Abstract
CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) is caused by NOTCH3 mutations affecting the number of cysteines. The pathogenic role of cysteine-sparing NOTCH3 mutations with typical clinical CADASIL syndrome is still debated. This review aimed to characterize NOTCH3 cysteine-sparing mutations in patients with clinical suspicion of CADASIL. Articles on NOTCH3 cysteine-sparing mutations with clinical suspicion of CADASIL were reviewed. Clinical and radiological cerebral phenotypes data were extracted and characterized across regions and compared with phenotypes of typical CADASIL patients. We screened 298 NOTCH3 cysteine-sparing mutation individuals from 20 publications, and mutations in exon 3 were the most frequently reported (21.46%). Gait impairment (76.47%), cognitive impairment (67.47%), and stroke (62.37%) were the three most common clinical phenotypes; the most frequent radiological cerebral phenotypes were lacunes (74.29%) and cerebral microbleeds (72.73%). Compared with CADASIL patients, cognitive impairment and cerebral microbleed frequencies were significantly higher in patients with NOTCH3 cysteine-sparing mutations, while the white matter hyperintensities in anterior temporal polar and external capsule were rarely observed. Compared with Western patients, radiological phenotypes were more common than clinical phenotypes in cysteine-sparing Asian patients. More than half of cysteine-sparing patients had positive granular osmiophilic material deposits. NOTCH3 cysteine-sparing mutations in patients with clinical suspicion of CADASIL mainly manifested with gait and cognitive impairment but rare white matter hyperintensities in anterior temporal pole and external capsule. Further studies are warranted to pay attention to atypical NOTCH3 variants, which could guide specific diagnosis and help unravel underlying mechanisms.
1. Introduction
CADASIL, which stands for Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, is the most frequently observed and widely recognized form of monogenetic hereditary cerebral small vessel disease [1,2]. It occurs due to over 220 mutations identified so far in the NOTCH3 gene [2,3]. NOTCH3 contains 33 exons and encodes a transmembrane receptor constituted by three domains. Of these, the extracellular domain (ECD) with 34 epidermal growth factor-like repeats (EGFrs) is the most critical contributor to maintaining protein tertiary structure [4]. A definitive diagnosis of CADASIL is established through genetic testing that identifies mutations in the NOTCH3 gene, resulting in an abnormal number of cysteine residues (typically five or seven) in one of the EGFr domains of the NOTCH3 protein, which is particularly prominent in the vascular smooth muscle cells in the brain, confirming the consensus on CADASIL diagnosis [5]. NOTCH3 mediates the NOTCH signaling pathway, which is important in cell–cell communication and plays a major role in blood vessel development. Pathologically, the odd number of cysteines leads to NOTCH3 misfolding and ECD aggregation, which is considered as the primary pathogenic mechanism triggering cerebral vascular smooth muscle cytopathology [6]. Increasing studies have also identified cysteine-sparing mutations as mutations that do not affect the number of cysteines in patients with granular osmiophilic material (GOM) accumulation which consists of parts of NOTCH3 ECD by skin biopsy and is considered as the typical pathological finding of CADASIL [7,8]. Cysteine-sparing mutations could also trigger significant loss of structure in NOTCH3, which challenges the traditional standard of pathogenic mutations [9].
Clinical hallmarks for CADASIL suspicion are constantly facing challenges. Migraine with aura, acute encephalopathy, recurrent lacunar strokes, cognitive decline, gait, and mood disorders have long been recognized as typical clinical manifestations of CADASIL [1]. However, community-dwelling studies suggested that participants harboring NOTCH3 cysteine-altering variants are associated with a comprehensive phenotypic spectrum, ranging from a severe CADASIL phenotype to non-penetrance [10]. Magnetic resonance imaging (MRI) features, including symmetrical subcortical white matter hyperintensities (WMHs) located in the anterior temporal lobe, and progressive lacunes, cerebral microbleeds, and enlarged perivascular spaces [11] can also be observed in other monogenic cerebral small-vessel diseases, increasing with age and disease severity [5]. Additionally, NOTCH3 cysteine-sparing variants can also present with CADASIL-like clinical and radiological phenotypes, indicating their potential pathogenic role in CADASIL. These findings make it unclear whether NOTCH3 cysteine-sparing mutations contribute to vasculopathy in patients with clinical suspicion of CADASIL.
Better characterization of NOTCH3 cysteine-sparing mutations in patients with clinical suspicion of CADASIL can offer insights into the pathomechanisms underlying CADASIL and aid in managing affected individuals. Given the considerations mentioned above, in the present study, a systematic literature review was performed to summarize genetic mutations and clinical manifestations of reported individuals with NOTCH3 cysteine-sparing nonsynonymous mutations, including missense, nonsense, and frameshift mutations, and compare both clinical and radiological phenotypes associated with those harboring typical mutations.
2. Results
2.1. Genetic Data and Patients
We included 20 publications from 298 identified for screening and extracted data on 268 NOTCH3 cysteine-sparing mutations individuals with clinical suspicion of CADASIL (Supplementary Table S3) [4,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. Genetic information was accessible for all the families involved in the study, with the exception of one family (consisting of five individuals), for which the precise mutation details were not disclosed, as the findings were only published in the form of a conference abstract.
With respect to reported 100 different mutations in all the 263 individuals, a total of 95 mutations in 263 individuals were in the exon region, including 89 missense mutations, 5 frameshift mutations, and 1 nonsense mutation, distributed across the 28 exons (Figure 1). Exon 3 was the most frequently involved (57/254, 22.44%), with the p.R75P being the most common mutation (41/57, 71.93% of Exon 3 mutation). Mutations in Exon 4 type were the most abundant, with 12 different kinds of amino acid alteration and p.P167S being the most common one (15/31, 48.39% of Exon 4 mutation). Exon 24, 25, 5, 11, and 33 mutations were also notable, as shown in Supplementary Table S3. With regard to Exon 8, 16, 23, and 27, no mutations were reported from previous studies in the reviewed literature.
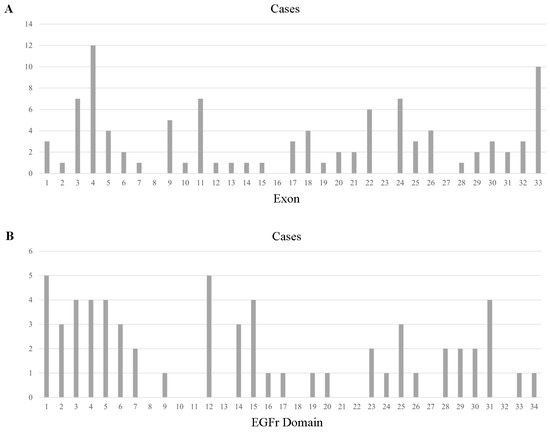
Figure 1.
