
Albiflorin Decreases Glutamate Release from Rat Cerebral Cortex Nerve Terminals (Synaptosomes) through Depressing P/Q-Type Calcium Channels and Protein Kinase A Activity
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Albiflorin Reduced Ca2+-Dependent Vesicular Exocytotic Glutamate Release from Cerebral Cortical Synaptosomes in Rats
2.2. Reduced Glutamate Release from Albiflorin Is Mediated through Ca2+ Channel Suppression
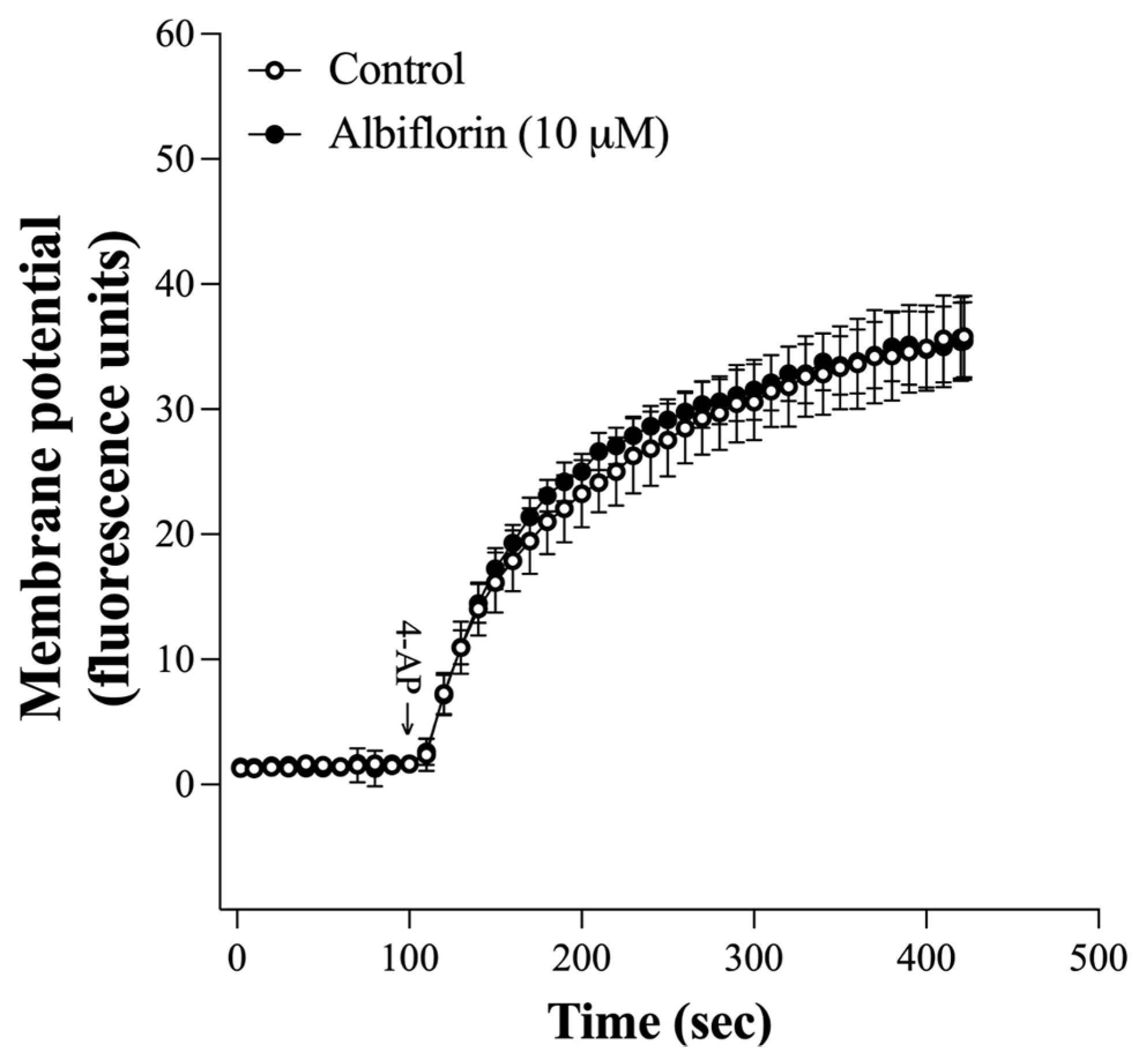
2.3. Albiflorin Failed to Affect the Synaptosomal Membrane Potential
2.4. The Albiflorin-Mediated Inhibition of Glutamate Release Involved the Suppression of the Protein Kinase A (PKA) Pathway
2.5. Albiflorin Reduces the Release Competence of Synaptic Vesicles Evoked by 4-AP in Synaptosomes
3. Discussion
3.1. The Mechanism by Which Albiflorin Inhibited the 4-AP-Evoked Glutamate Release
3.2. Therapeutic Implications
4. Materials and Methods
4.1. Animals
4.2. Preparation of Synaptosomes
4.3. Glutamate Release Assay
4.4. Plasma Membrane Potential
4.5. TEM
4.6. Western Blotting
4.7. Data Analysis
4.8. Chemicals
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef]
- Wu, S.H.; Wu, D.G.; Chen, Y.W. Chemical constituents and bioactivities of plants from the genus Paeonia. Chem. Biodivers. 2010, 7, 90–104. [Google Scholar] [CrossRef]
- He, D.Y.; Dai, S.M. Anti-inflammatory and immunomodulatory effects of paeonia lactiflora pall., a traditional chinese herbal medicine. Front. Pharmacol. 2011, 2, 10. [Google Scholar] [CrossRef]
- Fei, F.; Yang, H.; Peng, Y.; Wang, P.; Wang, S.; Zhao, Y.; Huang, J.; Yu, X.; Feng, S.; Sun, R.; et al. Sensitive analysis and pharmacokinetic study of the isomers paeoniflorin and albiflorin after oral administration of Total Glucosides of White Paeony Capsule in rats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1022, 30–37. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, L.; Wang, J.; Wang, C.; Yang, Z.; Wang, C.; Zhu, Y.; Zhang, J. Paeoniflorin and albiflorin attenuate neuropathic pain via MAPK pathway in chronic constriction injury rats. Evid. Based Complement. Alternat Med. 2016, 1, 8082753. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Xia, Y.; Lin, L.; Zhang, Z.; Tian, H.; Li, K. Next-generation metabolomics in the development of new antidepressants: Using albiflorin as an example. Curr. Pharm. Des. 2018, 24, 2530–2540. [Google Scholar] [CrossRef]
