Abstract
Haemonchus contortus (the barber’s pole worm)—a highly pathogenic gastric nematode of ruminants—causes significant economic losses in the livestock industry worldwide. H. contortus has become a valuable model organism for both fundamental and applied research (e.g., drug and vaccine discovery) because of the availability of well-defined laboratory strains (e.g., MHco3(ISE).N1 in the UK and Haecon-5 in Australia) and genomic, transcriptomic and proteomic data sets. Many recent investigations have relied heavily on the use of the chromosome-contiguous genome of MHco3(ISE).N1 in the absence of a genome for Haecon-5. However, there has been no genetic comparison of these and other strains to date. Here, we assembled and characterised the mitochondrial genome (14.1 kb) of Haecon-5 and compared it with that of MHco3(ISE).N1 and two other strains (i.e., McMaster and NZ_Hco_NP) from Australasia. We detected 276 synonymous and 25 non-synonymous single nucleotide polymorphisms (SNPs) within Haecon-5. Between the Haecon-5 and MHco3(ISE).N1 strains, we recorded 345 SNPs, 31 of which were non-synonymous and linked to fixed amino acid differences in seven protein-coding genes (nad5, nad6, nad1, atp6, nad2, cytb and nad4) between these strains. Pronounced variation (344 and 435 SNPs) was seen between Haecon-5 and each of the other two strains from Australasia. The question remains as to what impact these mitogenomic mutations might have on the biology and physiology of H. contortus, which warrants exploration. The high degree of mitogenomic variability recorded here among these strains suggests that further work should be undertaken to assess the nature and extent of the nuclear genomic variation within H. contortus.
1. Introduction
Haemonchus contortus, commonly known as the barber’s pole worm, is a highly pathogenic gastric nematode of ruminants, such as sheep and goats. It causes significant economic losses in the livestock industry by affecting animal health and productivity [1,2]. The control of this and related parasites has relied heavily on the use of anthelmintics. However, the excessive use of these drugs has led to widespread drug resistance, making it increasingly challenging to manage infections and disease [3]. The extent of this resistance problem emphasises the need to develop novel drug targets and interventions to control H. contortus and related parasites effectively.
H. contortus has become a valuable model organism for anthelmintic discovery because well-defined laboratory strains, such as MHco3(ISE).N1 and Haecon-5, are available, because screening platforms have been developed, and because extensive genomic, transcriptomic and proteomic resources are accessible [4,5,6,7,8] for in-depth investigations. The Haecon-5 strain of H. contortus is used for anthelmintic screening and evaluation as well as for molecular studies that rely on the chromosome-contiguous genome available in MHco3(ISE).N1 for the purpose of analyses and annotation [6]. However, given previous evidence of genetic variability in mitochondrial DNA within H. contortus among relatively large numbers of adult male specimens (n = 223) from different geographical regions around the world [9], there is a need to genetically characterise Haecon-5 and explore the nature and extent of the genetic variability between the well-defined laboratory strains (i.e., MHco3(ISE).N1 and Haecon-5) routinely utilised for ongoing fundamental and applied investigations.
As a first step, here we (i) assembled and characterised the mitochondrial genome of the Haecon-5 strain; (ii) evaluated mitogenomic sequence variability within this strain; and (iii) genetically compared the mitogenomes of Haecon-5 with MHco3(ISE).N1 and/or other strains of H. contortus. At the outset, we hypothesised that there is no mitogenomic variability within the Haecon-5 population and only minor differences between Haecon-5 and other currently used laboratory strains.
2. Results
2.1. The Haecon-5 Mitogenome, and Variation within an Individual and a Population
The complete mitochondrial genome of the Haecon-5 strain of H. contortus is 14,107 bp in length, has 36 genes (12 protein-coding genes, 22 transfer (t)RNA genes and 2 ribosomal (r)RNA genes) and is AT-rich (78%), consistent with the published mitogenomes of other strains (Figure 1A, Table 1).
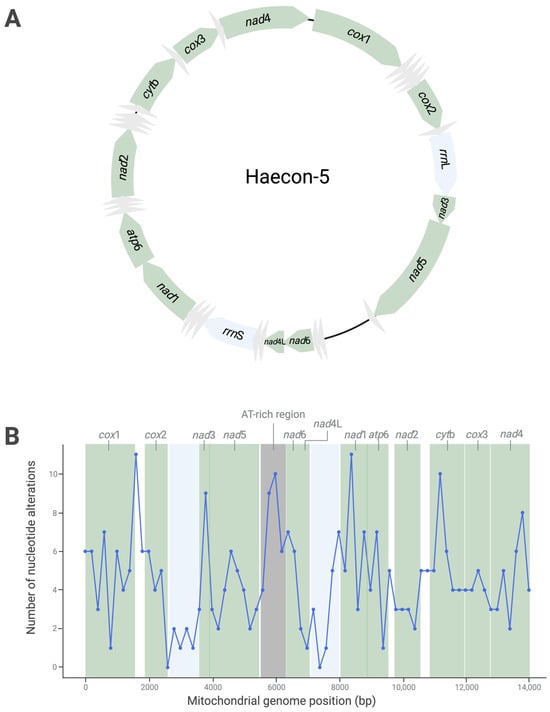
Figure 1.