Cysteine-sparing case number across exons of NOTCH3 (A) and EGFr domains of the NOTCH3 protein (B).
It is important to note that some cysteine-sparing mutations, including p.R75P, have been repeatedly confirmed as pathogenic in previous studies. However, it differs from the traditionally defined classic CADASIL pathogenic mutations as it does not alter the number of cysteines. Therefore, these mutations are still analyzed in this review, which can help us further explore the pathogenic mechanisms of NOTCH3 cysteine-sparing mutations.
2.2. Frequency of Clinical Phenotypes
The average age at neurological deficits onset was 53.64 ± 2.32 (100/262). The clinical features of 248 identified patients are summarized, including stroke, cognitive impairment, psychiatric disturbance, gait impairment, headaches, and seizures (Table 1).

Table 1.
Patient clinical and demographic information related to cysteine-sparing NOTCH3 mutations.
Clinical stroke attacks were reported in 62.37% (58/93) of patients with NOTCH3 cysteine-sparing mutations. Ischemic events (including transient ischemic attack) were more common than hemorrhagic events, affecting 19 cases and 1 case, respectively. Cognitive impairment prevalence was reported in 13 published studies, ranging from 37.50% to 100.00%, without detailed information on severity, impaired domain, etc. Gait impairment was also commonly reported (13/17, from six studies). Headache, including migraine, was reported in 43.48% (40/92) of individuals. Most headache cases were not specified as migraine. The frequency of psychiatric disturbance, mainly specified as apathy or irritability, was reported as 38.96% (30/77). Thirty percent of individuals (9/30) were reported to have suffered a seizure or have epilepsy. Two studies reported that livedo reticularis was not noted in patients (0/5).
For cysteine-sparing mutations in Exon 1–6, 72.22% of individuals (78/108) had typical clinical CADASIL symptoms, while only 38.24% of individuals (26/68) had mutations in Exon 7–34 (Supplementary Table S3).
2.3. Frequency of Radiological Cerebral Phenotypes
Table 1 provides an overview of the primary neuroimaging features observed in NOTCH3 cysteine-sparing mutations. This includes the involvement of white matter in the temporal pole and external capsule, the presence of lacunes, and cerebral microbleeds.
Ischemia was the most common radiological manifestation for individuals with NOTCH3 cysteine-sparing mutations, and lacunes were noted in 74.29% (26/35). Cerebral microbleeds were also notable and present in 72.73% (40/55). Given that most publications were generally lacking in detail, location and multiple lesions were less clear. Of note, this sample with WMHs in the temporal pole and external capsule was relatively milder. Typical anterior temporal polar lesions were only present in 10.50% (23/219), and the external capsule was severely involved in 25.11% (58/231) of patients.
2.4. Comparisons of Phenotypes between Patients with NOTCH3 Cysteine-Sparing Mutations and Typical CADASIL
Typical manifestations of CADASIL often include recurrent lacunar strokes, cognitive deterioration, migraine with aura, mood disturbances, symmetrical periventricular WMHs without a clear cause, and a positive family history for similar symptoms. Based on previous studies and reviews, the prevalence of clinical manifestations was quite different between typical CADASIL with NOTCH3 cysteine-altering mutations and suspected CADASIL with NOTCH3 cysteine-sparing mutations (Table 2). Compared with 3252 CADASIL patients from 22 studies, the prevalence of stroke was almost the same as in patients with NOTCH3 cysteine-sparing mutations (61.65% vs. 62.37%), and the prevalence of migraine (49.05% vs. 43.48%) as well. Interestingly, the proportion of cognitive impairment in patients with NOTCH3 cysteine-sparing mutations was significantly more prevalent (67.47% vs. 35.54%), similar to psychiatric disturbance (38.96% vs. 32.95%).
Table 2.
Comparison of phenotypes between NOTCH3 cysteine-sparing mutations and typical mutations.
WMHs in specific areas are highly suggestive and often considered a radiological hallmark for CADASIL, particularly in the anterior temporal lobe and external capsule. In nine studies (519 typical CADASIL patients) with specific mention of these two radiological features, the prevalence of WMHs in the anterior temporal lobe was 57.42% and that in the external capsule was 77.84%, which are significantly higher than those of patients with NOTCH3 cysteine-sparing mutations (10.50% and 25.11%, respectively). In contrast, the lacunes and cerebral microbleeds in patients with NOTCH3 cysteine-sparing mutations were more severe than those in typical CADASIL (74.29% vs. 62.00%, 72.73% vs. 35.80%, respectively), particularly for cerebral microbleeds with a significantly statistically higher proportion.
2.5. Differences between Asians and Patients in Western Countries
In comparison with patients with NOTCH3 cysteine-sparing mutations from Western countries (Europe, North America, and Australia), several differences were observed in Asian patients. For mutations in different exons, NOTCH3 mutations were more frequently detected in Exon 3 (23.98%), Exon 4 (10.05%), Exon 24 (9.59%), and Exon 25 (9.13%) in Asian populations; in contrast, for patients in Western countries, NOTCH3 mutations were observed mostly in Exon 4 (21.43%), followed by Exon 3 (9.52%), Exon 9 (7.14%), and Exon 26 (7.14%) (Figure S1). Of note, p.R75P in Exon 3 was the leading mutation in Asians (40/53, 75.47%), particularly in South Korea and Japan.
