 | 98.95 ± 8.48 | 65.22 ± 6.36 | 67.33 ± 5.03 | 98.26 ± 4.16 | 68.78 ± 7.21 | 71.34 ± 5.34 | 99.01 ± 12.44 |
| DSS-LC-MS/MS [n = 6] IS:ACV | ||||||||||||
| ME [%] | −14.83 ± 6.12 | −12.37 ± 4.81 | 0.79 ± 0.31 | −14.39 ± 6.41 | −12.22 ± 4.89 | 1.02 ± 0.91 | −16.21 ± 5.90 | −14.05 ± 6.16 | 0.95 ± 0.64 | −10.11 ± 6.32 | −9.99 ± 4.78 | 1.45 ± 1.05 |
| PE [%] | 66.57 ± 4.78 | 63.39 ± 4.57 | 101.21 ± 8.76 | 64.75 ± 5.09 | 67.98 ± 10.11 | 96.01 ± 4.01 | 63.33 ± 8.39 | 67.21 ± 5.78 | 96.06 ± 8.91 | 68.98 ± 7.94 | 70.77 ± 7.35 | 100.01 ± 6.23 |
| AR [%] | 60.01 ± 7.45 | 58.11 ± 9.54 | 98.99 ± 10.12 | 72.33 ± 8.43 | 70.14 ± 12.72 | 97.05 ± 5.48 | 64.67 ± 7.01 | 67.00 ± 6.23 | 98.01 ± 7.86 | 67.57 ± 6.78 | 70.98 ± 5.34 | 97.97 ± 3.22 |
| s-VAMS-LC-MS/MS [n = 6] IS:ACV | ||||||||||||
| ME [%] | −11.32 ± 7.00 | −10.11 ± 7.81 | −1.09 ± 0.82 | −10.09 ± 6.21 | −8.96 ± 5.21 | 1.00 ± 0.57 | −11.66 ± 4.72 | −11.09 ± 5.76 | 0.89 ± 0.42 | −10.01 ± 2.87 | −11.87 ± 6.79 | 1.22 ± 0.72 |
| PE [%] | 71.31 ± 8.12 | 72.07 ± 14.22 | 102.62 ± 10.23 | 70.77 ± 5.98 | 72.42 ± 7.01 | 97.01 ± 5.89 | 68.01 ± 7.52 | 69.87 ± 8.01 | 98.94 ± 6.12 | 70.54 ± 7.12 | 71.07 ± 5.55 | 97.56 ± 8.56 |
| AR [%] | 70.33 ± 6.66 | 75.12 ± 5.99 | 99.99 ± 12.01 | 76.53 ± 7.01 | 78.05 ± 6.43 | 98.52 ± 6.04 | 70.78 ± 5.29 | 70.13 ± 6.86 | 98.01 ± 3.31 | 73.24 ± 5.87 | 75.12 ± 6.31 | 96.831 ± 5.92 |
| GCV Concentration [mg/L] | Calculated Concentration [mg/L] and Stability at RT [%] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DSS (IS: ACV) [n = 6] | s-VAMS (IS: ACV) [n = 6] | |||||||||||
| T = 0 | 7 Days | 14 Days | 28 Days | 35 Days | 49 Days | T = 0 | 7 Days | 14 Days | 28 Days | 35 Days | 49 Days | |
| MQC1 (0.75) | 0.74 ± 0.05 100.00% | 0.75 ± 0.08 98.95% | 0.70 ± 0.11 95.16% | 0.68 ± 0.09 91.44% | 0.67 ± 0.06 89.76% | 0.66 ± 0.09 87.79% | 0.75 ± 0.04 100.00% | 0.75 ± 0.06 99.33% | 0.73 ± 0.05 96.57% | 0.70 ± 0.13 93.08% | 0.68 ± 0.19 89.76% | 0.67 ± 0.23 85.86% |
| MQC2 (7.50) | 7.56 ± 0.28 100.00% | 7.54 ± 0.18 97.89% | 7.31 ± 0.22 95.93% | 7.16 ± 0.54 93.61% | 6.89 ± 0.41 92.01% | 6.69 ± 0.72 89.41% | 7.52 ± 0.19 100.00% | 7.49 ± 0.25 100.29% | 7.43 ± 0.31 97.17% | 7.37 ± 0.36 96.01% | 7.24 ± 0.31 90.27% | 7.03 ± 0.57 87.28% |
| Statistical Tool | Compared Methods | |||||
|---|---|---|---|---|---|---|
| Serum-LC-MS/MS (GCV-d5 as IS) versus Serum-LC-MS/MS (ACV as IS) | Serum-LC-MS/MS (ACV as IS) versus DSS-LC-MS/MS (ACV as IS) | Serum-LC-MS/MS (ASC as IS) versus s-VAMS-LC-MS/MS (ACV as IS) | DSS-LC-MS/MS (ACV as IS) versus s-VAMS-LC-MS/MS (ACV as IS) | Serum-LC-MS/MS (GCV-d5 as IS) versus Serum-HPLC-UV (ACV as IS) | Serum-LC-MS/MS (ACV as IS) versus Serum-HPLC-UV (ACV as IS) | |
| Passing-Bablok regression formula | y = 0.996x + 0.003 | y = 1.048x − 0.013 | y = 1.072x − 0.011 | y = 1.019x + 0.019 | y = 0.997x + 0.021 | y = 1.009x − 0.075 |
| Intercept (A) | 0.003 (−0.032 to 0.053) | −0.013 (−0.073 to 0.066) | −0.011 (−0.058 to 0.029) | 0.019 (−0.029 to 0.058) | 0.021 (−0.010 to 0.083) | −0.075 (−0.183 to −0.008) |
