
Activation of the 5-HT1A Receptor by Eltoprazine Restores Mitochondrial and Motor Deficits in a Drosophila Model of Fragile X Syndrome
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
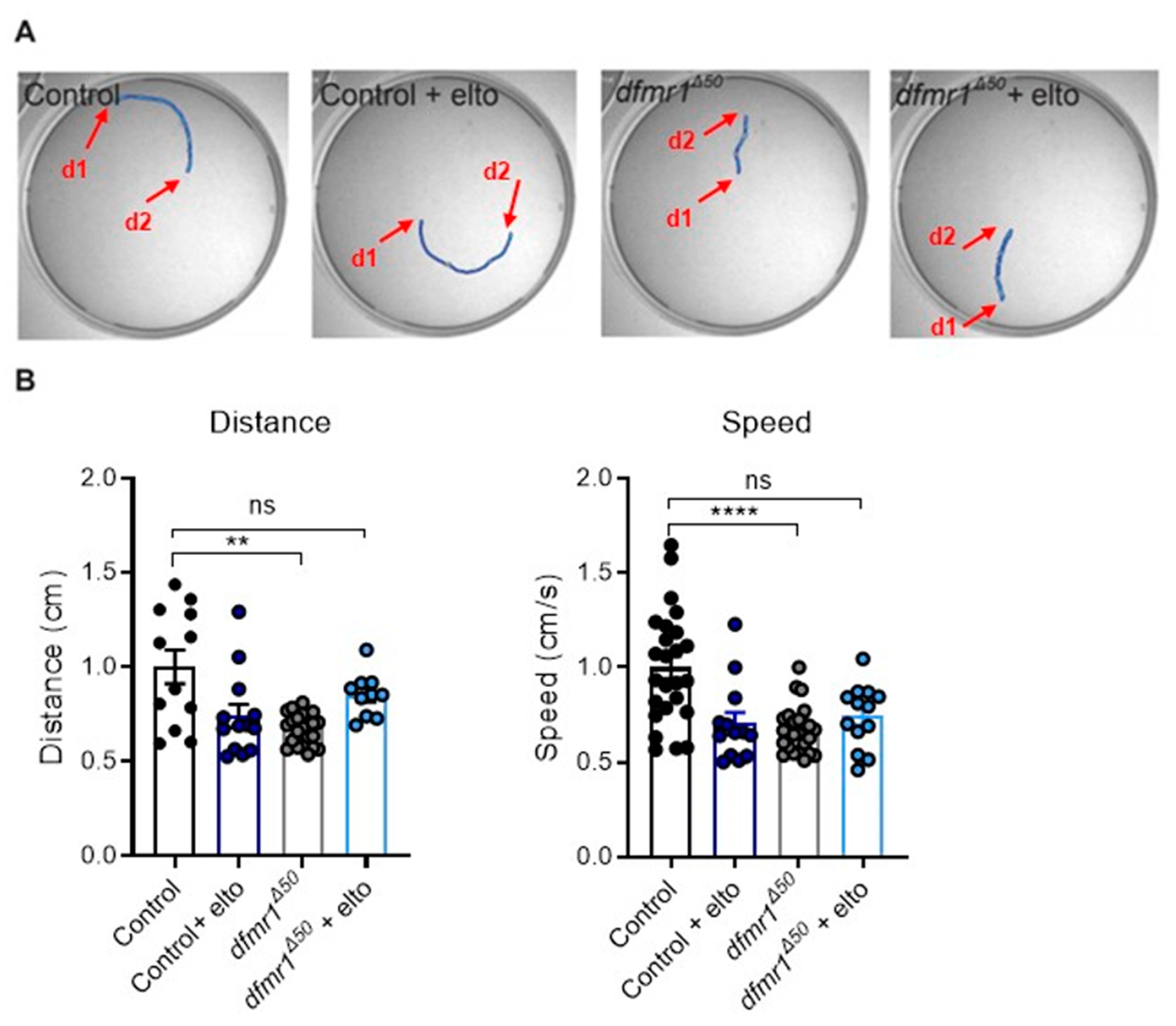
2.1. dfmr1Δ50 Mutants Have Locomotion Deficits That Are Rescued upon Eltoprazine Treatment
2.2. Eltoprazine Restores the Synaptic Transmission at the NMJ of dfmr1Δ50 Mutants
2.3. Eltoprazine Restores Mitochondrial Dysregulations in dfmr1Δ50 Mutants
2.4. Mitochondrial Biogenesis Regulates Locomotor Behavior in dfmr1Δ50 Mutants
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks
4.2. Larval Collection and Treatment
4.3. Larval Crawling Behavior
4.4. Electrophysiology
4.5. Immunohistochemistry, Confocal Microscopy, and Image Analysis
4.6. Mitochondrial Function Assays
4.7. RNA Extraction and RT-qPCR
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kann, O.; Kovacs, R. Mitochondria and Neuronal Activity. AJP Cell Physiol. 2006, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Flippo, K.H.; Strack, S. Mitochondrial Dynamics in Neuronal Injury, Development and Plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef]
- Raefsky, S.M.; Mattson, M.P. Adaptive Responses of Neuronal Mitochondria to Bioenergetic Challenges: Roles in Neuroplasticity and Disease Resistance. Free Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef]
- Khacho, M.; Slack, R.S. Mitochondrial Dynamics in the Regulation of Neurogenesis: From Development to the Adult Brain. Dev. Dyn. 2018, 247, 47–53. [Google Scholar] [CrossRef]
- Morella, I.M.; Brambilla, R.; Morè, L. Emerging Roles of Brain Metabolism in Cognitive Impairment and Neuropsychiatric Disorders. Neurosci. Biobehav. Rev. 2022, 142, 104892. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.; Redondo-Flórez, L.; Beltrán-Velasco, A.; Ramos-Campo, D.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Iwata, R.; Vanderhaeghen, P. Regulatory Roles of Mitochondria and Metabolism in Neurogenesis. Curr. Opin. Neurobiol. 2021, 69, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Casimir, P.; Erkol, E.; Boubakar, L.; Planque, M.; Gallego Lopez, I.M.; Ditkowska, M.; Gaspariunaite, V.; Beckers, S.; Remans, D.; et al. Mitochondria Metabolism Sets the Species-Specific Tempo of Neuronal Development. Science 2023, 379, eabn4705. [Google Scholar] [CrossRef]
- Sachdev, S.; Ansari, S.A.; Ansari, M.I. Generation and Fate of ROS in Mitochondria. In Reactive Oxygen Species in Plants; Springer Nature: Singapore, 2023; pp. 93–106. ISBN 978-981-19988-3-6. [Google Scholar]
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Rangaraju, V.; Tom Dieck, S.; Schuman, E.M. Local Translation in Neuronal Compartments: How Local Is Local? EMBO Rep. 2017, 18, 693–711. [Google Scholar] [CrossRef] [PubMed]
- Bapat, O.; Purimetla, T.; Kruessel, S.; Shah, M.; Fan, R.; Thum, C.; Rupprecht, F.; Langer, J.D.; Rangaraju, V. VAP Spatially Stabilizes Dendritic Mitochondria to Locally Support Synaptic Plasticity. Nat. Commun. 2024, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Winkle, C.C.; Taylor, K.L.; Dent, E.W.; Gallo, G.; Greif, K.F.; Gupton, S.L. Beyond the Cytoskeleton: The Emerging Role of Organelles and Membrane Remodeling in the Regulation of Axon Collateral Branches. Dev. Neurobiol. 2016, 76, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Semperboni, S.; Cavaletti, G.; Scuteri, A. Neurons: The Interplay between Cytoskeleton, Ion Channels/Transporters and Mitochondria. Cells 2022, 11, 2499. [Google Scholar] [CrossRef]