The typical phenotypes of suspicious CADASIL with NOTCH3 cysteine-sparing mutations varied significantly between ethnic groups (Table 3). A key factor contributing to these differences is the prevalence of Asian-specific CADASIL-causing mutations, such as p.R75P and p.R544C. For clinical phenotypes, Asians had a significantly lower proportion of headaches (29.58% vs. 66.67%), cognitive impairment (57.35% vs. 91.67%), and seizures (19.05% vs. 55.56%). Although proportions of stroke and psychiatric disturbance were not significantly different between the two groups, comparisons indicated that the stroke and psychiatric disturbance burden was lower in Asian patients (60.27% vs. 75.86%, 31.82% vs. 52.63%, respectively). In contrast, radiological cerebral phenotypes presented with more substantial burdens in Asians. In Asian patients, lacunae and cerebral microbleeds were twice as frequent as those in Western patients (88.00% vs. 40.00%, 80.44% vs. 33.33%, respectively). Similarly, Asian patients had a higher prevalence of anterior temporal pole and external capsule involvement with WMHs than those of European ancestry; however, no significant difference was found between these two groups (10.88% vs. 7.69%, and 26.39% vs. 6.67%, respectively). Notably, the presence of GOM deposits in skin biopsy was observed in 92.31% of Asian patients, significantly higher than in Western patients (42.86%).
Table 3.
Comparison of clinical and radiological cerebral phenotypes according to ancestry.
3. Discussion
CADASIL diagnosis is definitively confirmed by NOTCH3 cysteine-affecting mutations using genetic testing. However, patients with cysteine-sparing mutations who have typical clinical CADASIL features, extensive WMH, and even GOM deposits in skin biopsy are also highly suspected of CADASIL. In this study, we summarized the genetic spectrum of the NOTCH3 cysteine-sparing mutations and found that mutations were more frequently detected in Exon 3, with p.R75P being the most common type. The overall burden of clinical phenotypes in patients with cysteine-sparing mutations is comparable with that in CADASIL subjects, except for a relatively milder involvement of WMHs in anterior temporal pole and external capsule. As a result, regional differences in genetic spectrums and clinical features are also observed, suggesting possible ethnicity-based mechanisms of NOTCH3 cysteine-sparing mutations causing CADASIL.
3.1. Pathological Hallmarks and Molecular Insights
The accumulation and deposition of NOTCH3 ECD within the walls of small blood vessels are crucial pathological hallmarks of CADASIL. These phenomena are observed in patients and in a transgenic mouse model specifically designed to study this disease. For typical CADASIL patients, the odd number of cysteines may promote NOTCH3 ECD aggregation via their unpaired sulfhydryl groups [31]. The pathogenicity of cysteine-sparing mutations, including mutations outside EGFr, is still ambiguous. Some cysteine-sparing mutations, such as R75P, D80G, and A564T, showed significantly enhanced aggregation behavior in vitro similar to that of typical cysteine-affecting mutants associated with conformational changes in the protein [13,32,33]. Therefore, these mutations seem to cause a similar phenotype. For R61W and R213K, multimerization was absent and clinical symptoms were untypical [34,35]. According to these findings, additional mechanisms other than NOTCH3 ECD aggregation might be involved in NOTCH3 cysteine-sparing mutations. Notch3 and other molecules/pathways have recently been supposed to underlie the pathogenesis of NOTCH-related diseases [36,37,38,39]. Although these mutations, such as R103X and L1515P, did not directly contribute to NOTCH3 protein accumulation, the canonical NOTCH3 signaling was dysregulated through destabilization of the heterodimer [36,39]. Additionally, an insertion mutation found within the intracellular domain of NOTCH3 in a patient suspected of having CADASIL was anticipated to cause premature termination of the protein. This termination occurs without triggering the nonsense-mediated mRNA decay process, potentially impacting the downstream components of the Notch signaling pathway [40]. Notably, some other cysteine-sparing mutations without multimerization were still questionable about the possibility of polymorphism or other arteriopathy-causing mutations. Specifically, the mutations mentioned in the studies, p.Arg10Hisfs*16 [10], involving NOTCH3 homozygous nonsense mutations are likely to lead to impaired NOTCH3 signaling. This could play a critical role in CADASIL progression. However, the contribution of loss-of-function effects of NOTCH3 variants is still debated, and it remains unclear whether decreased NOTCH3 receptor activity also plays a central role. Delving into the functional characterization of these mutants could illuminate the pathogenetic mechanisms at the heart of CADASIL. This could provide deeper insights into how the disease develops and progresses.
3.2. Challenges in Confirming the Pathogenicity of Cysteine-Sparing NOTCH3 Mutations in CADASIL
Although cysteine-sparing NOTCH3 mutations have been studied more extensively, no consensus has been reached regarding their pathogenicity. Muiño reviewed 34 cases with a clinical suspicion of CADASIL and cysteine-sparing NOTCH3 missense mutations and concluded the following criteria for pathogenic identification: (1) the patients had typical clinical CADASIL syndrome; (2) the patients had diffuse WMHs in MRI; (3) the study analyzed the 33 exons of the NOTCH3 gene to rule out other pathogenic mutations; (4) the mutation had a MAF < 0.1%; and lastly (5) the patients had GOM deposits in the skin biopsy [4]. However, pathogenic criteria for clinical practice still face problems. Clinical and genetic diversity is a notable feature among CADASIL patients, with variations significantly influenced by the target population, study duration, and stage of the disease [41]. Conversely, the effectiveness of detecting GOM as a diagnostic marker for CADASIL is debatable. Recent findings have highlighted instances where GOM was absent in skin biopsies of CADASIL cohorts, particularly in patients carrying mutations in the EGFr 7–34 regions of the NOTCH3 gene. This inconsistency calls into question the reliability of GOM presence as a universal diagnostic criterion for CADASIL [42,43]. Therefore, the absence of GOM does not definitely exclude a CADASIL diagnosis, since genotype characteristics and different biopsy locations might greatly influence its positivity. In this review, participants with suspicious clinical CADASIL syndrome and a cysteine-sparing NOTCH3 mutation were selected. According to MAF and database searching, mutations identified as polymorphisms were also excluded to collect data about potential pathogenic cysteine-sparing NOTCH3 mutations associated with typical clinical CADASIL syndrome.
Muiño considered the cysteine-sparing NOTCH3 missense mutations p.R61W, p.R75P, p.D80G, and p.R213K as potentially pathogenic. This indicates a belief that these specific genetic alterations could contribute to disease processes despite not affecting cysteine residues directly [4]. Consistently, these patients presented typical clinical CADASIL syndrome, MRI profiles, GOM deposits, and family history in our study. In addition, we also found that p.G73A, p.G73S, p.R107W, p.R75Q, p.A202V, p.G149V, p.H170R, p.D239N, p.V252M, p.E309K, p.P496L, p.A564T, p.V644D, p.D887A, p.T1098S, p.H1133Q, p.R1175W, p.G1347R, p.S1418L, p.R1761H, p.V1922L, and p.V1952M also presented typical CADASIL phenotypes, positive family history, or GOM deposits by skin biopsy except for some cases in which examination are not available (Supplementary Table S3). PolyPhen-2 predictions were conducted on the above variants, and all indicated probably damaging or possibly damaging mutations. Additionally, according to Muiño’s criteria, the newly identified mutations p.G73A, p.R75Q, p.S1418L, and p.R1761H in this study are potentially pathogenic. Based on the clinical data available in the literature, these mutations can be viewed as potential pathogenic mutations. This assessment suggests that they warrant additional investigation to provide mechanistic evidence of their role in disease processes.
