Abstract
Clinical trials with treatments inhibiting myostatin pathways to increase muscle mass are currently ongoing in spinal muscular atrophy. Given evidence of potential myostatin pathway downregulation in Spinal Muscular Atrophy (SMA), restoring sufficient myostatin levels using disease-modifying treatments (DMTs) might arguably be necessary prior to considering myostatin inhibitors as an add-on treatment. This retrospective study assessed pre-treatment myostatin and follistatin levels’ correlation with disease severity and explored their alteration by disease-modifying treatment in SMA. We retrospectively collected clinical characteristics, motor scores, and mysotatin and follistatin levels between 2018 and 2020 in 25 Belgian patients with SMA (SMA1 (n = 13), SMA2 (n = 6), SMA 3 (n = 6)) and treated by nusinersen. Data were collected prior to treatment and after 2, 6, 10, 18, and 30 months of treatment. Myostatin levels correlated with patients’ age, weight, SMA type, and motor function before treatment initiation. After treatment, we observed correlations between myostatin levels and some motor function scores (i.e., MFM32, HFMSE, 6MWT), but no major effect of nusinersen on myostatin or follistatin levels over time. In conclusion, further research is needed to determine if DMTs can impact myostatin and follistatin levels in SMA, and how this could potentially influence patient selection for ongoing myostatin inhibitor trials.
1. Introduction
Spinal muscular atrophy (SMA) is a recessively inherited neuromuscular condition caused by mutations in the survival motoneuron gene (SMN1), resulting in the absence of survival motor neuron protein (SMN) [1]. The absence of SMN protein is partially compensated by an autologous gene, SMN2, that is responsible for the production of functional SMN protein, and directly influences SMA disease severity [2,3]. SMA primarily affects motor neurons in both the central and peripheral nervous system, leading to proximal muscle weakness, hypotonia, and muscle atrophy [4,5]. Traditionally, SMA has been divided into five main phenotypes based on age of symptom onset and highest level of motor function achieved [6,7], although these phenotypes tend to evolve with the increasing use of disease-modifying therapies (DMTs) and improved standard of care [8,9,10]. The SMA therapeutic landscape has undergone tremendous progress in the last 10 years, with the development of three DMTs and the implementation of newborn screening programs (NBSs) in several countries [11]. DMTs for SMA differ in their administration route and tissue distribution. Nusinersen, an antisense oligonucleotide, targets the central nervous system through intrathecal delivery [12,13,14]. Onasemnogene abeparvovec (Zolgensma), a gene replacement therapy [15], and risdiplam, a small-molecule splicing modifier [16,17], achieve systemic distribution through intravenous and oral routes, respectively. A large body of evidence has demonstrated a better impact of treatment when initiated within the first days of life, which has paved the way for implementation of NBS [18,19]. While older patients can still benefit from these treatments, the effect is less pronounced [20]. Importantly, even among early-treated patients, treatment benefits can vary, and some patients may still experience varying degrees of motor dysfunction. Beyond muscle function, other issues may persist despite treatment, including cognitive and language impairments [21,22,23]. All these remaining challenges justify ongoing efforts to develop new therapies and add-on therapies to further improve patient outcomes [24,25].
Myostatin, or growth differentiation factor-8 (GDF8), is a member of the transforming growth factor-β (TGFβ) family, that acts as a negative regulator of skeletal muscle bulk and inhibits skeletal muscle growth [26]. Although there is interest in leveraging myostatin inhibition to enhance muscle mass and function in several muscle-wasting diseases, the efficacy of anti-myostatin drugs in clinical trials remains limited [27,28]. Some evidence supports a down-regulation of myostatin pathways in SMA, leading to intrinsically low blood levels of myostatin and high levels of antagonist follistatin (FSTN) [29]. Higher myostatin predicted better anti-myostatin treatment outcomes in animals [30,31], suggesting that restoring sufficient levels of myostatin may be required prior to the inhibition of the myostatin pathways [29]. This information taken together raises relevant questions in the era of ongoing clinicals trials with several anti-myostatin drugs in SMA, as they could impact patient selection: (I) Does myostatin and its antagonist follistatin correlate with disease severity in SMA patients before and after DMTs? (II) Do DMTs influence the levels of myostatin and follistatin in treated patients? (III) Could myostatin and follistatin levels potentially guide patient selection in anti-myostatin therapy? Here, we investigated whether myostatin and follisatin levels correlate with phenotype and motor function prior to, and after, initiation and whether administration of nusinersen in SMA patients impacts myostatin or follistatin levels over time.
2. Results
2.1. Patients’ Demographics and Clinical Characteristics
We included 13 patients with SMA 1, six patients with SMA 2, and six patients with SMA 3. The median age at treatment initiation was 10.3 years (1 month—59.5 years). SMN2 copy number was unavailable for two patients; four patients had two copies; 13 had three copies; and six had four copies. At baseline, myostatin and follistatin levels were available for 22 patients (220–3036.4 pg/mL) and 11 patients (834.5–2927.0 pg/mL), respectively. One patient’s follistatin levels were excluded due to aberrant values that could not be verified or explained by any identifiable confounding factor. Median clinical and biological follow-ups were 18.5 months (1–33 months) and 15.5 months (1–29 months), respectively (Supplementary Materials Table S1).