The circular mitochondrial genome of the Haecon-5 strain of Haemonchus contortus determined from a single adult male worm, with protein-coding (green), tRNA (grey) and rRNA (blue) genes, as well as non-coding regions (black), indicated (A). Nucleotide variability in the mitochondrial genome within a population of third-stage larvae (L3s; n = 200,000) of Haecon-5, established by a sliding window analysis of the linearised map (using a 200 bp window) (B). Table 2 details the nature and extent of this variability.

Table 1.
Features of the mitochondrial genomes of the Haecon-5 strain (determined herein), and of the MHco3(ISE).N1, McMaster and NZ_Hco_NP strains (published previously).
Although there was no evidence of heteroplasmy within the single adult male of Haecon-5, marked variability (319 variants) was detected within the L3 population characterised by 301 (94.4%) SNPs, 15 (4.7%) indels and 3 (0.9%) MNPs (Figure 1; Table S1). Of all SNPs, 268 (89.1%) were transition (A↔G or C↔T) and 33 (10.9%) were transversion polymorphisms (A↔C, A↔T, G↔C or G↔T) (Figure 1; Table S1). Most SNPs (n = 238; 79%) were in the protein-coding genes (Figure 1), with 25 being non-synonymous and linked to amino acid alterations in nad5, nad6, nad1, atp6, nad2, cytb and nad4 (Figure 1; Table 2). All indels were in non-coding regions, and MNPs resulted in amino acid alterations in the genes nad6 (ATA→GTT, leading to M→V) and nad2 (GT→AC, leading to V→T) (Table 2 and Table S1); small numbers of SNPs were located in non-coding regions (n = 63; 21.0%), tRNAs (n = 9) and rRNA regions (n = 9) (Figure 1).
Table 2.
Nucleotide variation in the protein-coding, tRNA and rRNA genes, as well as the AT-rich (“control”) region of the mitochondrial genome, within a population of third-stage larvae (L3s) of the Haecon-5 strain of Haemonchus contortus.
The sliding window analysis revealed an uneven distribution of nucleotide alterations across the genome (Figure 1B), with four main peaks. Three of these peaks were in protein-coding gene (cox1, nad1 and cytb) regions. The fourth peak was in the AT-rich (“control”) region (Figure 1B). The highest number of SNPs, all of which were synonymous, were within the cox1 gene. In contrast, 13 alterations were recorded in nad6, 6 of which were non-synonymous—the most for any protein-coding gene (Table 2). As expected, variability in the AT-rich region was linked to 26 nucleotide alterations (Table 2 and Figure 1B).