3.3. Clinical Phenotypes of Cysteine-Sparing Mutations
The average age of presenting neurological symptoms or signs in CADASIL patients ranges from 45 to 51.3 years [30,44,45,46], specifically approximately 50 years for males and 53 for females [47], and that of NOTCH3 cysteine-sparing carriers is 53.64 years in the current review. Given the fact that the a number of cases reported onset age, as well as the fact that limited information was provided on corresponding gender and onset symptoms, the actual age may be different.
Radiological findings in patients with NOTCH3 cysteine-sparing mutations exhibit significant variability, reflecting a spectrum of clinical manifestations. WMHs in anterior temporal pole and external capsule are highly suggestive of CADASIL, observed in about 80% of all CADASIL patients [48,49]. Of patients with NOTCH3 cysteine-sparing mutations, 91% did not have anterior temporal pole involvement [4]. Similarly, we found a lower proportion of anterior temporal pole WMHs (10.50%) and that of external capsule (25.11%). Altogether, these findings suggest that a relatively milder white matter involvement in these two specific locations might be a characteristic of NOTCH3 cysteine-sparing mutations. The observed variation in temporal pole involvement between CADASIL cases with cysteine-sparing mutations and those with cysteine-affecting mutations could be attributed to the distinct convolutional structure and vascularization of the temporal pole. This area might be particularly susceptible to the effects of myelin depletion or edema, which can result from the aggregation of NOTCH3 ECD and changes in the microvasculature. These unique anatomical and physiological characteristics of the temporal pole could explain the differential impact observed in CADASIL phenotypes depending on the type of mutation [50]. In patients with cysteine-sparing mutations, these mutations are less involved in GOM deposits around vascular smooth muscle cells and may have less impact on the drainage of the interstitial fluid and white matter rarefaction. This hypothesis underscores the need for further research to validate the speculated effects and mechanisms. CMBs were prevalent in 72.73% of cases, with a marked increase in older patients, indicating a correlation between age and the burden of vascular lesions. The spatial distribution of CMBs, predominantly in the basal ganglia, thalamus, and brainstem, aligns with patterns seen in typical CADASIL but with potentially greater severity. Advanced MRI sequences and higher magnetic field strengths allowed for more sensitive detection of these lesions. However, variations in imaging protocols across studies may have contributed to inconsistencies in reporting, highlighting the need for standardized imaging criteria in future research.
The present result also suggests that compared with cysteine-sparing mutations in Exon 7–34, mutations in Exon 1–6 presented a high risk of typical CADASIL phenotype, somewhat in line with previous findings that the position of NOTCH3 cysteine-affecting variants plays a crucial role in determining CADASIL clinical and radiological severity. Specifically, variants in EGFr 1–6 of NOTCH3 are associated with an earlier onset of stroke and a more significant accumulation of WMHs, indicating a more severe disease progression [10,51,52,53]. Previous evidence speculated that NOTCH3 EGFr 7–34 were likely associated with attenuated CADASIL or late-onset phenotype. Therefore, the manifestations were relatively mild at currently considered normal age for cerebrovascular disease [52]. Therefore, NOTCH3 cysteine-sparing mutations located in the higher EGFr domains might represent an underrecognized risk factor for cerebral small vessel disease, suggesting the need for increased awareness and investigation into these variants’ impact on the disease’s pathology. Given the fact that the number of patients in different EGFr domains in the current sample was too low for further reliable analysis, we could not assess the associations with onset age, clinical phenotypes, and imaging features. Large-scale studies including more detailed phenotypic information are warranted.
Like typical CADASIL, the characteristics of clinical and radiological phenotypes vary across different regions in patients with NOTCH3 cysteine-sparing mutations. The clinical manifestation burden of Asian patients was milder than Western patients; however, the radiological cerebral phenotypes of Asian patients were more notable, especially for lacunes and cerebral microbleeds. Previous research suggests that involvement of the white matter in the temporal pole was less common in Chinese and Korean populations, while TIA and stroke occurrences were more frequent in Chinese and Japanese populations. These groups are characterized by a high prevalence of the R544C mutation. This pattern hints at a potential mechanism influenced by varying genotype frequencies across different ethnicities, indicating that specific genetic variants like R544C might have distinct pathological impacts in diverse populations [54,55]. Additionally, vascular risk factors, including hypertension and diabetes mellitus, are more prevalent in Asians due to the notable lacune presence [56]. The radiological cerebral findings summarized here indicate that other genetic background effects should be considered as possible mechanisms.
Concerning limitations, the small sample size and a low number of NOTCH3 cysteine-sparing mutants could skew the frequencies of different phenotypes. In addition, the number of studies in the Asian population is significantly higher than that in the Caucasian one. Moreover, due to lacking unified standards and definitions of various clinical phenotypes, the specific prevalence of sub-phenotypes, such as ischemic or hemorrhage stroke, migraine, and apathy, could not be evaluated further in our review. Finally, some population-based studies just analyzed the 33 exons of the NOTCH3 gene, leaving the other atypical mutations such as small deletions or mutations involving the intron to be assessed.
3.4. Future Directions
Given the findings of this systematic review, several areas warrant further investigation. First, future studies should aim to standardize MRI protocols, including the use of specific sequences and magnetic field strengths, to ensure consistent detection and characterization of white matter lesions and vascular abnormalities across different patient populations. Advanced imaging techniques, such as high-resolution MRI and quantitative imaging biomarkers, could provide more detailed information about the extent and nature of vascular lesions in CADASIL. These techniques may help identify subtle changes not detectable with conventional imaging methods.
Longitudinal studies are needed to track radiological and clinical feature progression over time in patients with NOTCH3 cysteine-sparing mutations. Such studies could provide insights into the natural history of the disease and identify early markers of disease progression.
