
Effect of Mifepristone on Migration and Proliferation of Oral Cancer Cells
Abstract
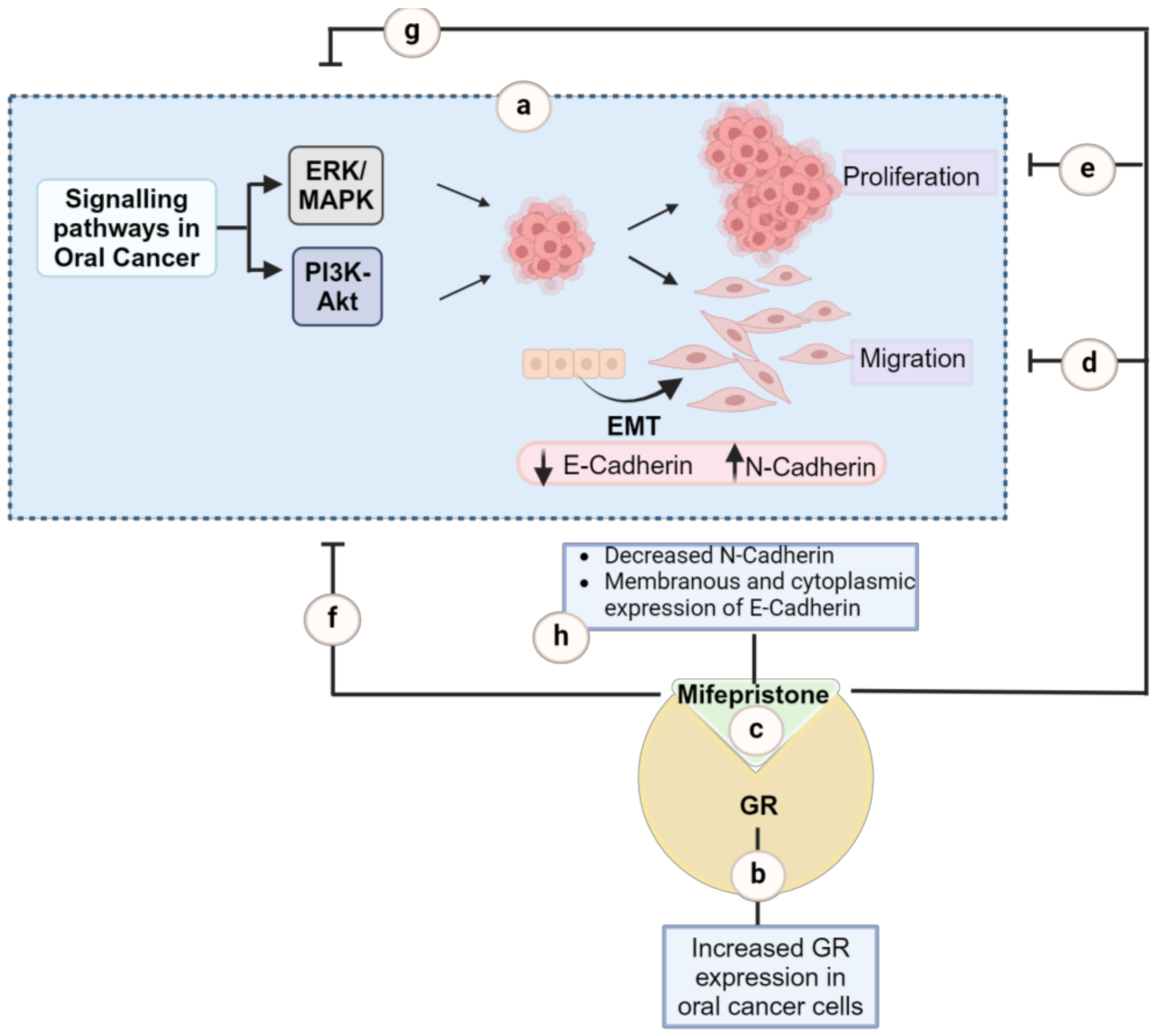
:1. Introduction
2. Results
2.1. TYS and SAS-H1 Cells Showed Increased Expression of GR with a Pronounced Nuclear Presence in Comparison to HaCaT Cells
2.2. Effect of Mifepristone on Expression and Localisation of the GR
2.3. Mifepristone Resulted in Reduced Cell Proliferation in a Dose-Dependent Manner in All Cell Lines
2.4. High Concentrations of Mifepristone Resulted in Decreased Collective Cell Migration and Compact Colonies of Oral Cancer Cells
2.5. Effect of Mifepristone on the PI3K-Akt Signalling Pathway
2.6. Effect of Mifepristone on the Phospho p44/42 MAPK Signalling Pathway
2.7. Effect of Mifepristone on EMT Markers
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture, and Conditions
4.2. Experimental Conditions
4.3. Scratch Assay
4.4. Scatter Assay
4.5. Cell Lysis, SDS-PAGE, and Western Blot
4.6. Proliferation Assay (MTT)
4.7. Immunofluorescence (IF)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Wang, J.; Shao, J.; Gao, Y.; Xu, J.; Yu, S.; Liu, Z.; Jia, L. The Unique Pharmacological Characteristics of Mifepristone (RU486): From Terminating Pregnancy to Preventing Cancer Metastasis. Med. Res. Rev. 2014, 34, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.; Ganatra, B.; Althabe, F. WHO Essential Medicines For Reproductive Health. BMJ Glob. Health 2019, 4, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Dalm, S.; Karssen, A.M.; Meijer, O.C.; Belanoff, J.K.; de Kloet, E.R. Resetting the Stress System with a Mifepristone Challenge. Cell. Mol. Neurobiol. 2019, 39, 503–522. [Google Scholar] [CrossRef]
- Llaguno-Munive, M.; Vazquez-Lopez, M.I.; Jurado, R.; Garcia-Lopez, P. Mifepristone Repurposing in Treatment of High-Grade Gliomas. Front. Oncol. 2021, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Lu, Y.; Li, Y.; Xie, H.; Zhou, G.; Jia, L. Similarities and Differences Between Embryonic Implantation and CTC Invasion: Exploring the Roles of Abortifacients in Cancer Metastasis Chemoprevention. Eur. J. Med. Chem. 2022, 237, 114416. [Google Scholar] [CrossRef] [PubMed]
- Pinski, J.; Halmos, G.; Shirahige, Y.; Wittliff, J.L.; Schally, A.V. Inhibition of Growth of the Human Malignant Glioma Cell Line (U87MG) by the Steroid Hormone Antagonist RU486. J. Clin. Endocrinol. Metab. 1993, 77, 1388–1392. [Google Scholar] [PubMed]
- Brandhagen, B.A.N.; Tieszen, C.R.; Ulmer, T.M.; Tracy, M.S.; Goyeneche, A.A.; Telleria, C.M. Cytostasis and Morphological Changes Induced by Mifepristone in Human Metastatic Cancer Cells Involve Cytoskeletal Filamentous Actin Reorganization and Impairment of Cell Adhesion Dynamics. BMC Cancer 2013, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Wempe, S.L.; Gamarra-Luques, C.D.; Telleria, C.M. Synergistic Lethality of Mifepristone and LY294002 in Ovarian Cancer Cells. Cancer Growth Metastasis 2013, 6, 1–13. [Google Scholar] [CrossRef]
