
Protein Language Models and Machine Learning Facilitate the Identification of Antimicrobial Peptides

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
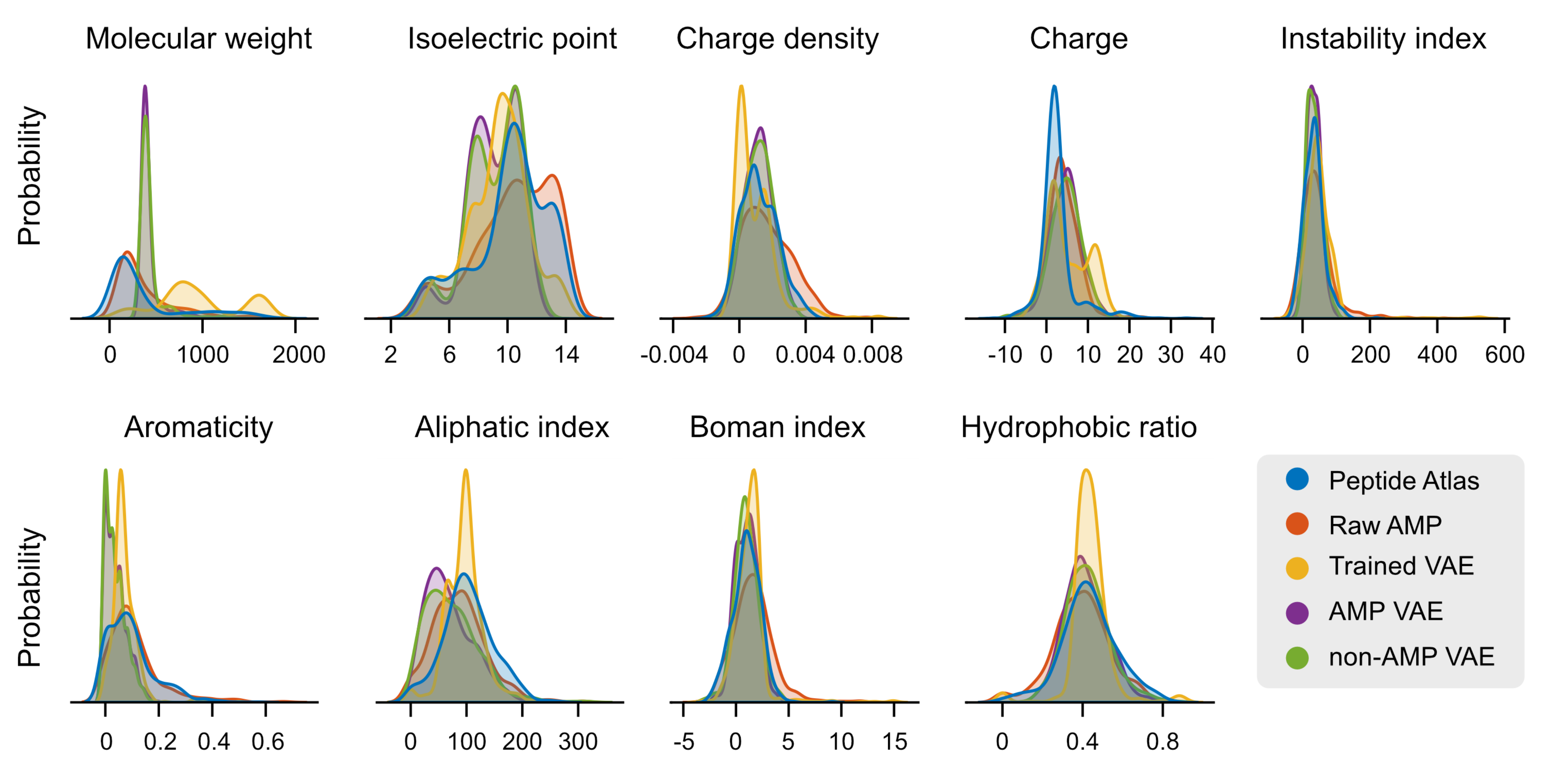
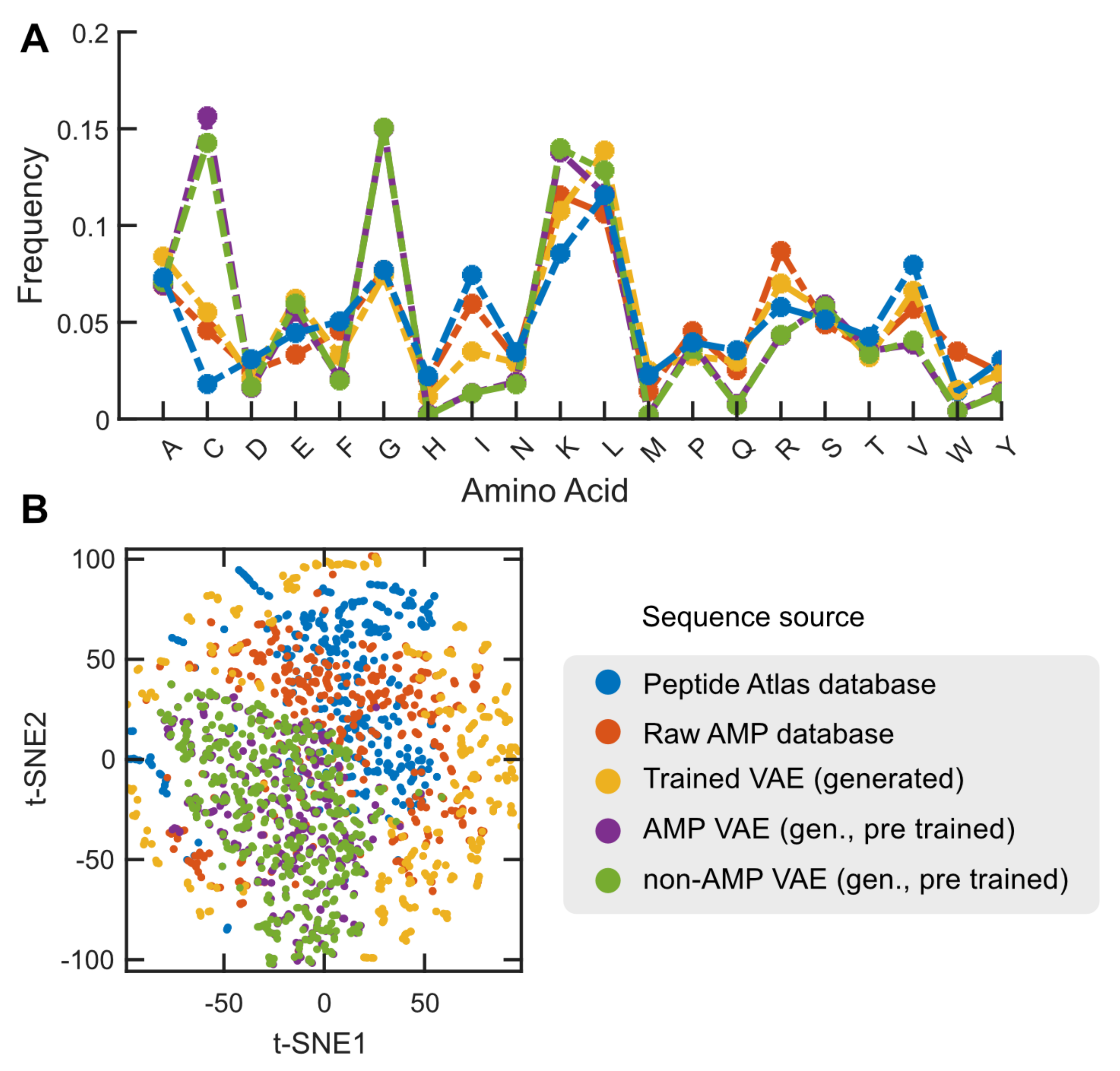
2.1. Main Features of the Studied Datasets
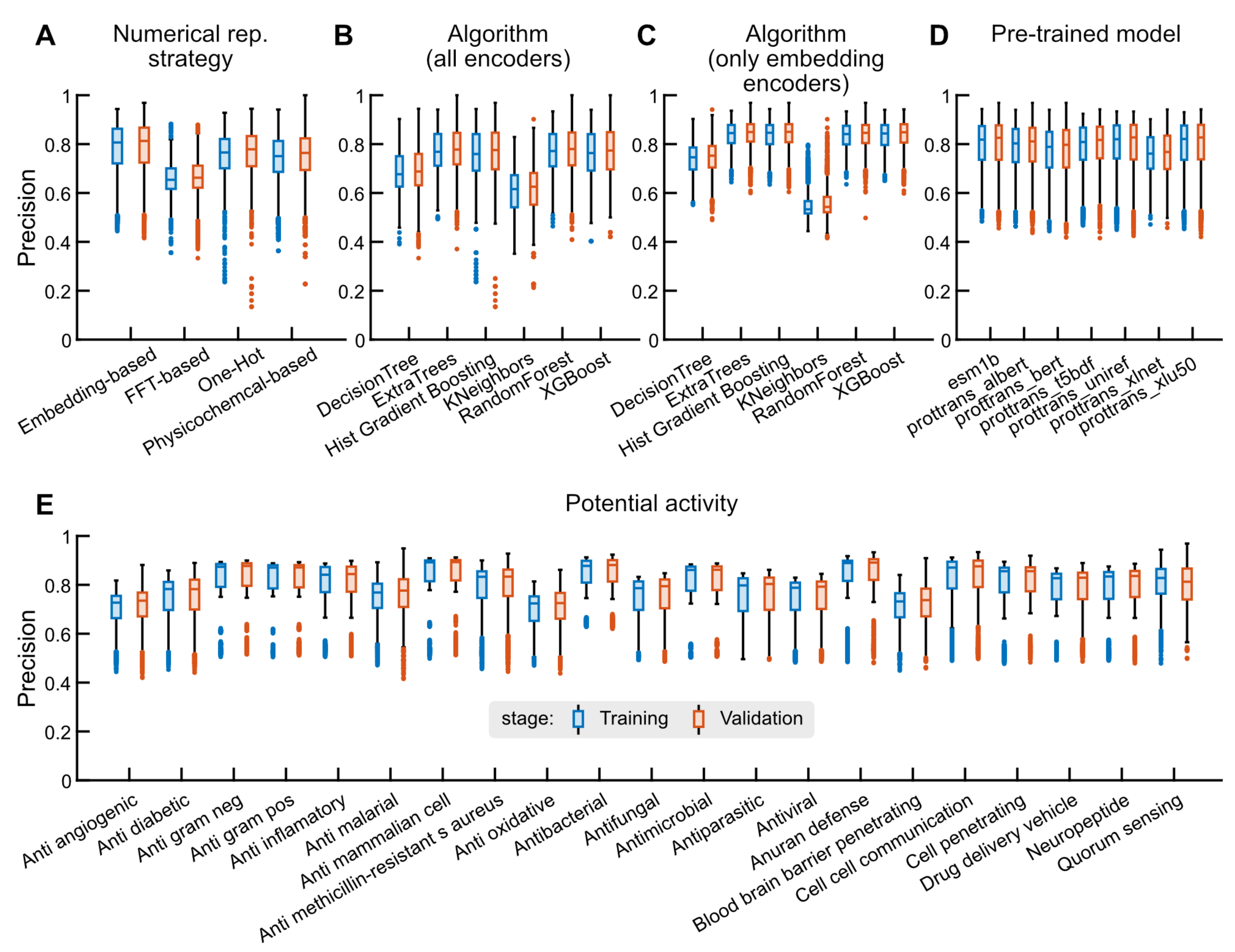
2.2. Binary Classification Tasks
2.2.1. Performance Statistics
2.2.2. Optimizing the Hyperparameters of Selected Models
2.2.3. Benchmark Analysis
2.3. Case Study: Antimicrobial Peptide Discovery and De Novo Peptide Generation
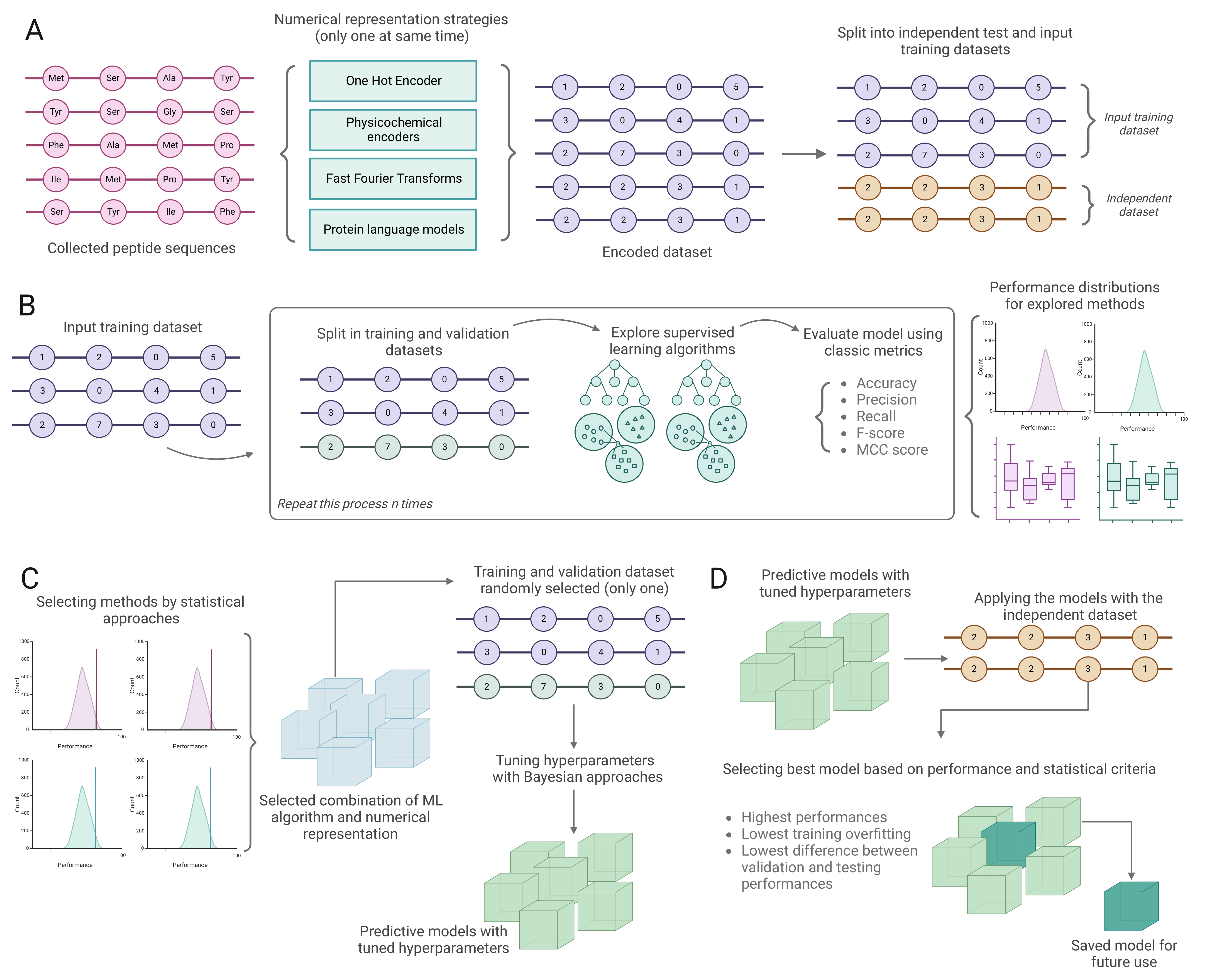
3. Materials and Methods
3.1. Data Collection and Preprocessing
3.2. Encoding Peptide Sequences
3.3. Training and Validating Classification Models
3.4. Benchmark Analysis
3.5. Discovering Potential Peptides with Desirable Biological Activity from the Peptide Atlas
3.6. De Novo Design of Antimicrobial Peptides Using VAE
3.7. Implementation Strategies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fu, J.; Nguyen, K. Reduction of promiscuous peptides-enzyme inhibition and aggregation by negatively charged biopolymers. ACS Appl. Bio Mater. 2022, 5, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Lowman, H.B. Therapeutic peptides. Trends Biotechnol. 