2.2. Marked Mitogenomic Variability between Haecon-5 and Other Strains
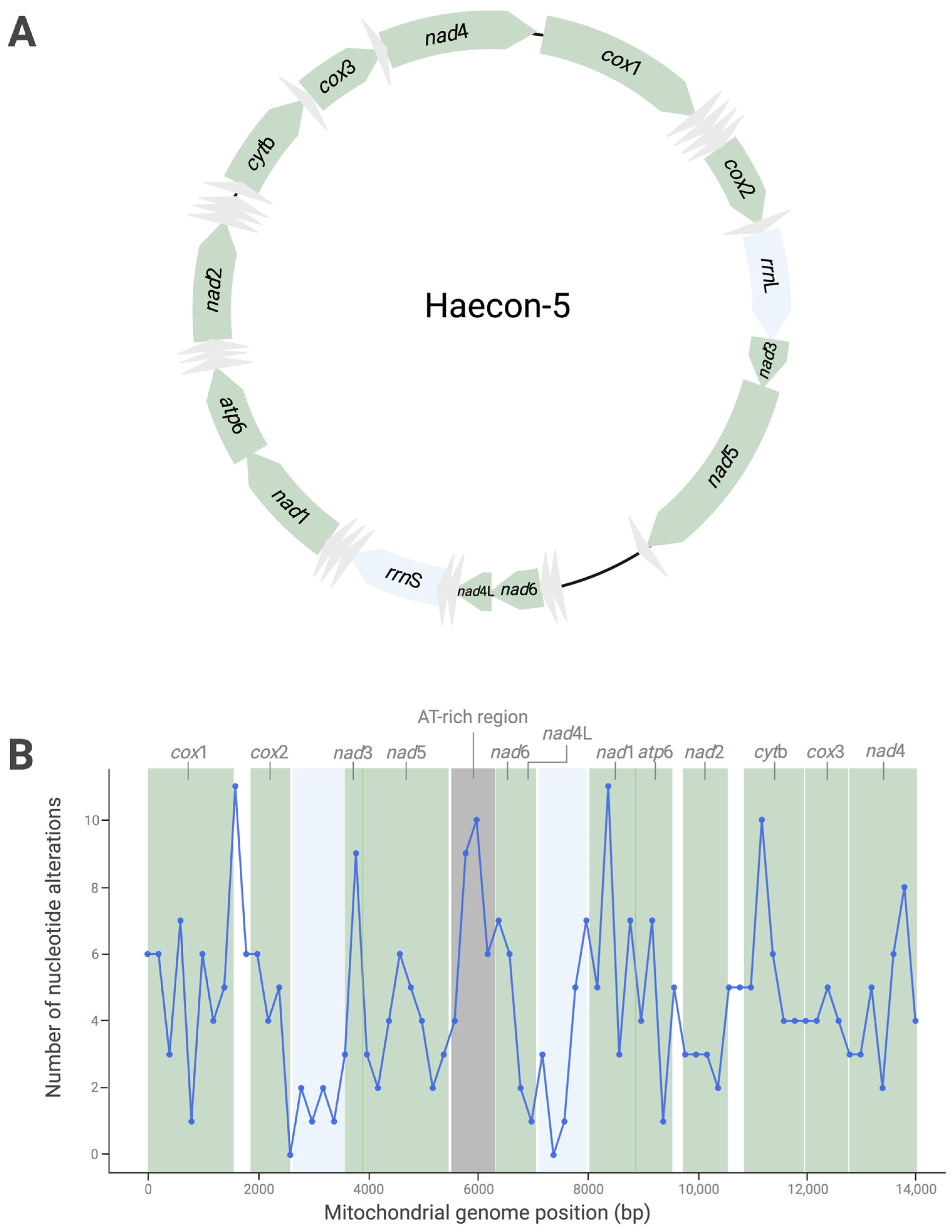
Pairwise mitogenomic comparisons of Haecon-5 with MHco3(ISE).N1, McMaster and NZ_Hco_NP revealed extensive nucleotide variability (Figure 2; Tables S2–S4). In total, 368 alterations were recorded between Haecon-5 and MHco3(ISE).N1, most (n = 278) of which were in coding regions, while 35 were linked to amino acid substitutions (Table 3). Most alterations (n = 345) were SNPs, and a minority were represented by indels (n = 16) and MNPs (n = 7) (Table 3).

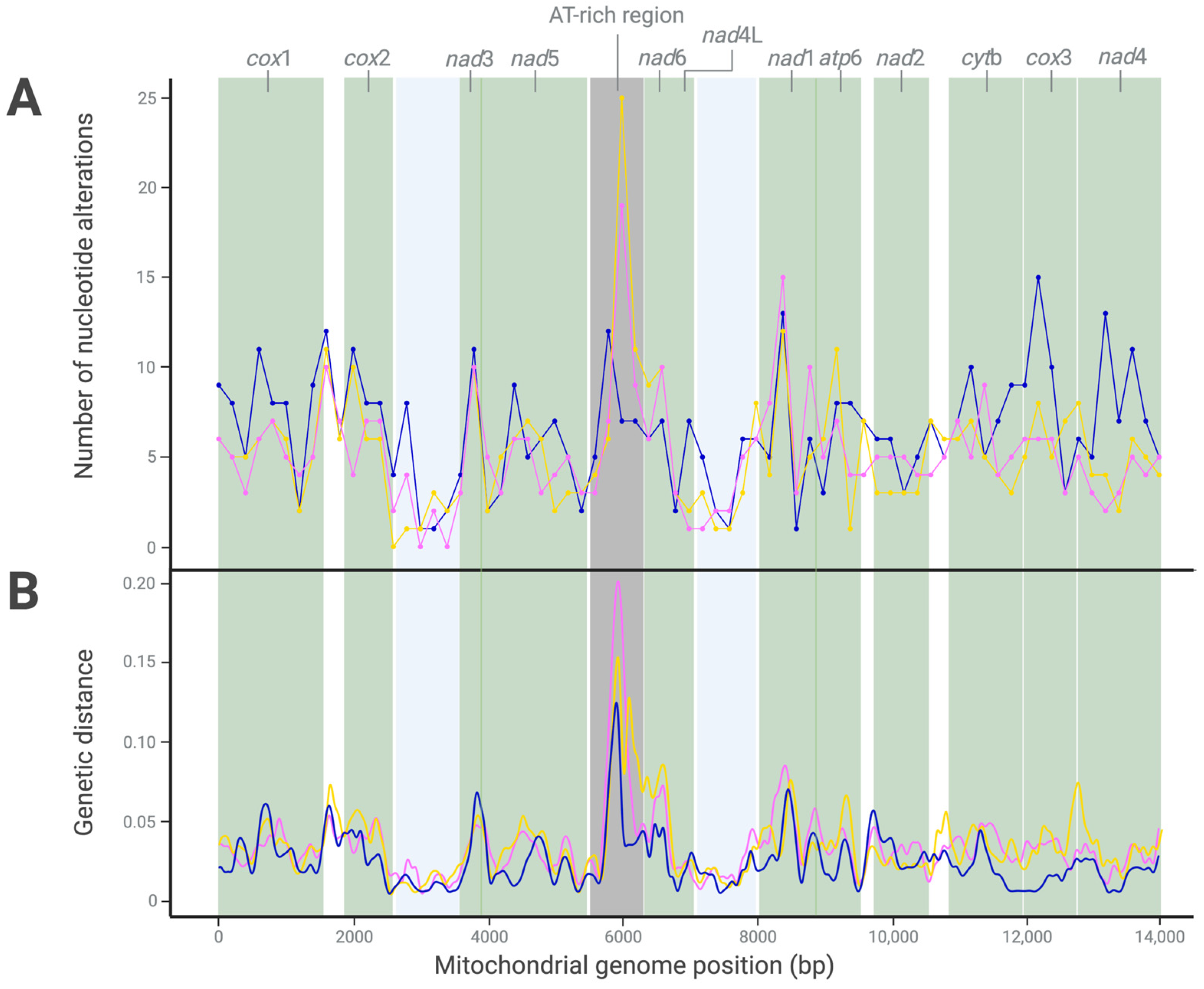
Figure 2.
Nucleotide variability (A) and genetic distances (B) recorded upon pairwise comparison of the mitochondrial genomes of the Haecon-5 strain (Figure 1) with the MHco3(ISE).N1, McMaster and NZ_Hco_NP strains of Haemonchus contortus (pink, yellow and blue, respectively; refs. [6,10,11]) following a sliding window analysis (200 bp window). Table 3 details the nature and extent of this variability.
Table 3.
Nucleotide variability recorded upon pairwise comparison of the mitochondrial genomes of the Haecon-5 (this study) and other strains MHco3(ISE).N1, McMaster or NZ_Hco_NP; refs. [6,10,11]).
Similarly, the mitogenomic comparison between the Haecon-5 and McMaster strains revealed 378 alterations, including 283 in coding regions, 36 of which were non-synonymous (Table 3). The variants identified were classified as 344 SNPs, 14 MNPs and 20 indels (Figure 2). Most alterations (n = 460) were recorded between Haecon-5 and NZ_Hco_NP; 355 were located in coding regions and 30 were linked to amino acid alterations, almost half of which were in the genes nad1 and nad5 (Table 3).
For all mitogenomic comparisons, the cox1 gene was the most variable, with 41 alterations in MHco3(ISE).N1, 42 in McMaster and 60 in NZ_Hco_NP. None of these alterations was linked to an amino acid change (Table 3). Conversely, the nad5 gene, which had the second highest number of nucleotide alterations, had the most non-synonymous SNPs (n = 6–8) (Table 3). The nad4L gene was least variable, with eight alterations—one of which was linked to an amino acid change (Table 3).