- Wang, X.; Su, L.; Tan, J.; Ding, T.; Yue, Y. Albiflorin alleviates DSS-induced ulcerative colitis in mice by reducing inflammation and oxidative stress. Iran. J. Basic. Med. Sci. 2023, 26, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Wang, J.X.; Hu, X.X.; Chen, L.; Qiu, Z.K.; Zhao, N.; Yu, Z.D.; Sun, S.Z.; Xu, Y.Y.; Guo, Y.; et al. Antidepressant-like effects of albiflorin extracted from Radix paeoniae Alba. J. Ethnopharmacol. 2016, 179, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.K.; He, J.L.; Liu, X.; Zeng, J.; Chen, J.S.; Nie, H. Anti-PTSD-like effects of albiflorin extracted from Radix paeoniae Alba. J. Ethnopharmacol. 2017, 198, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Mei, Y.; Shi, X.Q.; Zhang, Y.F.; Wang, X.Y.; Guan, L.; Wang, Q.; Pan, H.F. Albiflorin ameliorates memory deficits in APP/PS1 transgenic mice via ameliorating mitochondrial dysfunction. Brain Res. 2019, 1719, 113–123. [Google Scholar] [CrossRef]
- Wang, Q.S.; Yan, K.; Li, K.D.; Gao, L.N.; Wang, X.; Liu, H.; Zhang, Z.; Li, K.; Cui, Y.L. Targeting hippocampal phospholipid and tryptophan metabolism for antidepressant-like effects of albiflorin. Phytomedicine 2021, 92, 153735. [Google Scholar] [CrossRef]
- Lee, S.; Ryu, S.M.; Kim, D.H.; Lee, Y.E.; Lee, S.J.; Kang, S.; Kim, J.S.; Lee, S.I. Neuroprotective effect of Geijigadaehwang-tang against trimethyltin-induced hippocampal neurodegeneration: An in vitro and in vivo study. J. Ethnopharmacol. 2022, 296, 115451. [Google Scholar] [CrossRef]
- Zhu, F.; Xiong, J.; Yi, F.; Luo, E.; Huang, C.; Li, R. Albiflorin relieves cerebral ischemia-reperfusion injury by activating Nrf2/HO-1 pathway. Histol. Histopathol. 2023, 38, 233–245. [Google Scholar] [CrossRef]
- Fang, P.; Wang, Y.; Sun, F.; Lin, H.; Zhang, X. Effects of albiflorin on oxidative stress and inflammatory responses in rats with acute spinal cord injury. Immun. Inflamm. Dis. 2023, 11, e1015. [Google Scholar] [CrossRef]
- Lazarevic, V.; Mantas, I.; Flais, I.; Svenningsson, P. Fluoxetine suppresses glutamate- and GABA-mediated neurotransmission by altering SNARE complex. Int. J. Mol. Sci. 2019, 20, 4247. [Google Scholar] [CrossRef] [PubMed]
- Mdzinarishvili, A.; Sumbria, R.; Lang, D.; Klein, J. Ginkgo extract EGb761 confers neuroprotection by reduction of glutamate release in ischemic brain. J. Pharm. Pharm. Sci. 2012, 15, 94–102. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Jarvie, P.E.; Robinson, P.J. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat. Protoc. 2008, 3, 1718–1728. [Google Scholar] [CrossRef]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef]