2.2. Myostatin and Follistatin Levels at Baseline in Nusinersen-Naïve Patients
2.2.1. Myostatin and Follistatin Levels Per SMA Type and SMN2 Copy Number
We observed a significant difference in myostatin levels across the three SMA types (n = 22, η2 = 0.729, p = 0.03). SMA type 3 exhibited significantly higher (p = 0.03) myostatin levels (Md = 1818.4 pg/mL) compared to type 1 (Md = 517.8 pg/mL). The difference between type 3 and type 2 (Md = 802.6 pg/mL) was not significant (p = 0.19), although the small sample size may underestimate the difference (Figure 1A). There was no significant difference in myostatin levels between SMA type 1 and type 2 (p = 1). Despite significant overall group differences in myostatin levels across the different SMN2 copy number (n = 20, η2 = 0.76, p = 0.04), pairwise comparison remained non-significant. No significant difference was observed in the follistatin levels across SMA type (n = 11, p = 0.52) and SMN2 copy number (n = 11, p = 0.37).
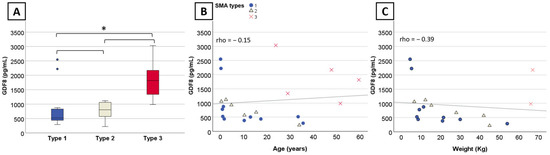
Figure 1.
Myostatin levels per SMA type and correlation with age and weight in nusinersen-naïve patients. (A) Boxplot displaying myostatin levels for SMA types with outliers indicated by dots. (B) Significant correlation between myostatin levels, (B) age, and (C) weight within SMA1 and SMA2. Significant results are indicated by an asterisk (*) and dots indicate outliers.
2.2.2. Correlation between Myostatin and Follistatin Levels and Baseline Characteristics
There was no significant correlation between myostatin levels and age (n = 22, rho = −0.15, 95%CI [−0.58; 0.35], p = 0.5) (Figure 1B) or weight (n = 18, rho = −0.39, 95%CI [−0.85; 0.22], p = 0.11) (Figure 1C) when considering the three SMA types altogether. However, myostatin levels negatively correlated with age and weight at baseline in SMA type 1 (n = 11, rho = −0.84, 95%CI [−1; −0.38], p = 0.001 and n = 10, rho = −0.81, 95%CI [−1; −0.30], p = 0.005, respectively) and type 2 (n = 6, rho = −0.89, 95%CI [−1; −0.34], p = 0.02 and n = 6, rho = −0.94, 95%CI [−1; −0.51], p = 0.05, respectively). Myostatin correlated with several motor scores (32-item Motor Function Measure (MFM32) (n = 12, rho = 0.83, 95%CI [0.31; 0.99], p < 0.001) (Figure 2A), Children’s Hospital of Philadelphia Infant Test of Neuromuscular-Disorders (CHOP-INTEND) (n = 9, rho = 0.80, 95%CI [0.13; 1], p = 0.01) (Figure 2B), Hammersmith Functional Motor Scale Expanded (HFMSE) (n = 6, rho = 0.89, 95%CI [0.20; 1], p = 0.02) (Figure 2C), and Six-Minute Walk Test (6MWT) (n = 5, rho = 0.90, 95%CI [0.11; 1], p = 0.04) (Figure 2D). No significant correlation was observed with the Hammersmith Infant Neurological Examination (HINE-2) (n = 12, rho = 0.36, 95%CI [−0.35; 0.84) (Figure 2G), although two outliers treated within two months of life might have skewed the results. No correlation was found with left (n = 5, rho = 0.7, 95%CI [−0.875; 1 pg/mL]) (Figure 2E) and right grip score (n = 5, rho = 0.7, 95%CI [−0.936; 1) (Figure 2F), and follistatin levels (n = 11, rho = −0.045, 95%CI [−0.628; 0.653]) (Figure 2H).
2.3. Change in Myostatin and Follistatin Levels over Time in Treated Patients
SMA types were analyzed altogether given the small sample size. We observed no significant changes in myostatin (n = 10, r = 0.11, estimate of 61.2 pg/mL, 95%CI: [−194.2; 316.8 pg/mL], p = 0.77) and follistatin levels (n = 10, r = 0.11, estimate of 92.5 pg/mL, 95%CI: [−469.85; 437.15 pg/mL], p = 0.77) at 18 months of treatment (Figure 3D). Change were non-significant at additional time points of 2 months (n = 10, r = 0.56, estimate of 151.6 pg/mL, 95%CI: [−33.4; 305.8 pg/mL] and r = 0.16, estimate of 20 pg/mL, 95%CI [−544.6; 360.5 pg/mL]) (Figure 3A), 6 months (n = 11, r = −0.05, estimate of −16.2 pg/mL, 95%CI: [−185.2; 192.0 pg/mL] (Figure 3B) and r = −0.08, estimate of −50.25 pg/mL, 95%CI [−243.5; 649.2 pg/mL]), and 10 months (n = 9, r = −0.14, estimate of −25.2 pg/mL, 95%CI [−125.6; 98.2 pg/mL] and r = 0.33, estimate of 70 pg/mL, 95%CI [−178; 506 pg/mL]) (Figure 3C). Statistical analysis at 30 months was not possible due to the small sample size (n = 4). Aligning with these findings, the line graph confirms no specific trend in myostatin level changes following up to 30 months of nusinersen. (Figure 4A). We observed a correlation at 18 months of treatment between myostatin and MFM32 (n = 14, rho = 0.9, 95%CI [0.62; 0.99], p = <0.001) (Figure 3B), HFMSE (n = 5, rho = 0.75, 95%CI [0.98; 0.99], p = 0.01) (Figure 4C), and 6MWT (n = 5, rho = 0.7, 95%CI [0.04–1], p = 0.01) (Figure 4D). There was no correlation with follistatin, nor between follistatin and motor scores (Supplementary Materials Table S2).