- Spitz, I.M.; Grunberg, S.M.; Chabbert-Buffet, N.; Lindenberg, T.; Gelber, H.; Sitruk-Ware, R. Management of Patients Receiving Long-Term Treatment with Mifepristone. Fertil. Steril. 2005, 84, 1719–1726. [Google Scholar] [CrossRef]
- Grunberg, S.M.; Weiss, M.H.; Russell, C.A.; Spitz, I.M.; Ahmadi, J.; Sadun, A.; Sitruk-Ware, R. Long-Term Administration of Mifepristone (RU486): Clinical Tolerance During Extended Treatment of Meningioma. Cancer Investig. 2009, 24, 727–733. [Google Scholar] [CrossRef]
- Check, J.; Check, D. Mifepristone Extends Both Length and Quality of Life in a Patient With Advanced Non-small Cell Lung Cancer that Has Progressed Despite Chemotherapy and a Check-point Inhibitor. Anticancer Res. 2019, 39, 1923–1926. [Google Scholar] [CrossRef] [PubMed]
- Check, J.; Dix, E. Mifepristone May Halt Progression of Extensively Metastatic Human Adenocarcinoma of the Colon: Case report. Anticancer Res. 2009, 29, 1611–1613. [Google Scholar] [PubMed]
- Fauvet, R.; Etienne, C.D.; Poncelet, C.; Bringuier, A.F.; Feldmann, G.; Daraï, E. Effects of Progesterone and Anti-progestin (mifepristone) Treatment on Proliferation and Apoptosis of the Human Ovarian Cancer Cell Line, OVCAR-3. Oncol. Rep. 2006, 15, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Lombardi, G.; Farina, P.; Kalamarides, M.; Sanson, M. Successful Treatment of Multiple Intracranial Meningiomas with the Antiprogesterone Receptor Agent Mifepristone (RU486). Acta Neurochir. 2014, 156, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Ponikwicka-Tyszko, D.; Chrusciel, M.; Stelmaszewska, J.; Bernaczyk, P.; Chrusciel, P.; Sztachelska, M.; Scheinin, M.; Bidzinski, M.; Szamatowicz, J.; Huhtaniemi, I.T.; et al. Molecular Mechanisms Underlying Mifepristone’s Agonistic Action on Ovarian Cancer Progression. EBioMedicine 2019, 47, 170–183. [Google Scholar] [CrossRef]
- Tieszen, C.R.; Goyeneche, A.A.; Brandhagen, B.A.N.; Ortbahn, C.T.; Telleria, C.M. Antiprogestin Mifepristone Inhibits the Growth of Cancer Cells of Reproductive and Non-Reproductive Origin Regardless of Progesterone Receptor Expression. BMC Cancer 2011, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Li, Q.; Tie, Y.; Alu, A.; Ma, X.; Shi, H. Targeted Therapy for Head and Neck Cancer: Signaling Pathways and Clinical Studies. Signal. Transduct. Target. Ther. 2023, 8, 31. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.J.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A. Pembrolizumab Alone or with Chemotherapy Versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S. Causes of Oral Cancer—An Appraisal of Controversies. Br. Dent. J. 2009, 207, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Semple, C.; Parahoo, K.; Norman, A.; Mccaughan, E.; Humphris, G.; Mills, M. Psychosocial Interventions for Patients with Head and Neck Cancer. Cochrane Database Syst. Rev. 2013, 7, CD009441. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.A.C.; Wand, G. Stress and the HPA Axis: Role of Glucocorticoids in Alcohol Dependence. Alcohol Res. 2012, 34, 468–483. [Google Scholar] [PubMed]