As expected, the AT-rich region (between 5531 and 6194 bp) was the most variable between Haecon-5 and each of the three other strains, reflected in the genetic distance results (Figure 2). Taken together, the variation was extensive among all four mitochondrial genomes, including 554 SNPs in coding regions, 63 of which were non-synonymous (Table 3 and Figure 2).
3. Discussion
Here, we characterised the mitochondrial genome of the Haecon-5 strain, assessed the mitogenomic sequence variability within this strain and compared the mitogenome of Haecon-5 with those of the strains MHco3(ISE).N1 from the UK, McMaster from Australia and NZ_Hco_NP from New Zealand. Based on our findings, we clearly rejected the hypotheses that (i) there is no mitogenomic sequence variability within the Haecon-5 population and that (ii) there are only minor differences between Haecon-5 and other currently used laboratory strains.
Although no heteroplasmy was detected within an individual adult male worm of Haecon-5, significant sequence variability was detected within the L3s population analysed—equating to 91.7% synonymous SNPs and 8.3% non-synonymous SNPs in all protein-coding genes. This magnitude of non-synonymous SNPs was unexpected and inferred to be associated with the amino acid changes at 27 positions in seven protein-coding genes. The nature and extent of the variability recorded (overall) supports a high mutation rate in this parasitic nematode [12,13,14] and explains the marked genetic diversity recorded in previous studies (e.g., [15,16,17]), particularly that of Sallé et al. [9], which revealed mitogenomic nucleotide sequence variation among 223 individual adult males of H. contortus from distinct countries and regions around the world. The results of the latter study were a major reason for assessing the extent of the genetic variability between laboratory strains, such as Haecon-5 and MHco3(ISE).N1, which are routinely employed for fundamental and applied investigations [6,18,19,20,21,22,23,24,25,26,27,28,29,30].