Further research is needed to explore ethnic and geographic variability in the prevalence and phenotypic expression of NOTCH3 cysteine-sparing mutations. Understanding these differences could inform region-specific diagnostic and management guidelines.
4. Materials and Methods
We registered a PREPARE (Practice guideline Registration for transPAREncy, No. IPGRP-2022CN186) at http://guidelines-registry.cn “http://guidelines-registry.cn/guid/1560?lang=en (accessed on 16 November 2023)” for the Chinese guideline for diagnosing and managing CADASIL. In addressing the challenges of diagnosing CADASIL, we adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines to review literature on phenotypes linked to NOTCH3 cysteine-sparing mutations in individuals suspected of having CADASIL. Drawing on data from existing studies, this systematic review does not necessitate ethical standards committee approval. The authors confirm that all the supporting data can be found within the article and its Supplementary Materials.
4.1. Search Strategy
An extensive systematic literature search was conducted to identify all published CADASIL pedigrees, families, and individual patients up to September 2022 and was performed independently by three reviewers (C.Y., H.F., and J.N.). We searched the MEDLINE, EMBASE, China National Knowledge Internet, Wanfang Data, and the Cochrane Library using search strings that included the following terms: “CADASIL”, “Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy”, “cysteine”, “dementia”, “cognition”, “lacune”, “white matter”, “cerebral microbleeds”, “skin biopsy”, and “granular osmiophilic material”. The search string used for literature search is provided in Supplementary Table S1. The PICO (Population, Intervention, Comparison, Outcomes) framework was applied to structure our search strategy (Supplementary Table S2). We did not restrict the search by language; we limited it to human studies and included conference abstracts.
Two reviewers, C.Y. and H.F., independently evaluated the titles and abstracts of all identified publications in our search, without knowledge of each other’s decisions. Subsequently, full texts of studies selected at this initial phase were further examined for eligibility by these two reviewers, who also noted any reasons for exclusion. The inter-rater reliability was assessed using Cohen’s kappa coefficient to ensure consistency in study selection, and Cohen’s kappa was 0.92. Any discrepancies were addressed and resolved through discussion and agreement, involving a third reviewer (Y.M.) when necessary.
Our inclusion criteria encompassed studies presenting case reports, case series, or any other research design (excluding review articles) that described the clinical or cerebral radiological phenotype of one or more individuals. These descriptions ranged from simply noting an individual’s health status to providing detailed case reports. Furthermore, the study had to regard the rare variant as likely or definitively pathogenic. The process of our literature review is illustrated in the flow diagram provided in Figure 2.
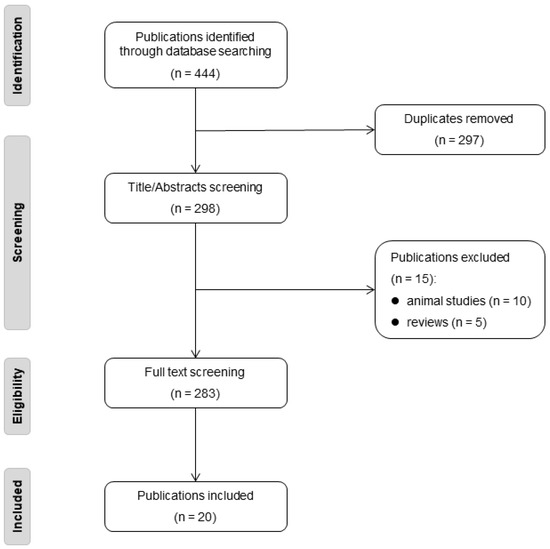
Figure 2.
The flow diagram of literature review.
4.2. Data Extraction
The reference lists of all included studies were screened to identify and assess other potentially relevant publications not captured in the initial search. The quality of included studies was assessed using the Newcastle–Ottawa Scale (NOS). Publication bias was evaluated through visual inspection of funnel plots and statistical tests where appropriate. We also assessed the quality of the conclusions drawn in the included studies to ensure the robustness of our findings.
We gathered information on the first author, year of publication, journal name, and the count of eligible individuals and pedigrees from each selected publication. For articles in languages other than English, we obtained a full translation whenever the English abstract failed to offer adequate details or was unavailable. We meticulously collected data for every individual deemed eligible using a pre-defined form. This included clinical cerebral phenotype aspects such as stroke, headache, cognitive symptoms, gait issues, seizures, and psychiatric conditions, as well as radiological cerebral phenotype elements like the occurrence of lacunes, white matter lesions, and cerebral microbleeds. These elements are recognized indicators of cerebral small vessel disease.
4.3. Data Synthesis
To evaluate the apparent frequency of each identified cerebral phenotype, both clinical and radiological, we compiled and summarized data concerning the presence or absence of these features. Additionally, we gathered cumulative evidence of any vascular radiological characteristics to provide a comprehensive overview of their occurrence. Specifically, descriptive statistics were used to summarize the clinical and demographic characteristics of the patients. For categorical variables, frequencies and percentages were calculated. Mean and standard errors (SEs) were reported for continuous variables. We analyzed the outcomes for patients with suspicious clinical CADASIL syndrome and a cysteine-sparing NOTCH3 mutation, focusing on identifying common patterns and variations in the prevalence of associated phenotypes. This assessment aimed to elucidate the shared characteristics and distinctive features in the manifestation of these mutations. Specifically, the frequency of each clinical phenotype was calculated. Frequencies of phenotypes in NOTCH3 cysteine-sparing mutation patients were compared with those of typical CADASIL patients using chi-square tests. Similarly, clinical and radiological phenotypes between Asian and Western patients were compared.
5. Conclusions
Patients with NOTCH3 cysteine-sparing mutations manifested with typical clinical CADASIL syndrome and radiological profile, mostly with gait and cognitive impairment but rare WMHs in the anterior temporal pole and external capsule. The characterized phenotype summarized here could aid in further diagnosing and managing CADASIL-like patients with NOTCH3 cysteine-sparing mutations. Evidence from clinical, radiological, and pathological GOM deposits indicated that most NOTCH3 cysteine-sparing mutations have a phenotype similar to that of CADASIL. This implied the hidden pathogenesis of NOTCH3 mutations, which needs further investigation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25168796/s1.