2003, 21, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Sun, H.; Wu, Y.; Li, C.; Wang, Y.; Liu, Y.; Li, Y.; Nie, J.; Sun, D.; Zhang, Y.; et al. A cyclic heptapeptide-based hydrogel boosts the healing of chronic skin wounds in diabetic mice and patients. NPG Asia Mater. 2022, 14, 99. [Google Scholar] [CrossRef]
- Khan, M.M.; Filipczak, N.; Torchilin, V.P. Cell penetrating peptides: A versatile vector for co-delivery of drug and genes in cancer. J. Control Release 2021, 330, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.I. Rational design of peptide agonists of cell-surface receptors. Trends Pharmacol. Sci. 2000, 21, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A global review on short peptides: Frontiers and perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.; Cândido, E.S.; Franco, O.L. Computer-aided design of antimicrobial peptides: Are we generating effective drug candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Hussain, W. sAMP-PFPDeep: Improving accuracy of short antimicrobial peptides prediction using three different sequence encodings and deep neural networks. Briefings Bioinform. 2022, 23, bbab487. [Google Scholar] [CrossRef]
- Lertampaiporn, S.; Vorapreeda, T.; Hongsthong, A.; Thammarongtham, C. Ensemble-amppred: Robust amp prediction and recognition using the ensemble learning method with a new hybrid feature for differentiating amps. Genes 2021, 12, 137. [Google Scholar] [CrossRef]
- Lin, T.-T.; Sun, Y.-Y.; Wang, C.-T.; Cheng, W.-C.; Lu, I.-H.; Lin, C.-Y.; Chen, S.-H. Ai4avp: An antiviral peptides predictor in deep learning approach with generative adversarial network data augmentation. Bioinform. Adv. 2022, 2, vbac080. [Google Scholar] [CrossRef] [PubMed]
- Lissabet, J.F.B.; Belén, L.H.; Farias, J.G. Antivpp 1.0: A portable tool for prediction of antiviral peptides. Comput. Biol. Med. 2019, 107, 127–130. [Google Scholar] [CrossRef]
- Pang, Y.; Yao, L.; Jhong, J.-H.; Wang, Z.; Lee, T.-Y. Avpiden: A new scheme for identification and functional prediction of antiviral peptides based on machine learning approaches. Briefings Bioinform. 2021, 22, bbab263. [Google Scholar] [CrossRef] [PubMed]