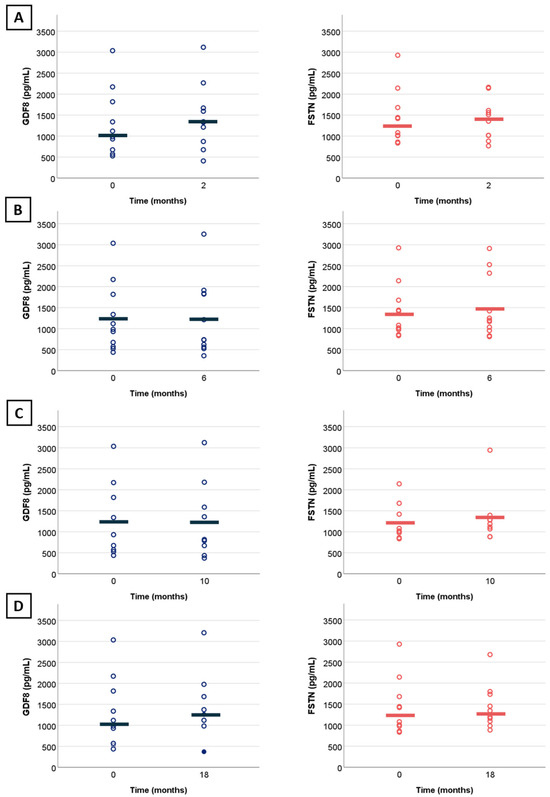
Figure 3.
Change in myostatin and follistatin levels from baseline in treated patients. (A) Change in myostatin (blue) and follistatin (red) levels over a period of 2 months with treatment by nusinersen (N = 10); (B) over a period of 6 months with treatment by nusinersen (N = 11); (C) over a period of 10 months with treatment by nusinersen (N = 9); (D) over a period of 18 months with treatment by nusinersen (N = 10). Full dots indicate overlapping points. Horizontal lines illustrate mean values.
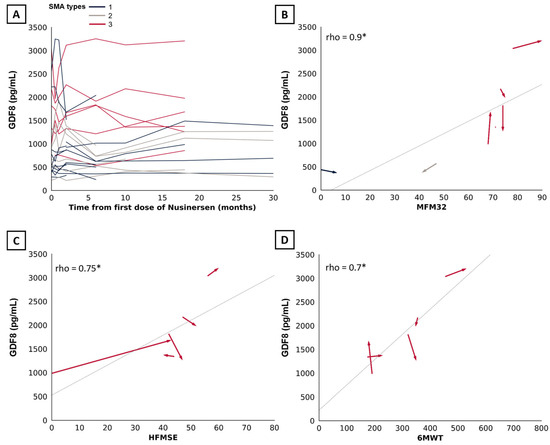
Figure 4.
Association between myostatin and motor scores over time. (A) Line graph displaying the trend in myostatin levels following up to 30 months of treatment with nusinersen (n = 25). (B) Arrow graph showing the association between myostatin and MFM32, (C) HFMSE, and (D) 6MWT from baseline to 18 months of treatment with nusinersen. Arrow indicates temporality. SMA types 1, 2, and 3 are indicated in blue, beige, and red, respectively. Significant results are indicated by an asterisk (*).

Figure 2.
Correlation of myostatin levels with motor scores and follistatin blood levels in nusinersen-naïve patients. Graphs showing the significant correlation of myostatin with (A) MFM32, (B) CHOP-INTEND, (C) HFMSE, (D) 6MWT. No significant correlation was observed between myostatin and left grip score (E), right grip score (F), HINE-2 (G), and follistatin (H). Significant results are indicated by an asterisk (*).
3. Discussion
In a small patient sample, we observed an inverse correlation between myostatin levels, age, and weight at baseline in both SMA1 and SMA2 patients. Additionally, our findings support prior research, showing correlations between myostatin levels and disease severity, as evidenced by lower myostatin levels in SMA1 patients, and correlation with several motor scores [32,33].
Myostatin and follistatin are growth and differentiation factors belonging to the transforming growth factor-beta (TGF-beta) superfamily. Myostatin RNA is specifically expressed in developing and mature muscle tissues in mice [34]. During embryogenesis, expression is restricted to the myotome, with more widespread muscular expression observed in adult animals [34]. Myostatin, like other TGF-β members, is initially expressed as a precursor protein that undergoes cleavage into an N-terminal propeptide and a disulfide-linked C-terminal dimer, which is the biologically active molecule. The circulating form of myostatin in the bloodstream consists of a latent complex of the myostatin C-terminal dimer and other proteins, including the inhibitory myostatin propeptide [35,36]. Cleavage of the propeptide by a metalloproteinase activates latent myostatin, allowing it to bind to its receptors, ActRIIA and ActRIIB [37,38]. Follistatin acts as a myostatin antagonist through direct protein–protein interaction that sequesters myostatin, preventing its binding to the activin type IIB receptor [39,40]. Myostatin’s role as a negative regulator of skeletal muscle mass was initially highlighted in GDF8-null mice, which displayed a significant increase in weight and muscle mass due to increased fiber size and fiber number [34]. This increase in muscle bulk was then observed as a result of MSTN mutations across different animal species [41,42,43] and humans [44]. Concomitantly, agents designed to block myostatin production in adult mouse models [45] confirmed the increase in muscle mass following myostatin inhibition, including in disease models of Becker (BMD) and Duchenne muscular dystrophy (DMD) [31,46,47], dysferlinopathy [48], limb-girdle muscular dystrophy type 2A (LGMD2A) [49], calpainopathy [50], and SMA [29,51,52,53,54]. Altogether, these results have generated enthusiasm regarding the potential of inhibiting myostatin and its pathway to increase muscle mass and motor function in muscle-wasting conditions.