- Bustamante-Barrientos, F.A.; Luque-Campos, N.; Araya, M.J.; Lara-Barba, E.; De Solminihac, J.; Pradenas, C.; Molina, L.; Herrera-Luna, Y.; Utreras-Mendoza, Y.; Elizondo-Vega, R.; et al. Mitochondrial Dysfunction in Neurodegenerative Disorders: Potential Therapeutic Application of Mitochondrial Transfer to Central Nervous System-Residing Cells. J. Transl. Med. 2023, 21, 613. [Google Scholar] [CrossRef] [PubMed]
- Weissman, J.R.; Kelley, R.I.; Bauman, M.L.; Cohen, B.H.; Murray, K.F.; Mitchell, R.L.; Kern, R.L.; Natowicz, M.R. Mitochondrial Disease in Autism Spectrum Disorder Patients: A Cohort Analysis. PLoS ONE 2008, 3, e3815. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef]
- Hollis, F.; Kanellopoulos, A.K.; Bagni, C. Mitochondrial Dysfunction in Autism Spectrum Disorder: Clinical Features and Perspectives. Curr. Opin. Neurobiol. 2017, 45, 178–187. [Google Scholar] [CrossRef]
- McDonald, T.; Puchowicz, M.; Borges, K. Impairments in Oxidative Glucose Metabolism in Epilepsy and Metabolic Treatments Thereof. Front. Cell. Neurosci. 2018, 12, 274. [Google Scholar] [CrossRef]
- Kim, Y.; Vadodaria, K.C.; Lenkei, Z.; Kato, T.; Gage, F.H.; Marchetto, M.C.; Santos, R. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Signal. 2019, 31, 275–317. [Google Scholar] [CrossRef]
- Kanellopoulos, A.K.; Mariano, V.; Spinazzi, M.; Woo, Y.J.; McLean, C.; Pech, U.; Li, K.W.; Armstrong, J.D.; Giangrande, A.; Callaerts, P.; et al. Aralar Sequesters GABA into Hyperactive Mitochondria, Causing Social Behavior Deficits. Cell 2020, 180, 1178–1197.e20. [Google Scholar] [CrossRef]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism Open Access 2016, 6, 1000190. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Gu, F.; Chauhan, V. Mitochondrial Dysfunction in Autism. In Studies on Psychiatric Disorders; Dietrich-Muszalska, A., Chauhan, V., Grignon, S., Eds.; Oxidative Stress in Applied Basic Research and Clinical Practice; Springer: New York, NY, USA, 2015; pp. 355–372. ISBN 978-1-4939-0439-6. [Google Scholar]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-González, X.R. Mitochondrial Dysfunction: A Common Denominator in Neurodevelopmental Disorders? Dev. Neurosci. 2021, 43, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Bailey, A.M.; Matthies, H.J.G.; Renden, R.B.; Smith, M.A.; Speese, S.D.; Rubin, G.M.; Broadie, K. Drosophila Fragile X-Related Gene Regulates the MAP1B Homolog Futsch to Control Synaptic Structure and Function. Cell 2001, 107, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Mariano, V.; Achsel, T.; Bagni, C.; Kanellopoulos, A.K. Modelling Learning and Memory in Drosophila to Understand Intellectual Disabilities. Neuroscience 2020, 445, 12–30. [Google Scholar] [CrossRef]