Author Contributions
Y.C. and M.Y. conceptualized, designed and wrote the primary draft of the manuscript. D.-D.Z., F.H., N.J. and Y.-C.Z. edited and revised the final version manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the CAMS Innovation Fund for Medical Sciences, grant number CIFMS #2021-I2M-1-025.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviation List
| Abbreviation | Definition |
| CADASIL | Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy |
| NOTCH3 | Neurogenic Locus Notch Homolog Protein 3 |
| ECD | Extracellular Domain |
| EGFr | Epidermal Growth Factor-like Repeats |
| GOM | Granular Osmiophilic Material |
| MRI | Magnetic Resonance Imaging |
| WMHs | White Matter Hyperintensities |
| CMBs | Cerebral Microbleeds |
| MAR | Missing at Random |
| MICE | Multiple Imputation by Chained Equations |
| SE | Standard Error |
| TIA | Transient Ischemic Attack |
References
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, I.; Bianchi, S.; De Stefano, N.; Dichgans, M.; Dotti, M.T.; Duering, M.; Jouvent, E.; Korczyn, A.D.; Lesnik-Oberstein, S.A.; Malandrini, A.; et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: Update on clinical, diagnostic, and management aspects. BMC Med. 2017, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Tournier-Lasserve, E.; Joutel, A.; Melki, J.; Weissenbach, J.; Lathrop, G.M.; Chabriat, H.; Mas, J.L.; Cabanis, E.A.; Baudrimont, M.; Maciazek, J.; et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat. Genet. 1993, 3, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Muiño, E.; Gallego-Fabrega, C.; Cullell, N.; Carrera, C.; Torres, N.; Krupinski, J.; Roquer, J.; Montaner, J.; Fernández-Cadenas, I. Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. Int. J. Mol. Sci. 2017, 18, 1964. [Google Scholar] [CrossRef]
- Mancuso, M.; Arnold, M.; Bersano, A.; Burlina, A.; Chabriat, H.; Debette, S.; Enzinger, C.; Federico, A.; Filla, A.; Finsterer, J.; et al. Monogenic cerebral small-vessel diseases: Diagnosis and therapy. Consensus recommendations of the European Academy of Neurology. Eur. J. Neurol. 2020, 27, 909–927. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Karpinska, A.; Rosner, S.; Hopfner, F.; Zechmeister, M.; Peters, N.; Kremmer, E.; Haffner, C.; Giese, A.; Dichgans, M.; et al. Co-aggregate formation of CADASIL-mutant NOTCH3: A single-particle analysis. Hum. Mol. Genet. 2011, 20, 3256–3265. [Google Scholar] [CrossRef]
- Adachi, Y.; Nakashima, K. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) characteristics in Japan: Variety of clinical features. Intern. Med. 2000, 39, 681–682. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Hayashi, M.; Nakashima, K.J.J.J.S. Examination of notch3 mutations in Japanese patients with leukoaraiosis without hypertension. Jpn. J. Stroke 2006, 28, 585–589. [Google Scholar] [CrossRef]
- Vlachakis, D.; Tsaniras, S.C.; Ioannidou, K.; Papageorgiou, L.; Baumann, M.; Kossida, S. A series of Notch3 mutations in CADASIL; insights from 3D molecular modelling and evolutionary analyses. J. Mol. Biochem. 2014, 3, 134. [Google Scholar]
- Rutten, J.W.; Hack, R.J.; Duering, M.; Gravesteijn, G.; Dauwerse, J.G.; Overzier, M.; van den Akker, E.B.; Slagboom, E.; Holstege, H.; Nho, K.; et al. Broad phenotype of cysteine-altering NOTCH3 variants in UK Biobank: CADASIL to nonpenetrance. Neurology 2020, 95, e1835–e1843. [Google Scholar] [CrossRef]
- Ling, Y.; De Guio, F.; Jouvent, E.; Duering, M.; Hervé, D.; Guichard, J.P.; Godin, O.; Dichgans, M.; Chabriat, H. Clinical correlates of longitudinal MRI changes in CADASIL. J. Cereb. Blood Flow Metab. 2019, 39, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Stellingwerff, M.D.; Nulton, C.; Helman, G.; Roosendaal, S.D.; Benko, W.S.; Pizzino, A.; Bugiani, M.; Vanderver, A.; Simons, C.; van der Knaap, M.S. Early-Onset Vascular Leukoencephalopathy Caused by Bi-Allelic NOTCH3 Variants. Neuropediatrics 2022, 53, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, C.; Shang, J.; Sun, R.; Wang, W.; Li, W.; Gao, D.; Huo, X.; Shi, Y.; Wang, Y.; et al. De novo Mutation Enables NOTCH3ECD Aggregation and Mitochondrial Dysfunction via Interactions with BAX and BCL-2. J. Alzheimer’s Dis. 2022, 86, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.R.; Elias, I.; Fernandes, C.; Machado, R.; Galego, O.; Santo, G. NOTCH3 mutations in a cohort of Portuguese patients within CADASIL spectrum phenotype. Neurogenetics 2022, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Feng, S. Clinical and Imaging Characteristics and Genetic Analysis of 26 Cases of CADASIL in Shandong. Master’s Thesis, Shandong University, Jinan, China, 2021. [Google Scholar]
- Liu, J.Y.; Yao, M.; Dai, Y.; Han, F.; Zhai, F.F.; Zhang, D.D.; Zhou, L.X.; Ni, J.; Zhang, S.Y.; Cui, L.Y.; et al. Rare NOTCH3 Variants in a Chinese Population-Based Cohort and Its Relationship With Cerebral Small Vessel Disease. Stroke 2021, 52, 3918–3925. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Mizuta, I.; Ueda, A.; Nozaki, H.; Sakurai, K.; Onodera, O.; Ando, Y.; Yamada, K.; Yuasa, H.; Mizuno, T. Heterozygous Cysteine-sparing NOTCH3 Variant p.Val237Met in a Japanese Patient with Suspected Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy. Intern. Med. 2021, 60, 2479–2482. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, Q.; Zhou, Y.; Yi, F.; Tang, H.; Yao, L.; Tian, Y.; Xie, N.; Luo, M.; Wang, Z.; et al. NOTCH3 Variants and Genotype-Phenotype Features in Chinese CADASIL Patients. Front. Genet. 2021, 12, 705284. [Google Scholar] [CrossRef] [PubMed]