- Timmons, P.B.; Hewage, C.M. Ennavia is a novel method which employs neural networks for antiviral and anti-coronavirus activity prediction for therapeutic peptides. Briefings Bioinform. 2021, 22, bbab258. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yang, C.; Xie, Y.; Wang, Y.; Li, X.; Wang, K.; Huang, J.; Yan, W. Gm-pep: A high efficiency strategy to de novo design functional peptide sequences. J. Chem. Inf. Model. 2022, 62, 2617–2629. [Google Scholar] [CrossRef]
- Das, P.; Wadhawan, K.; Chang, O.; Sercu, T.; Santos, C.D.; Riemer, M.; Chenthamarakshan, V.; Padhi, I.; Mojsilovic, A. Pepcvae: Semi-supervised targeted design of antimicrobial peptide sequences. arXiv 2018, arXiv:1810.07743. [Google Scholar]
- Dean, S.N.; Alvarez, J.A.E.; Zabetakis, D.; Walper, S.A.; Malanoski, A.P. Pepvae: Variational autoencoder framework for antimicrobial peptide generation and activity prediction. Front. Microbiol. 2021, 12, 725727. [Google Scholar] [CrossRef]
- Surana, S.; Arora, P.; Singh, D.; Sahasrabuddhe, D.; Valadi, J. Pandoragan: Generating antiviral peptides using generative adversarial network. SN Comput. Sci. 2023, 4, 607. [Google Scholar] [CrossRef]
- Van Oort, C.M.; Ferrell, J.B.; Remington, J.M.; Wshah, S.; Li, J. Ampgan v2: Machine learning-guided design of antimicrobial peptides. J. Chem. Inf. Model. 2021, 61, 2198–2207. [Google Scholar] [CrossRef]