- Bagni, C.; Tassone, F.; Neri, G.; Hagerman, R. Fragile X Syndrome: Causes, Diagnosis, Mechanisms, and Therapeutics. J. Clin. Investig. 2012, 122, 4314–4322. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Zukin, R.S. A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef]
- Pasciuto, E.; Bagni, C. SnapShot: FMRP mRNA Targets and Diseases. Cell 2014, 158, 1446–1446.e1. [Google Scholar] [CrossRef]
- Saldarriaga, W.; Tassone, F.; González-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X Syndrome. Colomb. Medica 2014, 45, 190–198. [Google Scholar] [CrossRef]
- Protic, D.D.; Aishworiya, R.; Salcedo-Arellano, M.J.; Tang, S.J.; Milisavljevic, J.; Mitrovic, F.; Hagerman, R.J.; Budimirovic, D.B. Fragile X Syndrome: From Molecular Aspect to Clinical Treatment. Int. J. Mol. Sci. 2022, 23, 1935. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X Mental Retardation Protein and Synaptic Plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Dockendorff, T.C.; Su, H.S.; McBride, S.M.J.; Yang, Z.; Choi, C.H.; Siwicki, K.K.; Sehgal, A.; Jongens, T.A. Drosophila Lacking Dfmr1 Activity Show Defects in Circadian Output and Fail to Maintain Courtship Interest. Neuron 2002, 34, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Hutson, R.L.; Thompson, R.L.; Bantel, A.P.; Tessier, C.R. Acamprosate Rescues Neuronal Defects in the Drosophila Model of Fragile X Syndrome. Life Sci. 2018, 195, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.R.; Kanellopoulos, A.K.; Bagni, C. Learning and Behavioral Deficits Associated with the Absence of the Fragile X Mental Retardation Protein: What a Fly and Mouse Model Can Teach Us. Learn. Mem. 2014, 21, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Jin, S.; Li, X.; Liu, Z.; Ma, X.; Tang, J.; Zhang, Y.Q. Drosophila FMRP Regulates Microtubule Network Formation and Axonal Transport of Mitochondria. Hum. Mol. Genet. 2011, 20, 51–63. [Google Scholar] [CrossRef]
- Weisz, E.D.; Towheed, A.; Monyak, R.E.; Toth, M.S.; Wallace, D.C.; Jongens, T.A. Loss of Drosophila FMRP Leads to Alterations in Energy Metabolism and Mitochondrial Function. Hum. Mol. Genet. 2018, 27, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Bülow, P.; Wenner, P.A.; Faundez, V.; Bassell, G.J. Mitochondrial Structure and Polarity in Dendrites and the Axon Initial Segment Are Regulated by Homeostatic Plasticity and Dysregulated in Fragile X Syndrome. Front. Cell Dev. Biol. 2021, 9, 702020. [Google Scholar] [CrossRef]
- Licznerski, P.; Park, H.-A.; Rolyan, H.; Chen, R.; Mnatsakanyan, N.; Miranda, P.; Graham, M.; Wu, J.; Cruz-Reyes, N.; Mehta, N.; et al. ATP Synthase C-Subunit Leak Causes Aberrant Cellular Metabolism in Fragile X Syndrome. Cell 2020, 182, 1170–1185.e9. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.R.; Wynne, M.E.; Faundez, V. Human-Specific Translational Control of Neuronal Mitochondria and Excitability. Neuron 2023, 111, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Imlach, W.L.; Beck, E.S.; Choi, B.J.; Lotti, F.; Pellizzoni, L.; McCabe, B.D. SMN Is Required for Sensory-Motor Circuit Function in Drosophila. Cell 2012, 151, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Riemensperger, T.; Issa, A.R.; Pech, U.; Coulom, H.; Nguyễn, M.V.; Cassar, M.; Jacquet, M.; Fiala, A.; Birman, S. A Single Dopamine Pathway Underlies Progressive Locomotor Deficits in a Drosophila Model of Parkinson Disease. Cell Rep. 2013, 5, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Goles, N.I.; Varas, R.; Campusano, J.M. Serotonin Receptors Expressed in Drosophila Mushroom Bodies Differentially Modulate Larval Locomotion. PLoS ONE 2014, 9, e89641. [Google Scholar] [CrossRef]