Thus, given that the laboratory strains MHco3(ISE).N1 and Haecon-5 strains have been, and will continue to be, used for decades for research purposes, we elected to directly compare these strains at the mitogenomic level. We also wanted to assess the extent of mitogenomic difference between these strains because the nucleotide genome of MHco3(ISE).N1 [6] has been used routinely as a “heterologous” reference for Haecon-5 for some years now without our knowing how genetically distinct it might be from Haecon-5. The results here (Section 2.2) indicated clearly that the MHco3(ISE).N1 and Haecon-5 strains are indeed unequivocally distinct (9.5% in the proteome-coding part of the mitogenome), which could have significant implications in relation to interpreting findings from molecular genetic investigations and conclusions made regarding, for example, the conservation of genes or proteins proposed as anthelmintic targets or vaccine molecules, or the functions of genes and their products.
Indeed, the marked genetic differences in the mitogenome (at 368 nucleotide positions) and mitoproteome (at 35 amino acid positions) between the strains Haecon-5 and MHco3(ISE).N1 of H. contortus suggest that some biological and/or phenotypic distinctiveness could exist. For instance, to date, there has been no direct comparison of the biology (e.g., life cycle–nematode development time and prepatency and patency periods), virulence or pathogenicity of these two strains. Moreover, no study has yet genetically characterised the distinct morphotypes of adult female worms (considering vulva types, cuticular ridge patterns and cervical papillae; refs. [31,32,33,34,35,36,37]) of these strains or examined the prevalence/abundance of these morphotypes in populations representing these strains. Indeed, it is possible that these morphotypes are genetically distinct genetic sub-types of H. contortus and that the dominant morphotype in each strain determines the mitogenome for each strain. This proposal remains to be explored. However, it is also possible that variation in the mitogenome sequences of H. contortus is not associated with distinct morphotypes but rather is the result of differences in development and/or mitonuclear coevolution (e.g., [38,39,40,41]).
4. Materials and Methods
The Haecon-5 strain of H. contortus was maintained using established protocols [26] approved by the University of Melbourne’s animal ethics committee (permit no. 23983). Genomic DNA was isolated from a single adult male worm (0.8 cm in length) or from a pool of 200,000 third-stage larvae (L3s) using the Circulomics tissue kit (Baltimore, MD, USA). The quality and quantity of DNA were assessed using the 4200 TapeStation (Agilent, Santa Clara, CA, USA). For short-read sequencing, genomic DNA from L3s or an adult male worm was used to construct paired-end libraries, which were sequenced (150 paired-end nucleotides) on the Illumina NextSeq500 or NOVAseq6000 platforms, and the data were stored in the FASTQ format. For long-read sequencing, libraries were prepared using SQK-LSK110 and SQK-LSK114 kits and sequenced on platforms from Oxford Nanopore Technologies (Oxford, UK), employing an established protocol [42]. Bases were called using Guppy v.6.4.6 from raw Pod5 files, with the output saved in FASTQ.
The mitochondrial genome of Haecon-5 was assembled from nanopore long-read sequences from a single male worm using Canu v.2.2 [43]. Protein-coding genes (PCGs), transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) were predicted using MITOS2 (http://mitos.bioinf.uni-leipzig.de/; accessed on 2 May 2024) and curated using Geneious Prime v.2024.0.5 [44]. The final annotated mitochondrial genome data were in FASTA and GFF3 formats for consistency and subsequent analyses.
For Haecon-5, short-read data representing a single adult male worm (used for assembly) or from pooled L3s were separately mapped and aligned independently to the homologous mitogenome using BWA v.0.7.17 [45] and sorted using SAMtools v.1.17 [46], and all duplicate reads were removed. Subsequently, single nucleotide polymorphisms (SNPs), insertion/deletion events (indels) and multi-nucleotide polymorphisms (MNPs) were explored using Geneious Prime v.2024.0.5 [44]. Each nucleotide alteration was recorded as synonymous or non-synonymous [47], with individual variants saved in the Variant Call Format (VCF).
Subsequently, the mitochondrial genomes of the MHco3(ISE).N1, McMaster and NZ_Hco_NP strains [6,10,11] and corresponding annotation files were downloaded from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/; accessed on 2 May 2024). Subsequently, a pairwise mitogenomic comparison of Haecon-5 with each of the three other strains was conducted (gene order and content, GC/AT composition and nucleotide variability), and nucleotide alterations (SNPs, indels and MNPs) were recorded using Geneious Prime v.2024.0.5, linked to a location in the mitogenome and annotated and saved in VCF. SPIDER v.1.1-2 [48] was used to calculate the genetic distance between Haecon-5 and the other strains.