- Iftikhar, A.; Islam, M.; Shepherd, S.; Jones, S.; Ellis, I. What is Behind the Lifestyle Risk Factors for Head and Neck Cancer? Front. Psychol. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17–28. [Google Scholar] [CrossRef]
- Wu, J.-S.; Jiang, J.; Chen, B.-J.; Wang, K.; Tang, Y.-L.; Liang, X.-H. Plasticity of Cancer Cell Invasion: Patterns and Mechanisms. Transl. Oncol. 2021, 14, 100899. [Google Scholar] [CrossRef]
- Clark, A.G.; Vignjevic, D.M. Modes of Cancer Cell Invasion and the Role of the Microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef]
- Costa, L.C.M.C.; Leite, C.F.; Cardoso, S.V.; Loyola, A.M.; de Faria, P.R.; Souza, P.E.A.; Horta, M.C.R. Expression of Epithelial-Mesenchymal Transition Markers at the Invasive Front of Oral Squamous Cell Carcinoma. J. Appl. Oral Sci. 2015, 23, 169–178. [Google Scholar] [CrossRef]
- Li, D.-Q. Effects of Mifepristone on Invasive and Metastatic Potential of Human Gastric Adenocarcinoma Cell Line MKN-45 In Vitro and In Vivo. World J. Gastroenterol. 2004, 10, 1726–1729. [Google Scholar] [CrossRef]
- Llaguno-Munive, M.; León-Zetina, S.; Vazquez-Lopez, I.; Ramos-Godinez, M.D.P.; Medina, L.A.; Garcia-Lopez, P. Mifepristone as a Potential Therapy to Reduce Angiogenesis and P-Glycoprotein Associated With Glioblastoma Resistance to Temozolomide. Front. Oncol. 2020, 10, 581814. [Google Scholar] [CrossRef]
- Zhang, X.W.; Li, Y.; Liu, J.J.; Liu, X.; Wang, Z.L.; Hu, B. Glucocorticoid Receptor Subunit Gene Expression in Parotid Gland and Adenomas. Otolaryngol. Head Neck Surg. 2006, 135, 849–852. [Google Scholar] [CrossRef]
- Block, T.S.; Murphy, T.I.; Munster, P.N.; Nguyen, D.P.; Lynch, F.J. Glucocorticoid Receptor Expression in 20 Solid Tumour Types Using Immhunohistochemistry Assay. Cancer Manag. Res. 2017, 9, 65–72. [Google Scholar] [CrossRef]
- Riki, K.; Ota, M. Cytosol Glucocorticoid Receptor in the Neoplastic Epithelial Duct Cell Line From Human Salivary Gland (HSG). Biochem. Int. 1988, 13, 671–679. [Google Scholar]
- Kurokawa, R.; Kyakumoto, S.; Hatakeyama, S.; Ota, M. Subcellular Distribution of Glucocorticoid Receptor in the Neoplastic Epithelial Duct Cell Line From Human Salivary Gland. Biochem. Int. 1987, 14, 37–44. [Google Scholar] [PubMed]
- Kurokawa, R.; Ota, M. Nonactivated and Activated Glucocorticoid Receptor Complexes From Human Salivary Gland Adenocarcinoma Cell Line. BBA Mol. Cell Res. 1988, 970, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Chen, Y.; Huang, J.; Zhang, Y.; Sun, C.K.; Shen, Y.Q. Glucocorticoid Reduces the Efficacy of Afatinib on the Head and Neck Squamous Cell Carcinoma. Biocell 2023, 47, 329–338. [Google Scholar] [CrossRef]
- Pan, C.; Kang, J.; Hwang, J.S.; Li, J.; Boese, A.C.; Wang, X.; Yang, L.; Boggon, T.J.; Chen, G.Z.; Saba, N.F.; et al. Cisplatin-Mediated Activation of Glucocorticoid Receptor Induces Platinum Resistance via MAST1. Nat. Commun. 2021, 12, 4960. [Google Scholar] [CrossRef]