- Majeed, Z.R.; Abdeljaber, E.; Soveland, R.; Cornwell, K.; Bankemper, A.; Koch, F.; Cooper, R.L. Modulatory Action by the Serotonergic System: Behavior and Neurophysiology in Drosophila Melanogaster. Neural Plast. 2016, 7291438. [Google Scholar] [CrossRef]
- Hsu, C.T.; Bhandawat, V. Organization of Descending Neurons in Drosophila Melanogaster. Sci. Rep. 2016, 6, 20259. [Google Scholar] [CrossRef]
- Eriksson, A.; Raczkowska, M.; Navawongse, R.; Choudhury, D.; Stewart, J.C.; Tang, Y.L.; Wang, Z.; Claridge-Chang, A. Neuromodulatory Circuit Effects on Drosophila Feeding Behaviour and Metabolism. Sci. Rep. 2017, 7, 8839. [Google Scholar] [CrossRef]
- Clark, M.Q.; Zarin, A.A.; Carreira-Rosario, A.; Doe, C.Q. Neural Circuits Driving Larval Locomotion in Drosophila. Neural Dev. 2018, 13, 6. [Google Scholar] [CrossRef]
- Saraf, T.S.; Chen, Y.; Tyagi, R.; Canal, C.E. Altered Brain Serotonin 5-HT1A Receptor Expression and Function in Juvenile Fmr1 Knockout Mice. Neuropharmacology 2024, 245, 109774. [Google Scholar] [CrossRef]
- Costa, L.; Sardone, L.M.; Bonaccorso, C.M.; D’Antoni, S.; Spatuzza, M.; Gulisano, W.; Tropea, M.R.; Puzzo, D.; Leopoldo, M.; Lacivita, E.; et al. Activation of Serotonin 5-HT7 Receptors Modulates Hippocampal Synaptic Plasticity by Stimulation of Adenylate Cyclases and Rescues Learning and Behavior in a Mouse Model of Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Friedman, D.B.; Wang, Z.; Woodruff, E.; Pan, L.; O’Donnell, J.; Broadie, K. Protein Expression Profiling of the Drosophila Fragile X Mutant Brain Reveals Up-Regulation of Monoamine Synthesis. Mol. Cell. Proteom. 2005, 4, 278–290. [Google Scholar] [CrossRef]
- Okusawa, S.; Kohsaka, H.; Nose, A. Serotonin and Downstream Leucokinin Neurons Modulate Larval Turning Behavior in Drosophila. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 2544–2558. [Google Scholar] [CrossRef] [PubMed]
- Cardon, I.; Grobecker, S.; Jenne, F.; Jahner, T.; Rupprecht, R.; Milenkovic, V.M.; Wetzel, C.H. Serotonin Effects on Human iPSC-Derived Neural Cell Functions: From Mitochondria to Depression. Mol. Psychiatry 2024. [Google Scholar] [CrossRef] [PubMed]
- Scholpa, N.E.; Lynn, M.K.; Corum, D.; Boger, H.A.; Schnellmann, R.G. 5-HT1F Receptor-Mediated Mitochondrial Biogenesis for the Treatment of Parkinson’s Disease. Br. J. Pharmacol. 2018, 175, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Sola-Penna, M.; Paixão, L.P.; Branco, J.R.; Ochioni, A.C.; Albanese, J.M.; Mundim, D.M.; Baptista-de-Souza, D.; Figueiredo, C.P.; Coelho, W.S.; Marcondes, M.C.; et al. Serotonin Activates Glycolysis and Mitochondria Biogenesis in Human Breast Cancer Cells through Activation of the Jak1/STAT3/ERK1/2 and Adenylate Cyclase/PKA, Respectively. Br. J. Cancer 2020, 122, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Tiwari, P.; Ghai, U.; Gupta, S.; Figueiredo, D.