A sliding window analysis of aligned mitogenome sequences was conducted using GenomicRanges v.3.19 [49]. Alignment was achieved using MUSCLE v.5.1 [50] as implemented in Geneious Prime v.2024.0.5 [44]. A sliding window of 200 bases was used to estimate the nucleotide diversity (number of nucleotide alterations) and genetic distance between distinct strains of H. contortus. Nucleotide diversity was plotted against the midpoint of each window, and gene boundaries were defined.
5. Conclusions
The genetic distinctiveness between the strains MHco3(ISE).N1 and Haecon-5 of H. contortus at the mitogenomic level is pronounced. Given the utility of both of these strains for ongoing research in a wide range of areas, such as anthelmintic resistance, host–parasite interactions, drug discovery and vaccine development, the question remains as to how different these two strains are at the nuclear genomic and proteomic levels. This is the next question that we plan to address in the near future.
Supplementary Materials
The supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms25168765/s1.
Author Contributions
Conceptualisation, N.D.Y. and R.B.G.; methodology, Y.Z. and N.D.Y.; formal analysis, Y.Z. and N.D.Y.; investigation, Y.Z. and N.D.Y.; data curation, Y.Z. and N.D.Y.; writing—original draft preparation, Y.Z.; writing—review and editing, Y.Z., N.D.Y., J.S. and R.B.G.; visualisation, Y.Z.; supervision, N.D.Y., J.S. and R.B.G.; project administration, N.D.Y., J.S. and R.B.G.; funding acquisition, N.D.Y. and R.B.G. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by Australian Research Council (ARC) grants to R.B.G. (LP180101085 and LP220200614).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of the University of Melbourne (protocol code 23983; 2024).
Informed Consent Statement
Not applicable.
Data Availability Statement
The complete mitochondrial genome assembly is available in the National Center for Biotechnology Information (NCBI) database under accession numbers PRJNA1071864 and PQ149937.
Acknowledgments
Y.Z. was supported by a Melbourne Research Scholarship and a Rowden White Prize from the University of Melbourne. The authors thank Ross S. Hall for bioinformatic support. We acknowledge the use of BioRender for figure preparation.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Emery, D.L.; Hunt, P.W.; Le Jambre, L.F. Haemonchus contortus: The then and now, and where to from here? Int. J. Parasitol. 2016, 46, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Carson, A.; Reichel, R.; Bell, S.; Collins, R.; Smith, J.; Bartley, D. Haemonchus contortus: An overview. Vet. Rec. 2023, 192, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.B.; von Samson-Himmelstjerna, G. Haemonchus contortus and Haemonchosis—Past, Present and Future Trends; Academic Press: New York, NY, USA, 2016. [Google Scholar]
- Laing, R.; Kikuchi, T.; Martinelli, A.; Tsai, I.J.; Beech, R.N.; Redman, E.; Holroyd, N.; Bartley, D.J.; Beasley, H.; Britton, C.; et al. The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome Biol. 2013, 14, R88. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.M.; Korhonen, P.K.; Campbell, B.E.; Young, N.D.; Jex, A.R.; Jabbar, A.; Hall, R.S.; Mondal, A.; Howe, A.C.; Pell, J.; et al. The genome and developmental transcriptome of the strongylid nematode Haemonchus contortus. Genome Biol. 2013, 14, R89. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.R.; Tracey, A.; Laing, R.; Holroyd, N.; Bartley, D.; Bazant, W.; Beasley, H.; Beech, R.; Britton, C.; Brooks, K.; et al. Genomic and transcriptomic variation defines the chromosome-scale assembly of Haemonchus contortus, a model gastrointestinal worm. Commun. Biol. 2020, 3, 656. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ma, G.; Ang, C.S.; Korhonen, P.K.; Koehler, A.V.; Young, N.D.; Nie, S.; Williamson, N.A.; Gasser, R.B. High throughput LC-MS/MS-based proteomic analysis of excretory-secretory products from short-term in vitro culture of Haemonchus contortus. J. Proteom. 2019, 204, 103375. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ma, G.; Nie, S.; Williamson, N.A.; Reid, G.E.; Gasser, R.B. Lipid composition and abundance in the reproductive and alimentary tracts of female Haemonchus contortus. Parasit. Vectors 2020, 13, 338. [Google Scholar] [CrossRef] [PubMed]
- Sallé, G.; Doyle, S.R.; Cortet, J.; Cabaret, J.; Berriman, M.; Holroyd, N.; Cotton, J.A. The global diversity of Haemonchus contortus is shaped by human intervention and climate. Nat. Commun. 2019, 10, 4811. [Google Scholar] [CrossRef]