- Greengard, P.; Valtorta, F.; Czernik, A.J.; Benfenati, F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 1993, 259, 780–785. [Google Scholar] [CrossRef]
- Menegon, A.; Bonanomi, D.; Albertinazzi, C.; Lotti, F.; Ferrari, G.; Kao, H.T.; Benfenati, F.; Baldelli, P.; Valtorta, F. Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J. Neurosci. 2006, 26, 11670–11681. [Google Scholar] [CrossRef]
- Nagy, G.; Reim, K.; Matti, U.; Brose, N.; Binz, T.; Rettig, J.; Neher, E.; Sørensen, J.B. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 2004, 41, 417–429. [Google Scholar] [CrossRef]
- Tibbs, G.R.; Dolly, J.O.; Nicholls, D.G. Dendrotoxin, 4-aminopyridine, and beta-bungarotoxin act at common loci but by two distinct mechanisms to induce Ca2+-dependent release of glutamate from guinea-pig cerebrocortical synaptosomes. J. Neurochem. 1989, 52, 201–206. [Google Scholar] [CrossRef]
- Lu, C.W.; Yeh, K.C.; Chiu, K.M.; Lee, M.Y.; Lin, T.Y.; Wang, S.J. The effect of isosaponarin derived from wasabi leaves on glutamate release in rat synaptosomes and its underlying mechanism. Int. J. Mol. Sci. 2022, 23, 8752. [Google Scholar] [CrossRef]
- Lin, T.Y.; Chen, I.Y.; Lee, M.Y.; Lu, C.W.; Chiu, K.M.; Wang, S.J. Inhibition of glutamate release from rat cortical nerve terminals by dehydrocorydaline, an alkaloid from Corydalis yanhusuo. Molecules 2022, 27, 960. [Google Scholar] [CrossRef]
- Jarvis, S.E.; Zamponi, G.W. Interactions between presynaptic Ca2+ channels, cytoplasmic messengers and proteins of the synaptic vesicle release complex. Trends Pharmacol. Sci. 2001, 22, 519–525. [Google Scholar] [CrossRef]
- Vázquez, E.; Sánchez-Prieto, J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef]
- Millán, C.; Sánchez-Prieto, J. Differential coupling of N- and P/Q-type calcium channels to glutamate exocytosis in the rat cerebral cortex. Neurosci. Lett. 2002, 330, 29–32. [Google Scholar] [CrossRef]
- Li, M.; West, J.W.; Numann, R.; Murphy, B.J.; Scheuer, T.; Catterall, W.A. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science 1993, 261, 1439–1442. [Google Scholar] [CrossRef]