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; et al. Serotonin Regulates Mitochondrial Biogenesis and Function in Rodent Cortical Neurons via the 5-HT 2A Receptor and SIRT1–PGC-1α Axis. Proc. Natl. Acad. Sci. USA 2019, 116, 11028–11037. [Google Scholar] [CrossRef] [PubMed]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive Locomotion in a Drosophila Model Is a Functional Readout for the Synaptic Abnormalities Underlying Fragile X Syndrome. Sci. Signal. 2017, 10, eaai8133. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, B.R.; Longoria, R.A.; Shubeita, G.T. A High Throughput and Sensitive Method Correlates Neuronal Disorder Genotypes to Drosophila Larvae Crawling Phenotypes. Fly 2012, 6, 303–308. [Google Scholar] [CrossRef]
- Pan, L.; Broadie, K.S. Drosophila Fragile X Mental Retardation Protein and Metabotropic Glutamate Receptor A Convergently Regulate the Synaptic Ratio of Ionotropic Glutamate Receptor Subclasses. J. Neurosci. 2007, 27, 12378–12389. [Google Scholar] [CrossRef]
- Scholpa, N.E.; Schnellmann, R.G. Mitochondrial-Based Therapeutics for the Treatment of Spinal Cord Injury: Mitochondrial Biogenesis as a Potential Pharmacological Target. J. Pharmacol. Exp. Ther. 2017, 363, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Yao, C.-K.; Chen, K.; Jaiswal, M.; Donti, T.; Lin, Y.Q.; Bayat, V.; Xiong, B.; Zhang, K.; David, G.; et al. Mitochondrial Fusion but Not Fission Regulates Larval Growth and Synaptic Development through Steroid Hormone Production. eLife 2014, 3, e03558. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, K.; Hale, C.R.; Park, C.Y.; Fak, J.J.; Gresack, J.E.; Van Driesche, S.J.; Kang, J.J.; Darnell, J.C.; Darnell, R.B. FMRP Has a Cell-Type-Specific Role in CA1 Pyramidal Neurons to Regulate Autism-Related Transcripts and Circadian Memory. eLife 2019, 8, e46919. [Google Scholar] [CrossRef] [PubMed]
- Gershman, B.; Puig, O.; Hang, L.; Peitzsch, R.M.; Tatar, M.; Garofalo, R.S. High-Resolution Dynamics of the Transcriptional Response to Nutrition in Drosophila: A Key Role for dFOXO. Physiol. Genom. 2007, 29, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J. Testing for Fragile X Gene Mutations Throughout the Life Span. JAMA 2008, 300, 2419. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Lindemann, L.; Jønch, A.E.; Apostol, G.; Bear, M.F.; Carpenter, R.L.; Crawley, J.N.; Curie, A.; Des Portes, V.; Hossain, F.; et al. Drug Development for Neurodevelopmental Disorders: Lessons Learned from Fragile X Syndrome. Nat. Rev. Drug Discov. 2017, 17, 280–299. [Google Scholar] [CrossRef]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered Synaptic Plasticity in a Mouse Model of Fragile X Mental Retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Hanson, A.C.; Hagerman, R.J. Serotonin Dysregulation in Fragile X Syndrome: Implications for Treatment. Intractable Rare Dis. Res. 2014, 3, 110–117. [Google Scholar] [CrossRef]
- Muller, C.L.; Anacker, A.M.J.; Veenstra-VanderWeele, J. The Serotonin System in Autism Spectrum Disorder: From Biomarker to Animal Models. Neuroscience 2016, 321, 24–41. [Google Scholar] [CrossRef] [PubMed]