- Jex, A.R.; Hu, M.; Littlewood, D.T.; Waeschenbach, A.; Gasser, R.B. Using 454 technology for long-PCR based sequencing of the complete mitochondrial genome from single Haemonchus contortus (Nematoda). BMC Genom. 2008, 9, 11. [Google Scholar] [CrossRef]
- Palevich, N.; Maclean, P.; Baten, A.; Scott, R.; Leathwick, D.M. The complete mitochondrial genome of the New Zealand parasitic roundworm Haemonchus contortus (Trichostrongyloidea: Haemonchidae) field strain NZ_Hco_NP. Mitochondrial DNA B Resour. 2019, 4, 2208–2210. [Google Scholar] [CrossRef] [PubMed]
- Blouin, M.S.; Yowell, C.A.; Courtney, C.H.; Dame, J.B. Host movement and the genetic structure of populations of parasitic nematodes. Genetics 1995, 141, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genomics of parasitic nematodes of socio-economic importance: Recent progress, and implications for population genetics and systematics. Adv. Parasitol. 2004, 56, 133–212. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Gasser, R.B. Mitochondrial genomes of parasitic nematodes-progress and perspectives. Trends Parasitol. 2006, 22, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Brasil, B.S.; Nunes, R.L.; Bastianetto, E.; Drummond, M.G.; Carvalho, D.C.; Leite, R.C.; Molento, M.B.; Oliveira, D.A. Genetic diversity patterns of Haemonchus placei and Haemonchus contortus populations isolated from domestic ruminants in Brazil. Int. J. Parasitol. 2012, 42, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Gasser, R.B.; Li, F.; Bao, M.; Huang, W.; Zou, F.; Zhao, G.; Wang, C.; Yang, X.; Zhou, Y.; et al. Genetic variability within and among Haemonchus contortus isolates from goats and sheep in China. Parasit. Vectors 2013, 6, 279. [Google Scholar] [CrossRef] [PubMed]
- Pitaksakulrat, O.; Chaiyasaeng, M.; Artchayasawat, A.; Eamudomkarn, C.; Boonmars, T.; Kopolrat, K.Y.; Prasopdee, S.; Petney, T.N.; Blair, D.; Sithithaworn, P. Genetic diversity and population structure of Haemonchus contortus in goats from Thailand. Infect. Genet. Evol. 2021, 95, 105021. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Gasser, R.B.; Wang, T.; Korhonen, P.K.; Young, N.D. Toward integrative ‘omics of the barber’s pole worm and related parasitic nematodes. Infect. Genet. Evol. 2020, 85, 104500. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, T.; Korhonen, P.K.; Ang, C.S.; Williamson, N.A.; Young, N.D.; Stroehlein, A.J.; Hall, R.S.; Koehler, A.V.; Hofmann, A.; et al. Molecular alterations during larval development of Haemonchus contortus in vitro are under tight post-transcriptional control. Int. J. Parasitol. 2018, 48, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, T.; Korhonen, P.K.; Stroehlein, A.J.; Young, N.D.; Gasser, R.B. Dauer signalling pathway model for Haemonchus contortus. Parasit. Vectors 2019, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Redman, E.; Grillo, V.; Saunders, G.; Packard, E.; Jackson, F.; Berriman, M.; Gilleard, J.S. Genetics of mating and sex determination in the parasitic nematode Haemonchus contortus. Genetics 2008, 180, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Redman, E.; Sargison, N.; Whitelaw, F.; Jackson, F.; Morrison, A.; Bartley, D.J.; Gilleard, J.S. Introgression of ivermectin resistance genes into a susceptible Haemonchus contortus strain by multiple backcrossing. PLoS Pathog. 2012, 8, e1002534. [Google Scholar] [CrossRef] [PubMed]
- Rezansoff, A.M.; Laing, R.; Martinelli, A.; Stasiuk, S.; Redman, E.; Bartley, D.; Holroyd, N.; Devaney, E.; Sargison, N.D.; Doyle, S.; et al. The confounding effects of high genetic diversity on the determination and interpretation of differential gene expression analysis in the parasitic nematode Haemonchus contortus. Int. J. Parasitol. 2019, 49, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Sargison, N.D.; Redman, E.; Morrison, A.A.; Bartley, D.J.; Jackson, F.; Hoberg, E.; Gilleard, J.S. Mating barriers between genetically divergent strains of the parasitic nematode Haemonchus contortus suggest incipient speciation. Int. J. Parasitol. 2019, 49, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Taki, A.C.; Byrne, J.J.; Wang, T.; Sleebs, B.E.; Nguyen, N.; Hall, R.S.; Korhonen, P.K.; Chang, B.C.H.; Jackson, P.; Jabbar, A.; et al. High-throughput phenotypic assay to screen for anthelmintic activity on Haemonchus contortus. Pharmaceuticals 2021, 14, 616. [Google Scholar] [CrossRef] [PubMed]