- Petti, S. Lifestyle Risk Factors for Oral Cancer. Oral Oncol. 2009, 45, 340–350. [Google Scholar] [CrossRef]
- Buchmann, L.; Conlee, J.; Hunt, J.; Agarwal, J.; White, S. Psychosocial Distress is Prevalent in Head and Neck Cancer Patients. Laryngoscope 2013, 123, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Spiga, F.; Knight, D.M.; Droste, S.K.; Conway-Campbell, B.; Kershaw, Y.; MacSweeney, C.P.; Thomson, F.J.; Craighead, M.; Peeters, B.W.M.M.; Lightman, S.L. Differential Effect of Glucocorticoid Receptor Antagonists on Glucocorticoid Receptor Nuclear Translocation and DNA Binding. J. Psychopharmacol. 2011, 25, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jonklaas, J.; Danielsen, M. The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids 2007, 72, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Chen, J.; Liu, W.; Liu, J.; Li, T.; Chen, H.; Wang, J.; Jia, L. Mifepristone Inhibits Ovarian Cancer Metastasis by Intervening in SDF-1/CXCR4 Chemokine Axis. Oncotarget 2017, 8, 59123–59135. [Google Scholar] [CrossRef]
- Yu, S.; Yan, C.; Yang, X.; He, S.; Liu, J.; Qin, C.; Huang, C.; Lu, Y.; Tian, Z.; Jia, L. Pharmacoproteomic Analysis Reveals that Metapristone (RU486 metabolite) Intervenes E-cadherin and Vimentin to Realize Cancer Metastasis Chemoprevention. Sci. Rep. 2016, 6, 22388. [Google Scholar] [CrossRef] [PubMed]
- Borm, B.; Requardt, R.P.; Herzog, V.; Kirfel, G. Membrane Ruffles in Cell Migration: Indicators of Inefficient Lamellipodia Adhesion and Compartments of Actin Filament Reorganization. Exp. Cell Res. 2005, 302, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Chen, J.; Liu, W.; Liu, J.; Jia, L. Metapristone (RU486 derivative) Inhibits Cell Proliferation and Migration as Melanoma Metastatic Chemopreventive Agent. Biomed. Pharmacother. Pharmacother. 2017, 90, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.Z.; Yan, L.; Zhang, H.; Li, M.J.; Zhang, X.H.; Zhao, X.X. Mifepristone Inhibits the Migration of Endometrial Cancer Cells Through Regulating H19 Methylation. Zhonghua Zhong Liu Za Zhi. 2016, 38, 411–416. [Google Scholar] [PubMed]
- Devaraja, K.; Pillai, S.; Valiathan, M.; Geetha, V.; Pujary, K. E-Cadherin Expression Pattern in Head and Neck Squamous Cell Carcinoma and its Association with Clinico-Pathological Predictors. Egypt. J. Otolaryngol. 2023, 39, 138. [Google Scholar] [CrossRef]
- Ritch, S.J.; Brandhagen, B.N.; Goyeneche, A.A.; Telleria, C.M. Advanced Assessment of Migration and Invasion of Cancer Cells in Response to Mifepristone Therapy Using Double Fluorescence Cytochemical Labeling. BMC Cancer 2019, 19, 376. [Google Scholar] [CrossRef]
- Goyeneche, A.A.; Carón, R.W.; Telleria, C.M. Mifepristone Inhibits Ovarian Cancer Cancer Cell Growth In Vitro and In Vivo. Clin. Cancer Res. 2007, 13, 3370. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P.B.; Laskaris, A.; Goyeneche, A.A.; Chen, Y.; Telleria, C.M.; Burnier, J.V. Anticancer Effects of Mifepristone on Human Uveal Melanoma Cells. Cancer Cell Int. 2021, 21, 1–15. [Google Scholar] [CrossRef]