- Wu, L.G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Risinger, C.; Bennett, M.K. Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms. J. Neurochem. 1999, 72, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Henter, I.D.; Park, L.T.; Zarate, C.A., Jr. Novel glutamatergic modulators for the treatment of mood disorders: Current status. CNS Drugs 2021, 35, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Hsieh, P.W.; Kuo, J.R.; Wang, S.J. Rosmarinic Acid, a Bioactive Phenolic Compound, Inhibits Glutamate Release from Rat Cerebrocortical Synaptosomes through GABA(A) Receptor Activation. Biomolecules 2021, 11, 1029. [Google Scholar] [CrossRef]
- Chiu, K.M.; Lee, M.Y.; Lu, C.W.; Lin, T.Y.; Wang, S.J. Plantainoside D reduces depolarization-evoked glutamate release from rat cerebral cortical synaptosomes. Molecules 2023, 28, 1313. [Google Scholar] [CrossRef]
- Lu, C.W.; Wu, C.C.; Chiu, K.M.; Lee, M.Y.; Lin, T.Y.; Wang, S.J. Inhibition of Synaptic Glutamate Exocytosis and Prevention of Glutamate Neurotoxicity by Eupatilin from Artemisia argyi in the Rat Cortex. Int. J. Mol. Sci. 2022, 23, 13406. [Google Scholar] [CrossRef] [PubMed]
- Akerman, K.E.; Scott, I.G.; Heikkilä, J.E.; Heinonen, E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiS-C2-(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, C.J.; Hsieh, P.W.; Chiu, K.M.; Lee, M.Y.; Lin, T.Y.; Wang, S.J. An Anthranilate Derivative Inhibits Glutamate Release and Glutamate Excitotoxicity in Rats. Int. J. Mol. Sci. 2022, 23, 2641. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-W.; Lin, T.-Y.; Chang, Y.-Y.; Chiu, K.-M.; Lee, M.-Y.; Wang, S.-J. Albiflorin Decreases Glutamate Release from Rat Cerebral Cortex Nerve Terminals (Synaptosomes) through Depressing P/Q-Type Calcium Channels and Protein Kinase A Activity. Int. J. Mol. Sci. 2024, 25, 8846. https://doi.org/10.3390/ijms25168846
Lu C-W, Lin T-Y, Chang Y-Y, Chiu K-M, Lee M-Y, Wang S-J. Albiflorin Decreases Glutamate Release from Rat Cerebral Cortex Nerve Terminals (Synaptosomes) through Depressing P/Q-Type Calcium Channels and Protein Kinase A Activity. International Journal of Molecular Sciences. 2024; 25(16):8846. https://doi.org/10.3390/ijms25168846
Chicago/Turabian StyleLu, Cheng-Wei, Tzu-Yu Lin, Ya-Ying Chang, Kuan-Ming Chiu, Ming-Yi Lee, and Su-Jane Wang. 2024. "Albiflorin Decreases Glutamate Release from Rat Cerebral Cortex Nerve Terminals (Synaptosomes) through Depressing P/Q-Type Calcium Channels and Protein Kinase A Activity" International Journal of Molecular Sciences 25, no. 16: 8846. https://doi.org/10.3390/ijms25168846