Three drugs targeting the myostatin signaling pathway are currently being evaluated in clinical trials (Table 1). SRK-015 (Scholar Rock®,, Cambridge, MS, USA) is a monoclonal antibody that selectively inhibits pro- and latent myostatin. It has completed phase 1 (NCT02644777) and phase 2 (TOPAZ, NCT03921528) trials [55,56], as well as a 12-month randomized, controlled phase 3 trial (RCT) in patients with type 2 and type 3 SMA, undergoing treatment by nusinersen or risdiplam. An open-label extension study is currently ongoing for patients who completed the two aforementioned trials (ONYX, NCT05626855). The TOPAZ trial (NCT03921528) demonstrated sustained improvement with apitegromab and nusinersen in non-ambulatory patients, using HFMSE, Revised Upper Limb Module (RULM), and WHO motor development milestones as functional outcomes [57]. Taldefgrobep alfa (Biohaven®, New Haven, CT, USA) is a humanized recombinant protein designed to neutralize free myostatin and block the activin IIb receptor. This dual action inhibits the signaling of both myostatin and activin A [58]. Preclinical data and a well-established safety profile exist from studies in patients with neuromuscular diseases [59], including DMD (NCT03039686, NCT02515669) [58]. A randomized, double-blind, placebo-controlled phase 3 trial (RESILIENCE, NCT05337553) is currently underway in both ambulatory and non-ambulatory patients. Notably, unlike SRK-015 trials, RESILIENCE allows for the inclusion of patients with a history of onasemnogene abeparvovec-xioi treatment [60]. RO7204239 (Roche®, Basel, Switzerland), a monoclonal anti-myostatin antibody designed to eliminate myostatin from plasma and tissues, is currently being investigated in a phase 2 and phase 3 RCT (MANATEE, NCT05115110). This two-part study is evaluating the safety and efficacy of the antibody when used in combination with risdiplam in ambulatory and non-ambulatory patients [61].

Table 1.
Anti-myostatin therapies in spinal muscular atrophy. HFMSE: Hammersmith Functional Motor Scale Expanded, MFM32: 32-item Motor Function Measure, N/A: Not Applicable, RHS: Revised Hammersmith Scale, Y: years old.
Excluding a few small open-label studies and RCTs [62,63,64], therapies targeting the myostatin pathway have yielded inconclusive results in DMD [27,65] and other adult neuromuscular diseases [66,67,68]. One plausible explanation is the downregulation of the pathway itself in muscle-wasting diseases, limiting the availability of therapeutic targets [29]. Evidence for this comes from the reduced myostatin pathway mRNA expression observed in skeletal muscles of neuromuscular patients with severe muscle loss, such as those with SMA and DMD [29]. Higher myostatin predicted better anti-myostatin treatment outcomes in animals [28,29,30,31], suggesting that restoring sufficient levels of myostatin may be required prior to the inhibition of the myostatin pathways [29]. Moreover, SMA animal models suggest greater benefit from myostatin-targeting drugs combined with SMN-restoring therapies [51,52,69]. Several key questions remain regarding myostatin and its role in SMA treatment. Firstly, there is a need to determine whether myostatin levels accurately reflect the severity of the phenotype and the patient’s response to treatment. Secondly, only a few studies have investigated the impact of DMTs on myostatin levels so far. Finally, it is still unclear whether myostatin levels could serve as a biomarker for selecting patients who might respond best to anti-myostatin therapies. If achieving minimum blood levels of myostatin proves to be a prerequisite for the effectiveness of anti-myostatin therapies, then identifying which patients are likely to fall into this category becomes crucial. Thus, analyzing blood levels of myostatin in SMA patients across different SMA subtypes, SMN2 copies, and during DMTs could be highly informative for future trials.
Our data support previous findings, which demonstrated an inverted correlation between myostatin levels and disease severity in SMA [32,33] and other neuromuscular disorders [29,70,71]. This aligns with the observation of lower myostatin levels in neuromuscular patients compared to healthy controls [72]. Our findings suggest a trend of decreasing myostatin levels with age in SMA types I and II, but not in SMA type III, which has a milder phenotype. Similarly, a significant decrease in myostatin levels with age was shown in DMD patients, while the decrease was significant but less pronounced in BMD patients [72]. However, this relationship remains unclear due to inconsistent results across studies [33,70]. The correlation between myostatin levels and baseline age in our cohort might not reflect a true age-related decline. The lack of a clear trend in myostatin levels over time within individual patients could indicate either no correlation with age or a stabilization of myostatin levels following treatment. No correlation of follistatin with phenotype, type, or age and no trends in blood levels over time were observed, consistent with recent published studies in SMA [33].
Myostatin has been suggested as a potential biomarker for monitoring disease progression in several neuromuscular diseases, including SMA [30,32,33,73,74]. Our findings suggest that myostatin levels correlate with motor function, before and after treatment. However, the small sample size and potential confounding factors need to be addressed in future research. We did not observe significant changes in serum levels of follistatin and myostatin over time in patients treated with nusinersen, contrasting with a recent study reporting ongoing decreases in myostatin levels over time [33]. Limited nusinersen efficacy in SMA, small sample size, and intrathecal delivery’s potential lack of effect on systemic myostatin levels are possible explanations. Future studies with systemic treatments in SMA patients might offer valuable insights.
Several limitations of our study must be acknowledged. The retrospective design and small sample size limited statistical power. Given the mixed-age cohort and inclusion of three SMA subtypes, we may have missed subtle or transient changes, especially within specific SMA subgroups. Data gaps due to disruption in follow-up and unavailable samples during COVID-19 further reduced statistical power. Short follow-up periods in some patients might have missed slower changes. Additionally, we did not control for age at treatment, weight, or body lean mass, and the lack of robust statistical models to adjust for confounding variables limits our conclusions.