| Slope (B) | 0.996 (0.935 to 1.075) | 1.048 (0.930 to 1.155) | 1.072 (0.984 to 1.125) | 1.019 (0.981 to 1.067) | 0.997 (0.917 to 1.048) | 1.009 (0.920 to 1.112) |
| Bland-Altman bias [%] | 4.53% (−0.71 to 9.77) | 3.56% (−4.56 to 11.72) | −3.61% (−8.38 to 1.16) | −9.06% (−15.29 to −2.82) | 5.87% (−2.65 to 14.39) | 12.34% (2.20 to 22.48) |
| % of paired samples fulfilled EMA criteria (bias < 20%) | 67.25% | 72.50% | 82.50% | 86.25% | 63.25% | 61.25% |
| Pearson’s correlation coefficient (R2) | 0.96 (0.93 to 0.97) | 0.94 (0.90 to 0.96) | 0.94 (0.91 to 0.96) | 0.97 (0.95 to 0.98) | 0.98 (0.97 to 0.99) | 0.93 (0.89 to 0.95) |
| Spearman rank correlation coefficient (SRCC) | 0.99 (0.98 to 0.99) | 0.97 (0.96 to 0.98) | 0.98 (0.98 to 0.99) | 0.99 (0.98 to 1.00) | 0.98 (0.96 to 0.99) | 0.96 (0.93 to 0.97) |
| Analyte | Monitored Adduct | MRM Pairs (m/z) | CE [eV] | Dwell Time [ms] |
|---|---|---|---|---|
| GCV | [GCV + H]+ | 256.05 → 152.00 256.05 → 135.10 | −20 −12 | 17 17 |
| GCV-d5 | [(GCV-d5) + H]+ | 261.15 → 152.05 261.15 → 135.10 | −13 −12 | 17 17 |
| ACV | [ACV + H]+ | 226.10 → 152.00 226.10 → 135.00 | −15 −27 | 17 17 |
| Parameter | Value |
|---|---|
| electrospray voltage [kV] | 1.00 |
| nebulizing gas [L/min] | 2.50 |
| drying gas [L/min] | 5.00 |
| heating gas [L/min] | 7.00 |
| interface temperature [°C] | 200 |
| desolvation temperature [°C] | 355 |
| desolvation line temperature [°C] | 200 |
| heat block temperature [°C] | 300 |
| CID pressure [kPa] | 270 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocur, A.; Czajkowska, A.; Moczulski, M.; Kot, B.; Rubik, J.; Pawiński, T. Assessment of Dried Serum Spots (DSS) and Volumetric-Absorptive Microsampling (VAMS) Techniques in Therapeutic Drug Monitoring of (Val)Ganciclovir—Comparative Study in Analytical and Clinical Practice. Int. J. Mol. Sci. 2024, 25, 8760. https://doi.org/10.3390/ijms25168760
Kocur A, Czajkowska A, Moczulski M, Kot B, Rubik J, Pawiński T. Assessment of Dried Serum Spots (DSS) and Volumetric-Absorptive Microsampling (VAMS) Techniques in Therapeutic Drug Monitoring of (Val)Ganciclovir—Comparative Study in Analytical and Clinical Practice. International Journal of Molecular Sciences. 2024; 25(16):8760. https://doi.org/10.3390/ijms25168760
Chicago/Turabian StyleKocur, Arkadiusz, Agnieszka Czajkowska, Mateusz Moczulski, Bartłomiej Kot, Jacek Rubik, and Tomasz Pawiński. 2024. "Assessment of Dried Serum Spots (DSS) and Volumetric-Absorptive Microsampling (VAMS) Techniques in Therapeutic Drug Monitoring of (Val)Ganciclovir—Comparative Study in Analytical and Clinical Practice" International Journal of Molecular Sciences 25, no. 16: 8760. https://doi.org/10.3390/ijms25168760
APA StyleKocur, A., Czajkowska, A., Moczulski, M., Kot, B., Rubik, J., & Pawiński, T. (2024). Assessment of Dried Serum Spots (DSS) and Volumetric-Absorptive Microsampling (VAMS) Techniques in Therapeutic Drug Monitoring of (Val)Ganciclovir—Comparative Study in Analytical and Clinical Practice. International Journal of Molecular Sciences, 25(16), 8760. https://doi.org/10.3390/ijms25168760