- Ponandai-Srinivasan, S.; Lalitkumar, P.G.; Garcia, L.; Varghese, S.J.; Carlson, J.W.; Gemzell-Danielsson, K.; Radestad, A.F. Mifepristone Mediates Anti-Proliferative Effect on Ovarian Mesenchymal Stem/Stromal Cells From Female BRCA 1−/2− Carriers. Acta Obstet. Gynecol. Scand. 2019, 98, 250–261. [Google Scholar] [CrossRef]
- Casulari, L.A.; Dondi, D.; Pratesi, G.; Piva, F.; Milani, M.; Piccolella, M.; Maggi, R. Antiproliferative Effect of Mifepristone (RU486) on Human Neuroblastoma Cells (SK-N-SH): In Vitro and In Vivo Studies. Braz. J. Med. Biol. Res. 2020, 53, e10067. [Google Scholar] [CrossRef]
- Mitani, Y.; Lin, S.H.; Pytynia, K.B.; Ferrarotto, R.; El-Naggar, A.K. Reciprocal and Autonomous Glucocorticoid and Androgen Receptor Activation in Salivary Duct Carcinoma. Clin. Cancer Res. 2020, 26, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.A.; Golden, B.; Yan, L.; Carroll, W.R.; Helman, E.E.; Rosenthal, E.L. Disruption of the AKT pathway inhibits metastasis in an orthotopic model of head and neck squamous cell carcinoma. Laryngoscope 2011, 121, 2359–2365. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Alzawi, A.; Iftikhar, A.; Shalgm, B.; Jones, S.; Ellis, I.; Islam, M. Receptor, Signal, Nucleus, Action: Signals That Pass through Akt on the Road to Head and Neck Cancer Cell Migration. Cancers 2022, 14, 2606. [Google Scholar] [CrossRef] [PubMed]
- Thwe, A.M.; Mossey, P.; Ellis, I.R. Effect of tyrosine kinase inhibitors on cell migration and epithelial-to-mesenchymal transition in Asian head and neck cancer cell lines. J. Oral Pathol. Med. 2021, 50, 1031–1039. [Google Scholar] [CrossRef]
- Rosen, L.S.; LoRusso, P.; Ma, W.W.; Goldman, J.W.; Weise, A.; Colevas, A.D.; Adjei, A.; Yazji, S.; Shen, A.; Johnston, S.; et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 604–613. [Google Scholar] [CrossRef]
- Shao, J.; Zheng, G.; Chen, H.; Liu, J.; Xu, A.; Chen, F.; Li, T.; Lu, Y.; Xu, J.; Zheng, N. Metapristone (RU486 metabolite) Suppresses NSCLC by Targeting EGFR-Mediated PI3K/AKT Pathway. Oncotarget 2017, 8, 78351. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Shen, Z.; Chen, H.; Liu, J.; Jiang, K.; Fan, L.; Jia, L.; Shao, J. Metapristone Suppresses Non-Small Cell Lung Cancer Proliferation and Metastasis via Modulating RAS/RAF/MEK/MAPK Signaling Pathway. Biomed. Pharmacother. 2017, 90, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Lu, D.; Zhang, J.; Du, S.; Zhao, X. Mifepristone Inhibits Proliferation, Migration and Invasion of HUUA Cells and Promotes its Apoptosis by Regulation of FAK and PI3K/AKT Signaling Pathway. OncoTargets Ther. 2018, 11, 5441–5449. [Google Scholar] [CrossRef]
- Clarisse, D.; De Bosscher, K. How the Glucocorticoid Receptor Contributes to Platinum-Based Therapy Resistance in Solid Cancer. Nat. Commun. 2021, 12, 4959. [Google Scholar] [CrossRef] [PubMed]