Nevertheless, understanding myostatin pathways in treated SMA patients remains crucial in the era of newborn screening and ongoing clinical trials. Future research should explore the correlation of myostatin with clinical metrics such as standardized motor scores reflecting patient phenotypes. Criteria defining therapeutic response should be established upfront and tailored to each SMA subgroup or SMA copy number, as the boundaries between classical SMA types are blurring with the implementation of DMTs and newborn screening. These criteria should be standardized across studies to minimize variability. Statistical corrections for multiple comparisons [75], and robust statistical models for repeated measures, such as linear mixed-effects models [76] adjusted for age, lean mass, and weight, should be employed. Longer follow-up periods will help capture slower fluctuations, and we believe a 12-month period of stable nusinersen dosage might be considered for phenotype assessment, based on previous studies evaluating clinical response [77].
4. Methods
Patient Characteristics and Study Protocol
In this retrospective monocentric study, we gathered clinical data and serum myostatin and follistatin levels from 25 Belgian SMA patients treated with intrathecal nusinersen at a standard dosing regimen in Citadelle Hospital, Liège, Belgium, between 2018 and 2020. The study received approval from the institutional Ethical Committee of the Citadelle Hospital (reference: JL/rc/2105). Data were collected at multiple time points (baseline, 14 days, 1, 2, 6, 10, 18, and 30 months) and values were averaged within a ±25% interval around each specific time point. Clinical data collected from medical records included gender, SMA type, number of SMN2 copies, age at treatment initiation, weight, BMI, and number of SMN2 copies. When available, motor scores were collected for CHOP-INTEND, HINE-2, HFSME, MFM32, Six-Minute Walk Test (6MWT), and grip strength test. Peripheral venous samples were collected as part of the standard of care for nusinersen treatment, using serum separator tubes (10 mL). Patients’ parents agreed to have leftover fluids used for future research. Myostatin and follistatin levels were assessed at UCL Great Ormond Street Institute of Child Health. After 30 min at room temperature, tubes were centrifuged at 2000 rpm for 10 min at 4 °C. The collected serum (5 mL) was aliquoted and stored at −80 °C.
Myostatin and follistatin concentrations in the sera were measured using an ELISA kit following the manufacturer’s instructions and previous methodology [29]. Optical density was measured with a microplate reader (Infinite 200 Pro, Tecan Group Ltd., Männedorf, Switzerland).
Given the small sample size, we performed non-parametric tests (p < 0.05). Spearman correlations and bootstrapping for a 95% confidence interval (95%CI) were used for the correlation between myostatin and follistatin levels and clinical parameters. Myostatin and follistatin levels across SMA types and SMN2 copy numbers were compared using the Kruskal–Wallis test with Dunn’s post hoc tests for pairwise differences. Bonferroni correction was adjusted for multiple comparisons, and the effect size was assessed with eta squared (η2). We reported the Wilcoxon signed-rank test along with Wilcoxon effect size (r) [78], Hodges–Lehmann estimator, and 95%CI for changes in myostatin and follistatin levels over time (2, 6, 10, and 18 months).
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms25168763/s1.
Author Contributions
Conceptualization, J.D., V.M., L.S. and L.M.; methodology, J.D., V.M., L.S. and L.M.; software, L.M.; validation, L.M., J.D., V.M. and L.S.; formal analysis, L.M.; investigation, J.D., V.M., L.S., L.B. and L.M.; resources, L.B., J.D., V.M., L.S. and L.M.; data curation, L.M.; writing—original draft preparation, L.M.; writing—review and editing, L.M., J.D., V.M., L.S. and L.B.; visualization, L.M., L.S., J.D. and V.M.; supervision, J.D. and L.S.; project administration, J.D., V.M., L.S. and L.M.; funding acquisition, not applicable. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study received approval from the institutional Ethical Committee of the Citadelle Hospital (reference: JL/rc/2105).
Informed Consent Statement
Informed consent was obtained from all participants involved in the study, or their proxies.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors due to the General Data Protection Regulation.
Acknowledgments
We are grateful to Gita Thapaliya for her expertise in revising and validating the statistical plan. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Conflicts of Interest
V.M. and J.D. are named inventors of a patent entitled “Method Relating to Myostatin Pathway Inhibition” that has been filled by UCL (PCT/GB2018/050619). L.S. gave consultancy and/or took part in the board of Biogen, Novartis, Roche, Scholar Rock, BioHaven, and Zentech.