- Arnardottir, S.; Del Gaudio, F.; Klironomos, S.; Braune, E.B.; Lombraña, A.A.; Oliveira, D.V.; Jin, S.; Karlström, H.; Lendahl, U.; Sjöstrand, C. Novel Cysteine-Sparing Hypomorphic NOTCH3 A1604T Mutation Observed in a Family with Migraine and White Matter Lesions. Neurol. Genet. 2021, 7, e584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Qin, X.-M.; Wu, Y.-Y.; Shi, Y.-Y.; Li, G.; Zhao, J.-Y.; Gao, D.-D.; Qin, W.-W.; Zhang, J.-W. Clinical and imaging characteristics of five patients with autosomal dominant cerebral artery disease with subcortical infarction and white matter encephalopathy carrying a cysteine-sparing NOTCH3 gene mutation. Chin. J. Neurol. 2020, 03, 184–191. [Google Scholar]
- Mukai, M.; Mizuta, I.; Watanabe-Hosomi, A.; Koizumi, T.; Matsuura, J.; Hamano, A.; Tomimoto, H.; Mizuno, T. Genotype-phenotype correlations and effect of mutation location in Japanese CADASIL patients. J. Hum. Genet. 2020, 65, 637–646. [Google Scholar] [CrossRef]
- Kim, H.; Lim, Y.M.; Lee, E.J.; Oh, Y.J.; Kim, K.K. Clinical and imaging features of patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy and cysteine-sparing NOTCH3 mutations. PLoS ONE 2020, 15, e0234797. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, W.; Li, Y.; Song, C.; Wang, P.; Wang, H.; Sun, X. A novel cysteine-sparing G73A mutation of NOTCH3 in a Chinese CADASIL family. Neurogenetics 2020, 21, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Qualtieri, A.; Ungaro, C.; Bagalà, A.; Bianchi, S.; Pantoni, L.; Moccia, M.; Mazzei, R. Notch3 protein expression in skin fibroblasts from CADASIL patients. J. Neurol. Sci. 2018, 390, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Tachiyama, K.; Shiga, Y.; Shimoe, Y.; Mizuta, I.; Mizuno, T.; Kuriyama, M. CADASIL with cysteine-sparing NOTCH3 mutation manifesting as dissociated progression between cognitive impairment and brain image findings in 3 years: A case report. Rinsho Shinkeigaku Clin. Neurol. 2018, 58, 235–240. [Google Scholar] [CrossRef][Green Version]
- Gong, P.; Luo, Y.-H.; Han, S.; Wang, L. A case of suspected autosomal dominant cerebral artery disease with subcortical cerebral infarction and white matter encephalopathy in which NOTCH3 mutation does not involve cysteine alteration. Chin. J. Geriatr. Cardiovasc. Cerebrovasc. Dis. 2017, 19, 1100–1102. [Google Scholar]
- Qureshi, A.I.; Khan, M.T.; Naveed, O.; Saleem, M.A. Potential New Cysteine Sparing Mutation in the NOTCH3 Gene in a Patient with Nonfamilial CADASIL-like Disease. J. Vasc. Interv. Neurol. 2017, 9, 51–54. [Google Scholar] [PubMed]
- Matsushima, T.; Conedera, S.; Tanaka, R.; Li, Y.; Yoshino, H.; Funayama, M.; Ikeda, A.; Hosaka, Y.; Okuzumi, A.; Shimada, Y.; et al. Genotype-phenotype correlations of cysteine replacement in CADASIL. Neurobiol. Aging 2017, 50, 169.e7–169.e14. [Google Scholar] [CrossRef]
- Vincenzi, C.; Grisendi, I.; Assenza, F.; Napoli, M.; Moratti, C.; Pascarella, R.; Valzania, F.; Zedde, M. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (Cadasil): Description of a single center experience. Eur. Stroke J. 2021, 6, 423–424. [Google Scholar]
- Liu, X.; Zuo, Y.; Sun, W.; Zhang, W.; Lv, H.; Huang, Y.; Xiao, J.; Yuan, Y.; Wang, Z. The genetic spectrum and the evaluation of CADASIL screening scale in Chinese patients with NOTCH3 mutations. J. Neurol. Sci. 2015, 354, 63–69. [Google Scholar] [CrossRef]
- Opherk, C.; Duering, M.; Peters, N.; Karpinska, A.; Rosner, S.; Schneider, E.; Bader, B.; Giese, A.; Dichgans, M. CADASIL mutations enhance spontaneous multimerization of NOTCH3. Hum. Mol. Genet. 2009, 18, 2761–2767. [Google Scholar] [CrossRef]
- Wollenweber, F.A.; Hanecker, P.; Bayer-Karpinska, A.; Malik, R.; Bäzner, H.; Moreton, F.; Muir, K.W.; Müller, S.; Giese, A.; Opherk, C.; et al. Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke 2015, 46, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Xiromerisiou, G.; Marogianni, C.; Dadouli, K.; Zompola, C.; Georgouli, D.; Provatas, A.; Theodorou, A.; Zervas, P.; Nikolaidou, C.; Stergiou, S.; et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy revisited: Genotype-phenotype correlations of all published cases. Neurol. Genet. 2020, 6, e434. [Google Scholar] [CrossRef]
- Brass, S.D.; Smith, E.E.; Arboleda-Velasquez, J.F.; Copen, W.A.; Frosch, M.P. Case records of the Massachusetts General Hospital. Case 12-2009. A 46-year-old man with migraine, aphasia, and hemiparesis and similarly affected family members. N. Engl. J. Med. 2009, 360, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Uchino, M.; Hirano, T.; Uyama, E.; Hashimoto, Y. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and CADASIL-like disorders in Japan. Ann. N. Y. Acad. Sci. 2002, 977, 273–278. [Google Scholar] [CrossRef]
- Fouillade, C.; Chabriat, H.; Riant, F.; Mine, M.; Arnoud, M.; Magy, L.; Bousser, M.G.; Tournier-Lasserve, E.; Joutel, A. Activating NOTCH3 mutation in a patient with small-vessel-disease of the brain. Hum. Mutat. 2008, 29, 452. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Mosca, L.; Erro, R.; Cervasio, M.; Allocca, R.; Vitale, C.; Leonardi, A.; Caranci, F.; Del Basso-De Caro, M.L.; Barone, P.; et al. Hypomorphic NOTCH3 mutation in an Italian family with CADASIL features. Neurobiol. Aging 2015, 36, 547.e5–547.e11. [Google Scholar] [CrossRef]