- Veneris, J.T.; Hou, X.; Weroha, S.J.; Heinzen, E.P.; Maurer, M.J.; Oberg, A.L.; Conzen, S.D.; Fleming, G.F. Selective and Nonselective GR Antagonists in Combination with Chemotherapy in Ovarian Cancer PDX Models. JCO 2019, 37, e17039. [Google Scholar] [CrossRef]
- Yue, P.Y.K.; Leung, E.P.Y.; Mak, N.K.; Wong, R.N.S. A Simplified Method for Quantifying Cell Migration/Wound Healing in 96-Well Plates. J. Biomol. Screen. 2010, 15, 427–433. [Google Scholar] [CrossRef]
- Alkhadar, H.; Macluskey, M.; White, S.; Ellis, I. Nerve Growth Factor-Induced Migration in Oral and Salivary Gland Tumour Cells Utilises the PI3K/Akt Signalling Pathway: Is There a Link to Perineural Invasion? J. Oral Pathol. Med. 2020, 49, 227–234. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | |
|---|---|
| Cell lines |
|
| Mifepristone | # M8046, sourced from Sigma-Aldrich. Reconstituted at 20 mg/mL in DMSO. Used in µM concentrations of 1, 5, 10, and 20 in serum-free media, (SFM). |
| Primary antibodies | pAkt S473 # 4060 (WB 1:2000, ICC 1:400), pAkt T308 #2965L (WB 1:1000, ICC 1:1600), p42/p44 pMAPK #4370 (WB 1:2000, ICC 1:400), Glucocorticoid receptor #12041 (WB 1:1000, ICC 1:200), E-Cadherin #3195 S (WB 1:1000, ICC 1:200), N-Cadherin #13116 S (WB 1:1000, ICC 1:800) All antibodies sourced from Cell Signaling Technology (Danvers, MA, USA) |
| Secondary antibodies | #7074 for Western blot, (1:2000) IgG Fab 2 Alexa Fluor for IF (1:1000) (Sourced from Cell Signaling Technology, Danvers, MA, USA) |
| DAPI (4′,6-diamidino-2-phenylindole) | # 4083, To stain cell nuclei in Immunocytochemistry (ICC), sourced from Cell Signaling Technology (Danvers, MA, USA) |
| Methods | |
| Cell migration | Scratch assay Scatter assay |
| Cell Proliferation | MTT assay ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium) assay (24 and 48 h) |
| Protein expression and localisation | Western blot Immunofluorescence (IF) |
| Software and Microscope | |
| Image Lab | To visualise and quantify the SDS-PAGE and Western blot image |
| Image J | To quantify the scratch assay |
| Photoshop | To merge DAPI and labelled antibody images |
| Microscope | Light and fluorescence microscope IX70; digital camera XM10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iftikhar, A.; Shepherd, S.; Jones, S.; Ellis, I. Effect of Mifepristone on Migration and Proliferation of Oral Cancer Cells. Int. J. Mol. Sci. 2024, 25, 8777. https://doi.org/10.3390/ijms25168777
Iftikhar A, Shepherd S, Jones S, Ellis I. Effect of Mifepristone on Migration and Proliferation of Oral Cancer Cells. International Journal of Molecular Sciences. 2024; 25(16):8777. https://doi.org/10.3390/ijms25168777
Chicago/Turabian StyleIftikhar, Anem, Simon Shepherd, Sarah Jones, and Ian Ellis. 2024. "Effect of Mifepristone on Migration and Proliferation of Oral Cancer Cells" International Journal of Molecular Sciences 25, no. 16: 8777. https://doi.org/10.3390/ijms25168777