- AlOlaby, R.R.; Sweha, S.R.; Silva, M.; Durbin-Johnson, B.; Yrigollen, C.M.; Pretto, D.; Hagerman, R.J.; Tassone, F. Molecular Biomarkers Predictive of Sertraline Treatment Response in Young Children with Fragile X Syndrome. Brain Dev. 2017, 39, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Sardone, L.M.; Lacivita, E.; Leopoldo, M.; Ciranna, L. Novel Agonists for Serotonin 5-HT7 Receptors Reverse Metabotropic Glutamate Receptor-Mediated Long-Term Depression in the Hippocampus of Wild-Type and Fmr1 KO Mice, a Model of Fragile X Syndrome. Front. Behav. Neurosci. 2015, 9, 65. [Google Scholar] [CrossRef]
- Greiss Hess, L.; Fitzpatrick, S.E.; Nguyen, D.V.; Chen, Y.; Gaul, K.N.; Schneider, A.; Lemons Chitwood, K.; Eldeeb, M.A.A.A.; Polussa, J.; Hessl, D.; et al. A Randomized, Double-Blind, Placebo-Controlled Trial of Low-Dose Sertraline in Young Children with Fragile X Syndrome. J. Dev. Behav. Pediatr. 2016, 37, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Rosenblad, C.; af Edholm Arvidsson, K.; Wictorin, K.; Keywood, C.; Shankar, B.; Lowe, D.A.; Björklund, A.; Widner, H. Eltoprazine Counteracts L-DOPA-Induced Dyskinesias in Parkinson’s Disease: A Dose-Finding Study. Brain 2015, 138, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A.; Ko, W.K.D.; Costa, G.; Tronci, E.; Fidalgo, C.; Simola, N.; Li, Q.; Tabrizi, M.A.; Bezard, E.; Carta, M.; et al. Antidyskinetic Effect of A2A and 5HT1A/1B Receptor Ligands in Two Animal Models of Parkinson’s Disease. Mov. Disord. 2016, 31, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Mineo, D.; Vannelli, A.; Cacace, F.; Mancini, M.; Pendolino, V.; Napolitano, F.; di Maio, A.; Mellone, M.; Stanic, J.; et al. Modulation of Serotonergic Transmission by Eltoprazine in L-DOPA-Induced Dyskinesia: Behavioral, Molecular, and Synaptic Mechanisms. Neurobiol. Dis. 2016, 86, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.K.D.; Li, Q.; Cheng, L.Y.; Morelli, M.; Carta, M.; Bezard, E. A Preclinical Study on the Combined Effects of Repeated Eltoprazine and Preladenant Treatment for Alleviating L-DOPA-Induced Dyskinesia in Parkinson’s Disease. Eur. J. Pharmacol. 2017, 813, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Will, E.A.; Bishop, S.L.; Roberts, J.E. Developmental Divergence: Motor Trajectories in Children with Fragile X Syndrome with and without Co-Occurring Autism. J. Neurodev. Disord. 2019, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Sawacha, Z.; Spolaor, F.; Piątkowska, W.J.; Cibin, F.; Ciniglio, A.; Guiotto, A.; Ricca, M.; Polli, R.; Murgia, A. Feasibility and Reliability Assessment of Video-Based Motion Analysis and Surface Electromyography in Children with Fragile X during Gait. Sensors 2021, 21, 4746. [Google Scholar] [CrossRef]