Abbreviations
| 6MWT | Six-Minute Walk Test |
| BMD | Becker muscular dystrophy |
| CHOP-INTEND | Children’s Hospital of Philadelphia Infant-Test-of-Neuromuscular-Disorders |
| DMD | Duchenne muscular dystrophy |
| DMTs | Disease-modifying therapies |
| FSTN | Follistatin |
| GDF8 | Growth differentiation factor-8 |
| HFMSE | Hammersmith Functional Motor Scale Expanded |
| HINE-2 | Hammersmith Infant Neurological Examination 2 |
| LGMD2A | Limb-girdle muscular dystrophy type 2A |
| MFM32 | 32-item Motor Function Measure |
| NBS | Newborn screening |
| RCT | Randomized control trial |
| RHS | Revised Hammersmith Scale |
| RULM | Revised Upper Limb Module |
| SMA | Spinal muscular atrophy |
| SMN | Survival motor neuron |
| TGFβ | Transforming growth factor-β |
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Gavrilov, D.K.; Shi, X.; Das, K.; Gilliam, T.C.; Wang, C.H. Differential SMN2 expression associated with SMA severity. Nat. Genet. 1998, 20, 230–231. [Google Scholar] [CrossRef]
- Swoboda, K.J.; Prior, T.W.; Scott, C.B.; McNaught, T.P.; Wride, M.C.; Reyna, S.P.; Bromberg, M.B. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 704–712. [Google Scholar] [CrossRef]
- Annoussamy, M.; Seferian, A.M.; Daron, A.; Péréon, Y.; Cances, C.; Vuillerot, C.; De Waele, L.; Laugel, V.; Schara, U.; Gidaro, T.; et al. Natural history of Type 2 and 3 spinal muscular atrophy: 2-year NatHis-SMA study. Ann. Clin. Transl. Neurol. 2021, 8, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; McDermott, M.P.; Kaufmann, P.; Darras, B.T.; Chung, W.K.; Sproule, D.M.; Kang, P.B.; Foley, A.R.; Yang, M.L.; Martens, W.B. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014, 83, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Zerres, K.; Rudnik-Schöneborn, S. Natural history in proximal spinal muscular atrophy: Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch. Neurol. 1995, 52, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, A.; Seferian, A.M.; Daron, A.; Péréon, Y.; Cances, C.; Vuillerot, C.; De Waele, L.; Cuisset, J.-M.; Laugel, V.; Schara, U. Prospective and longitudinal natural history study of patients with type 2 and 3 spinal muscular atrophy: Baseline data NatHis-SMA study. PLoS ONE 2018, 13, e0201004. [Google Scholar] [CrossRef]
- Mercuri, E. Spinal muscular atrophy: From rags to riches. Neuromuscul. Disord. 2021, 31, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Tizzano, E.F.; Finkel, R.S. Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscul. Disord. 2017, 27, 883–889. [Google Scholar] [CrossRef]
- Chen, T.-H. New and developing therapies in spinal muscular atrophy: From genotype to phenotype to treatment and where do we stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef]
- Dangouloff, T.; Vrscaj, E.; Servais, L.; Osredkar, D.; Group, S.N.W.S. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul. Disord. 2021, 31, 574–582. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.D.; Chiriboga, C.A.M.D.; Vajsar, J.M.D.; Day, J.W.M.D.; Montes, J.E.; De Vivo, D.C.M.D.; Yamashita, M.M.D.; Rigo, F.P.; Hung, G.M.D.; Schneider, E.M.D.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Rose, K.; Xiong, H.; Zanoteli, E.; Baranello, G.; Bruno, C.; Vlodavets, D.; Wang, Y.; et al. Risdiplam-Treated Infants with Type 1 Spinal Muscular Atrophy versus Historical Controls. N. Engl. J. Med. 2021, 385, 427–435. [Google Scholar] [CrossRef]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Dangouloff, T.; Servais, L. Clinical Evidence Supporting Early Treatment Of Patients With Spinal Muscular Atrophy: Current Perspectives. Ther. Clin. Risk Manag. 2019, 15, 1153–1161. [Google Scholar] [CrossRef]
- Aragon-Gawinska, K.; Mouraux, C.; Dangouloff, T.; Servais, L. Spinal Muscular Atrophy Treatment in Patients Identified by Newborn Screening—A Systematic Review. Genes 2023, 14, 1377. [Google Scholar] [CrossRef]
- Darras, B.T.; Chiriboga, C.A.; Iannaccone, S.T.; Swoboda, K.J.; Montes, J.; Mignon, L.; Xia, S.; Bennett, C.F.; Bishop, K.M.; Shefner, J.M. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology 2019, 92, e2492–e2506. [Google Scholar] [CrossRef]
- Baranello, G.; Roy, S.Q.; Servais, L.; Munell, F.; Molinero, M.A.; de Benito, D.N.; Nascimento, A.; Gomez-Andres, D.; Comellas, L.C.; Exposito, J. The emerging spectrum of neurodevelopmental comorbidities in early-onset Spinal Muscular Atrophy. Eur. J. Paediatr. Neurol. 2024, 48, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Steffens, P.; Weiss, D.; Perez, A.; Appel, M.; Weber, P.; Weiss, C.; Stoltenburg, C.; Ehinger, U.; von der Hagen, M.; Schallner, J. Cognitive function in SMA patients with 2 or 3 SMN2 copies treated with SMN-modifying or gene addition therapy during the first year of life. Eur. J. Paediatr. Neurol. 2024, 51, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ngawa, M.; Dal Farra, F.; Marinescu, A.-D.; Servais, L. Longitudinal developmental profile of newborns and toddlers treated for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231154335. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, S.; Servais, L. New treatments in spinal muscular atrophy: An overview of currently available data. Expert Opin. Pharmacother. 2020, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Baranello, G.; Scoto, M.; Daron, A.; Oskoui, M. Therapeutic interventions for spinal muscular atrophy: Preclinical and early clinical development opportunities. Expert Opin. Investig. Drugs 2021, 30, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Orpí, M.; Busquets, S.; López-Soriano, F.J. Myostatin: More than just a regulator of muscle mass. Drug Discov. Today 2012, 17, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef] [PubMed]