- Erro, R.; Lees, A.J.; Moccia, M.; Picillo, M.; Penco, S.; Mosca, L.; Vitale, C.; Barone, P. Progressive parkinsonism, balance difficulties, and supranuclear gaze palsy. JAMA Neurol. 2014, 71, 104–107. [Google Scholar] [CrossRef]
- Coupland, K.; Lendahl, U.; Karlström, H. Role of NOTCH3 Mutations in the Cerebral Small Vessel Disease Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. Stroke 2018, 49, 2793–2800. [Google Scholar] [CrossRef]
- Hung, L.Y.; Ling, T.K.; Lau, N.K.C.; Cheung, W.L.; Chong, Y.K.; Sheng, B.; Kwok, K.M.; Mak, C.M. Genetic diagnosis of CADASIL in three Hong Kong Chinese patients: A novel mutation within the intracellular domain of NOTCH3. J. Clin. Neurosci. 2018, 56, 95–100. [Google Scholar] [CrossRef]
- Brice, S.; Reyes, S.; Jabouley, A.; Machado, C.; Rogan, C.; Gastellier, N.; Alili, N.; Guey, S.; Jouvent, E.; Hervé, D.; et al. Trajectory Pattern of Cognitive Decline in Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. Neurology 2022, 99, e1019–e1031. [Google Scholar] [CrossRef]
- Gravesteijn, G.; Hack, R.J.; Mulder, A.A.; Cerfontaine, M.N.; van Doorn, R.; Hegeman, I.M.; Jost, C.R.; Rutten, J.W.; Lesnik Oberstein, S.A.J. NOTCH3 variant position is associated with NOTCH3 aggregation load in CADASIL vasculature. Neuropathol. Appl. Neurobiol. 2022, 48, e12751. [Google Scholar] [CrossRef]
- Poganik, J.R.; Aye, Y. Electrophile Signaling and Emerging Immuno- and Neuro-modulatory Electrophilic Pharmaceuticals. Front. Aging Neurosci. 2020, 12, 1. [Google Scholar] [CrossRef]
- Moreton, F.C.; Razvi, S.S.; Davidson, R.; Muir, K.W. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol. Scand. 2014, 130, 197–203. [Google Scholar] [CrossRef]
- Chabriat, H.; Vahedi, K.; Iba-Zizen, M.T.; Joutel, A.; Nibbio, A.; Nagy, T.G.; Krebs, M.O.; Julien, J.; Dubois, B.; Ducrocq, X.; et al. Clinical spectrum of CADASIL: A study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet 1995, 346, 934–939. [Google Scholar] [CrossRef]
- Chen, S.; Ni, W.; Yin, X.Z.; Liu, H.Q.; Lu, C.; Zheng, Q.J.; Zhao, G.X.; Xu, Y.F.; Wu, L.; Zhang, L.; et al. Clinical features and mutation spectrum in Chinese patients with CADASIL: A multicenter retrospective study. CNS Neurosci. Ther. 2017, 23, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Juhosová, M.; Chandoga, J.; Cisárik, F.; Dallemule, S.; Ďurina, P.; Jarásková, D.; Jungová, P.; Kantarská, D.; Kvasnicová, M.; Mistrík, M.; et al. Influence of different spectra of NOTCH3 variants on the clinical phenotype of CADASIL—Experience from Slovakia. Neurogenetics 2023, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Bedini, G.; Markus, H.S.; Vitali, P.; Colli-Tibaldi, E.; Taroni, F.; Gellera, C.; Baratta, S.; Mosca, L.; Carrera, P.; et al. The role of clinical and neuroimaging features in the diagnosis of CADASIL. J. Neurol. 2018, 265, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Nannucci, S.; Pescini, F.; Bertaccini, B.; Bianchi, S.; Ciolli, L.; Valenti, R.; Dotti, M.T.; Federico, A.; Inzitari, D.; Pantoni, L. Clinical, familial, and neuroimaging features of CADASIL-like patients. Acta Neurol. Scand. 2015, 131, 30–36. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ihara, M.; Tham, C.; Low, R.W.; Slade, J.Y.; Moss, T.; Oakley, A.E.; Polvikoski, T.; Kalaria, R.N. Neuropathological correlates of temporal pole white matter hyperintensities in CADASIL. Stroke 2009, 40, 2004–2011. [Google Scholar] [CrossRef]
- Rutten, J.W.; Dauwerse, H.G.; Gravesteijn, G.; van Belzen, M.J.; van der Grond, J.; Polke, J.M.; Bernal-Quiros, M.; Lesnik Oberstein, S.A. Archetypal NOTCH3 mutations frequent in public exome: Implications for CADASIL. Ann. Clin. Transl. Neurol. 2016, 3, 844–853. [Google Scholar] [CrossRef]
- Rutten, J.W.; Van Eijsden, B.J.; Duering, M.; Jouvent, E.; Opherk, C.; Pantoni, L.; Federico, A.; Dichgans, M.; Markus, H.S.; Chabriat, H.; et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1-6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7-34 pathogenic variant. Genet. Med. 2019, 21, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Dupé, C.; Guey, S.; Biard, L.; Dieng, S.; Lebenberg, J.; Grosset, L.; Alili, N.; Hervé, D.; Tournier-Lasserve, E.; Jouvent, E.; et al. Phenotypic variability in 446 CADASIL patients: Impact of NOTCH3 gene mutation location in addition to the effects of age, sex and vascular risk factors. J. Cereb. Blood Flow Metab. 2023, 43, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Bae, J.S.; Lee, J.Y.; Song, H.K.; Lee, J.H.; Lee, M.; Kim, C.; Lee, S.H. Genotype and Phenotype Differences in CADASIL from an Asian Perspective. Int. J. Mol. Sci. 2022, 23, 11506. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Zhang, Y.; Zhang, L.; Xie, J.J.; Li, H.F.; Wu, Z.Y. Genetic spectrum of NOTCH3 and clinical phenotype of CADASIL patients in different populations. CNS Neurosci. Ther. 2022, 28, 1779–1789. [Google Scholar] [CrossRef]
- Jacob, M.A.; Ekker, M.S.; Allach, Y.; Cai, M.; Aarnio, K.; Arauz, A.; Arnold, M.; Bae, H.J.; Bandeo, L.; Barboza, M.A.; et al. Global Differences in Risk Factors, Etiology, and Outcome of Ischemic Stroke in Young Adults-A Worldwide Meta-analysis: The GOAL Initiative. Neurology 2022, 98, e573–e588. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).