- Repicky, S.; Broadie, K. Metabotropic Glutamate Receptor–Mediated Use–Dependent Down-Regulation of Synaptic Excitability Involves the Fragile X Mental Retardation Protein. J. Neurophysiol. 2009, 101, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.H.; Dani, N.; Rushton, E.; Broadie, K. Fragile X Mental Retardation Protein Regulates Trans-Synaptic Signaling in Drosophila. Dis. Models Mech. 2013, 6, 1400–1413. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Crossin, K.L.; Edelman, D.B. Serotonin Stimulates Mitochondrial Transport in Hippocampal Neurons. Mol. Cell. Neurosci. 2007, 36, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Gupta, S.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; Kolthur-Seetharam, U.; Vaidya, V.A. Serotonin Regulates Mitochondrial Biogenesis and Function in Rodent Cortical Neurons via the 5-HT2A Receptor and SIRT1-PGC-1α Axis Equal Senior Corresponding Author Title: Serotonin and Mitochondria. bioRxiv 2018. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic Mitochondria in Synaptic Transmission and Organization of Vesicle Pools in Health and Disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the Neuronal Presynapse in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Wang, F.; Li, M.; Sah, N.; Stockton, M.E.; Tidei, J.J.; Gao, Y.; Korabelnikov, T.; Kannan, S.; Vevea, J.D.; et al. Reduced Mitochondrial Fusion and Huntingtin Levels Contribute to Impaired Dendritic Maturation and Behavioral Deficits in Fmr1-Mutant Mice. Nat. Neurosci. 2019, 22, 386–400. [Google Scholar] [CrossRef] [PubMed]
- D’Antoni, S.; De Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.A.; Catania, M.V. Aberrant Mitochondrial Bioenergetics in the Cerebral Cortex of the Fmr1 Knockout Mouse Model of Fragile X Syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Maurin, T.; Lebrigand, K.; Castagnola, S.; Paquet, A.; Jarjat, M.; Popa, A.; Grossi, M.; Rage, F.; Bardoni, B. HITS-CLIP in Various Brain Areas Reveals New Targets and New Modalities of RNA Binding by Fragile X Mental Retardation Protein. Nucleic Acids Res. 2018, 46, 6344–6355. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.D.; Becnel, J.; Pandey, U.B. Methods to Assay Drosophila Behavior. J. Vis. Exp. 2012, 61, 3795. [Google Scholar] [CrossRef]
- Brent, J.R.; Werner, K.M.; McCabe, B.D. Drosophila Larval NMJ Dissection. J. Vis. Exp. 2009, 24, e1107. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Fontana-Ayoub, M.; Sumbalova, Z.; Heidler, J.; Gauper, K.; Fasching, M.; Gnaiger, E. Simultaneous High-Resolution Measurement of Mitochondrial Respiration and Hydrogen Peroxide Production. In Mitochondrial Medicine; Humana Press: New York, NY, USA, 2015; pp. 245–261. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannelli, A.; Mariano, V.; Bagni, C.; Kanellopoulos, A.K. Activation of the 5-HT1A Receptor by Eltoprazine Restores Mitochondrial and Motor Deficits in a Drosophila Model of Fragile X Syndrome. Int. J. Mol. Sci. 2024, 25, 8787. https://doi.org/10.3390/ijms25168787
Vannelli A, Mariano V, Bagni C, Kanellopoulos AK. Activation of the 5-HT1A Receptor by Eltoprazine Restores Mitochondrial and Motor Deficits in a Drosophila Model of Fragile X Syndrome. International Journal of Molecular Sciences. 2024; 25(16):8787. https://doi.org/10.3390/ijms25168787
Chicago/Turabian StyleVannelli, Anna, Vittoria Mariano, Claudia Bagni, and Alexandros K. Kanellopoulos. 2024. "Activation of the 5-HT1A Receptor by Eltoprazine Restores Mitochondrial and Motor Deficits in a Drosophila Model of Fragile X Syndrome" International Journal of Molecular Sciences 25, no. 16: 8787. https://doi.org/10.3390/ijms25168787