- Abati, E.; Manini, A.; Comi, G.P.; Corti, S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell. Mol. Life Sci. CMLS 2022, 79, 374. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Hourdé, C.; Féasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1859. [Google Scholar] [CrossRef]
- Mariot, V.; Le Guiner, C.; Barthélémy, I.; Montus, M.; Blot, S.; Torelli, S.; Morgan, J.; Muntoni, F.; Voit, T.; Dumonceaux, J. Myostatin Is a Quantifiable Biomarker for Monitoring Pharmaco-gene Therapy in Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2020, 18, 415–421. [Google Scholar] [CrossRef]
- Dumonceaux, J.; Marie, S.; Beley, C.; Trollet, C.; Vignaud, A.; Ferry, A.; Butler-Browne, G.; Garcia, L. Combination of Myostatin Pathway Interference and Dystrophin Rescue Enhances Tetanic and Specific Force in Dystrophic mdx Mice. Mol. Ther. 2010, 18, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Hellbach, N.; Karrer, T.; Ly-Le Moal, M.; King, N.; Glaab, W.; Daron, A.; Annoussamy, M.; Pereon, Y.; Cances, C.; Seferian, A. Characterizing novel exploratory biomarkers in a longitudinal natural history study in patients with Types 2 and 3 SMA. Change 2021, 7, 5. [Google Scholar]
- de Albuquerque, A.L.A.; Chadanowicz, J.K.; Giudicelli, G.C.; Staub, A.L.P.; Weber, A.C.; Silva, J.M.D.S.; Becker, M.M.; Kowalski, T.W.; Siebert, M.; Saute, J.A.M. Serum myostatin as a candidate disease severity and progression biomarker of spinal muscular atrophy. Brain Commun. 2024, 6, fcae062. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Tries, R.S.; Chen, T.; Da Vies, M.V.; Tomkinson, K.N.; Pearson, A.A.; Shakey, Q.A.; Wolfman, N.M. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors 2001, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.J.; Davies, M.V.; Pearson, A.A.; Wang, J.H.; Hewick, R.M.; Wolfman, N.M.; Qiu, Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 2002, 277, 40735–40741. [Google Scholar] [CrossRef]
- Wolfman, N.M.; McPherron, A.C.; Pappano, W.N.; Davies, M.V.; Song, K.; Tomkinson, K.N.; Wright, J.F.; Zhao, L.; Sebald, S.M.; Greenspan, D.S. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl. Acad. Sci. USA 2003, 100, 15842–15846. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.N.; Rejon, C.A.; McPherron, A.C.; Bernard, D.J.; Thompson, T.B. The structure of myostatin: Follistatin 288: Insights into receptor utilization and heparin binding. EMBO J. 2009, 28, 2662–2676. [Google Scholar] [CrossRef]
- Amthor, H.; Nicholas, G.; McKinnell, I.; Kemp, C.F.; Sharma, M.; Kambadur, R.; Patel, K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev. Biol. 2004, 270, 19–30. [Google Scholar] [CrossRef]
- Kambadur, R.; Sharma, M.; Smith, T.P.; Bass, J.J. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997, 7, 910–915. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lee, S.-J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Marcq, F.; Takeda, H.; Pirottin, D.; Tordoir, X.; Bibé, B.; Bouix, J.; Caiment, F.; Elsen, J.-M.; Eychenne, F. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006, 38, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef]
- Whittemore, L.-A.; Song, K.; Li, X.; Aghajanian, J.; Davies, M.; Girgenrath, S.; Hill, J.J.; Jalenak, M.; Kelley, P.; Knight, A. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res. Commun. 2003, 300, 965–971. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.-A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef]
- Qiao, C.; Li, J.; Jiang, J.; Zhu, X.; Wang, B.; Li, J.; Xiao, X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther. 2008, 19, 241–254. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lehar, A.; Sebald, S.; Liu, M.; Swaggart, K.A.; Talbot, C.C., Jr.; Pytel, P.; Barton, E.R.; McNally, E.M.; Lee, S.-J. Muscle hypertrophy induced by myostatin inhibition accelerates degeneration in dysferlinopathy. Hum. Mol. Genet. 2015, 24, 5711–5719. [Google Scholar] [CrossRef]
- Harish, P.; Malerba, A.; Lu-Nguyen, N.; Forrest, L.; Cappellari, O.; Roth, F.; Trollet, C.; Popplewell, L.; Dickson, G. Inhibition of myostatin improves muscle atrophy in oculopharyngeal muscular dystrophy (OPMD). J. Cachexia Sarcopenia Muscle 2019, 10, 1016–1026. [Google Scholar] [CrossRef]
- Bartoli, M.; Poupiot, J.; Vulin, A.; Fougerousse, F.; Arandel, L.; Daniele, N.; Roudaut, C.; Noulet, F.; Garcia, L.; Danos, O. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not α-sarcoglycan deficiency. Gene Ther. 2007, 14, 733–740. [Google Scholar] [CrossRef][Green Version]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef]
- Liu, M.; Hammers, D.W.; Barton, E.R.; Sweeney, H.L. Activin receptor type IIB inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS ONE 2016, 11, e0166803. [Google Scholar] [CrossRef] [PubMed]
- Rose, F.F., Jr.; Mattis, V.B.; Rindt, H.; Lorson, C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009, 18, 997–1005. [Google Scholar] [CrossRef]