- Taki, A.C.; Wang, T.; Nguyen, N.N.; Ang, C.S.; Leeming, M.G.; Nie, S.; Byrne, J.J.; Young, N.D.; Zheng, Y.; Ma, G.; et al. Thermal proteome profiling reveals Haemonchus orphan protein HCO_011565 as a target of the nematocidal small molecule UMW-868. Front. Pharmacol. 2022, 13, 1014804. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.A.; Chaudhry, U.; Andrews, L.; Melville, L.; Doyle, S.R.; Sargison, N.D.; Bartley, D.J. Phenotypic and genotypic analysis of benzimidazole resistance in reciprocal genetic crosses of Haemonchus contortus. Int. J. Parasitol. Drugs Drug Resist. 2022, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ma, G.; Wang, T.; Hofmann, A.; Song, J.; Gasser, R.B.; Young, N.D. Ubiquitination pathway model for the barber’s pole worm-Haemonchus contortus. Int. J. Parasitol. 2022, 52, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Young, N.D.; Song, J.; Chang, B.C.H.; Gasser, R.B. An informatic workflow for the enhanced annotation of excretory/secretory proteins of Haemonchus contortus. Comput. Struct. Biotechnol. J. 2023, 21, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Young, N.D.; Song, J.; Gasser, R.B. Genome-wide analysis of Haemonchus contortus proteases and protease inhibitors using advanced informatics provides insights into parasite biology and host-parasite interactions. Int. J. Mol. Sci. 2023, 24, 12320. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F. The anatomy and life-history of the Haemonchus contortus (Rud.). Rep. Dir. Vet. Res. 1915, 3, 347–500. [Google Scholar]
- Das, K.M.; Whitlock, J.H. Subspeciation in Haemonchus contortus (Rudolphi, 1803) Nemata, Trichostrongyloidea. Cornell. Vet. 1960, 50, 182–197. [Google Scholar] [PubMed]
- Lichtenfels, J.R.; Pilitt, P.A.; Hoberg, E.P. New morphological characters for identifying individual specimens of Haemonchus spp. (Nematoda: Trichostrongyloidea) and a key to species in ruminants of North America. J. Parasitol. 1994, 80, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Rahman, W.A.; Abd Hamid, S. Morphological characterization of Haemonchus contortus in goats (Capra hircus) and sheep (Ovis aries) in Penang, Malaysia. Trop. Biomed. 2007, 24, 23–27. [Google Scholar] [PubMed]
- Hunt, P.W.; Knox, M.R.; Le Jambre, L.F.; McNally, J.; Anderson, L.J. Genetic and phenotypic differences between isolates of Haemonchus contortus in Australia. Int. J. Parasitol. 2008, 38, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ortiz-de-Montellano, C.; Torres-Acosta, J.F.J.; Sandoval-Castro, C.A.; Fourquaux, I.; Hoste, H. Scanning electron microscopy of different vulval structures in a Mexican Haemonchus contortus isolate. Vet. Parasitol. Reg. Stud. Rep. 2021, 26, 100640. [Google Scholar] [CrossRef] [PubMed]
- Vongnady, K.; Rucksaken, R.; Mangkit, B. Cuticular ridge patterns applied for identifying adult female worms of Haemonchus species with various vulvar morphological types in infected native goats in Laos. Trop. Biomed. 2021, 38, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Lemire, B.D. Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 2002, 291, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Lemire, B.D. The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochim. Biophys. Acta 2003, 1638, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Estes, S.; Dietz, Z.P.; Katju, V.; Bergthorsson, U. Evolutionary codependency: Insights into the mitonuclear interaction landscape from experimental and wild Caenorhabditis nematodes. Curr. Opin. Genet. Dev. 2023, 81, 102081. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Thompson, O.; Waterston, R.H.; Moerman, D.G.; Keightley, P.D.; Bergthorsson, U.; Katju, V. Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol. Biol. Evol. 2017, 34, 1319–1334. [Google Scholar] [CrossRef]
- Kinkar, L.; Gasser, R.B.; Webster, B.L.; Rollinson, D.; Littlewood, D.T.J.; Chang, B.C.H.; Stroehlein, A.J.; Korhonen, P.K.; Young, N.D. Nanopore sequencing resolves elusive long tandem-repeat regions in mitochondrial genomes. Int. J. Mol. Sci. 2021, 22, 1811. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Tomiczek, B.; Sojo, V.; dos Reis, M. A beginners guide to estimating the non-synonymous to synonymous rate ratio of all protein-coding genes in a genome. Methods Mol. Biol. 2015, 1201, 65–90. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).