- Crawford, T.O.; Darras, B.T.; Day, J.W.; Dunaway Young, S.; Duong, T.; Nelson, L.L.; Barrett, D.; Song, G.; Bilic, S.; Cote, S. Safety and Efficacy of Apitegromab in Patients With Spinal Muscular Atrophy Types 2 and 3: The Phase 2 TOPAZ Study. Neurology 2024, 102, e209151. [Google Scholar] [CrossRef] [PubMed]
- Pokrzywinski, R.; Cutts, K.; Shah, H.; Kertesz, N.; Lesh, S.; Sadanowicz, M.; Song, G.; O’Neil, J.; Barrett, D.; Barnobi, E. Treatment Effects Among Patients with Type 2 and Type 3 SMA Directly Reported by Patients and Caregivers from the TOPAZ Clinical Trial (P2-8.010). Neurology 2023, 100, 3355. [Google Scholar] [CrossRef]
- Crawford, T.; Darras, B.; Day, J.; De Vivo, D.; Mercuri, E.; Nascimento, A.; Mazzone, E.; Waugh, A.; Song, G.; Evans, R. P224 Effect of apitegromab on motor function at 36 months in patients with nonambulatory spinal muscular atrophy aged 2-12 years old. Neuromuscul. Disord. 2023, 33, S91. [Google Scholar] [CrossRef]
- Muntoni, F.; Byrne, B.J.; McMillan, H.J.; Ryan, M.M.; Wong, B.L.; Dukart, J.; Bansal, A.; Cosson, V.; Dreghici, R.; Guridi, M. The Clinical Development of Taldefgrobep Alfa: An Anti-Myostatin Adnectin for the Treatment of Duchenne Muscular Dystrophy. Neurol. Ther. 2024, 13, 183–219. [Google Scholar] [CrossRef]
- Lair, L.; Qureshi, I.; Bechtold, C.; Heller, L.; Durham, S.; Campbell, D.; Marin, J.; Chen, K.; Coric, V. P04 Taldefgrobep alfa: Preclinical and clinical data supporting the phase 3 RESILIENT study in spinal muscular atrophy. Neuromuscul. Disord. 2023, 33, S163. [Google Scholar] [CrossRef]
- Lair, L.; Qureshi, I.; Bechtold, C.; Durham, S.; Campbell, D.; Marin, J.; Coric, V. The Phase 3 RESILIENT Study: Taldefgrobep Alfa in Spinal Muscular Atrophy (P2-11.008). In Neurology; Lippincott Williams & Wilkins: Hagerstown, MD, USA, 2024; p. 3255. [Google Scholar]
- Duong, T.; Darras, B.; Morrow, J.; Muntoni, F.; Servais, L.; Rabbia, M.; Gerber, M.; Kletzl, H.; Gaki, E.; Fletcher, S. MANATEE: GYM329 (RO7204239) in combination with risdiplam treatment in patients with spinal muscular atrophy (SMA). Neuromuscul. Disord. 2023, 33, S92. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Sahenk, Z.; Rodino-Klapac, L.R.; Kaspar, B.; Mendell, J.R. Follistatin gene therapy improves ambulation in Becker muscular dystrophy. J. Neuromuscul. Dis. 2015, 2, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.A.; Sivakumar, K.; Goyal, N.; David, W.S.; Salajegheh, M.; Praestgaard, J.; Lach-Trifilieff, E.; Trendelenburg, A.-U.; Laurent, D.; Glass, D.J. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 2014, 83, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef]
- Wagner, K.R. The elusive promise of myostatin inhibition for muscular dystrophy. Curr. Opin. Neurol. 2020, 33, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. No longer going to waste. Nat. Biotechnol. 2016, 34, 458–462. [Google Scholar] [CrossRef]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, J.; Marrosu, E.; Janghra, N.; Morgan, J.; Muntoni, F. Repeated low doses of morpholino antisense oligomer: An intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum. Mol. Genet. 2015, 24, 6265–6277. [Google Scholar] [CrossRef]
- van de Velde, N.M.; Koeks, Z.; Signorelli, M.; Verwey, N.; Overzier, M.; Bakker, J.A.; Sajeev, G.; Signorovitch, J.; Ricotti, V.; Verschuuren, J. Longitudinal Assessment of Creatine Kinase, Creatine/Creatinineratio, and Myostatin as Monitoring Biomarkers in Becker Muscular Dystrophy. Neurology 2023, 100, e975–e984. [Google Scholar] [CrossRef] [PubMed]
- Moore, U.; Fernández-Simón, E.; Schiava, M.; Cox, D.; Gordish-Dressman, H.; James, M.K.; Mayhew, A.; Wilson, I.; Guglieri, M.; Rufibach, L. Myostatin and follistatin as monitoring and prognostic biomarkers in dysferlinopathy. Neuromuscul. Disord. 2023, 33, 199–207. [Google Scholar] [CrossRef]
- Burch, P.M.; Pogoryelova, O.; Palandra, J.; Goldstein, R.; Bennett, D.; Fitz, L.; Guglieri, M.; Bettolo, C.M.; Straub, V.; Evangelista, T. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J. Neurol. 2017, 264, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Buono, S.; Menuet, A.; Robé, A.; Djeddi, S.; Kretz, C.; Gomez-Oca, R.; Depla, M.; Monseur, A.; Thielemans, L. Myostatin: A circulating biomarker correlating with disease in myotubular myopathy mice and patients. Molecular 2020, 17, 1178–1189. [Google Scholar] [CrossRef]
- Mahoudeau, A.; Anquetil, C.; Tawara, N.; Khademian, H.; Amelin, D.; Bolko, L.; Silvestro, M.; Cin, J.D.; Tendrel, B.; Tardif, V. Myostatin in idiopathic inflammatory myopathies: Serum assessment and disease activity. Neuropathol. Appl. Neurobiol. 2023, 49, e12849. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Schober, P.; Vetter, T.R. Repeated measures designs and analysis of longitudinal data: If at first you do not succeed—Try, try again. Anesth. Analg. 2018, 127, 569–575. [Google Scholar] [CrossRef]
- Lilien, C.; Vrscaj, E.; Thapaliya, G.; Deconinck, N.; De Waele, L.; Duong, T.; Haberlová, J.; Kumhera, M.; Peirens, G.; Szabo, L. Patients’ Perceptions of Nusinersen Effects according to Their Responder Status. J. Clin. Med. 2024, 13, 3418. [Google Scholar] [CrossRef]
- Li, G.; Taljaard, M.; Van den Heuvel, E.R.; Levine, M.A.; Cook, D.J.; Wells, G.A.; Devereaux, P.J.; Thabane, L. An introduction to multiplicity issues in clinical trials: The what, why, when and how. Int. J. Epidemiol. 2016, 46, 746–755. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).