- Wu, Z.; Johnston, K.E.; Arnold, F.H.; Yang, K.K. Protein sequence design with deep generative models. Curr. Opin. Chem. Biol. 2021, 65, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Valiente, P.A.; Kim, P.M. Helixgan a deep-learning methodology for conditional de novo design of α-helix structures. Bioinformatics 2023, 39, btad036. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Vure, P.; Pulugurta, R.; Chatterjee, P. Amp-diffusion: Integrating latent diffusion with protein language models for antimicrobial peptide generation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kong, X.; Huang, W.; Liu, Y. Full-atom peptide design with geometric latent diffusion. arXiv 2024, arXiv:2402.13555. [Google Scholar]
- Wang, Y.; Liu, X.; Huang, F.; Xiong, Z.; Zhang, W. A multi-modal contrastive diffusion model for therapeutic peptide generation. In Proceedings of the AAAI Conference on Artificial Intelligence, Vancouver, BC, Canada, 20–27 February 2024; Volume 38, pp. 3–11. [Google Scholar]
- Medina-Ortiz, D.; Cabas-Mora, G.; Moya-Barria, I.; Soto-Garcia, N.; Uribe-Paredes, R. Rudeus, a machine learning classification system to study dna-binding proteins. bioRxiv 2024. [Google Scholar] [CrossRef]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. The peptideatlas project. Nucleic Acids Res. 2006, 34 (Suppl. S1), D655–D658. [Google Scholar] [CrossRef] [PubMed]
- Cabas, G.E.; Daza, A.; Soto, N.; Garrido, V.; Alvarez, D.; Navarrete, M.; Sarmiento-Varon, L.; Sepulveda, J.; Davari Sr, M.E.; Cadet, F.; et al. Peptipedia v2. 0: A peptide sequence database and user-friendly web platform. A major update. bioRxiv 2024. [Google Scholar] [CrossRef]
- Biswas, S.; Khimulya, G.; Alley, E.C.; Esvelt, K.M.; Church, G.M. Low-n protein engineering with data-efficient deep learning. Nat. Methods 2021, 18, 389–396. [Google Scholar] [CrossRef]
- Lv, H.; Yan, K.; Liu, B. Tppred-le: Therapeutic peptide function prediction based on label embedding. BMC Biol. 2023, 21, 238. [Google Scholar] [CrossRef]
- Pinacho-Castellanos, S.A.; García-Jacas, C.R.; Gilson, M.K.; Brizuela, C.A. Alignment-free antimicrobial peptide predictors: Improving performance by a thorough analysis of the largest available data set. J. Chem. Inf. Model. 2021, 61, 3141–3157. [Google Scholar] [CrossRef]
- Li, C.; Warren, R.L.; Birol, I. Models and data of amplify: A deep learning tool for antimicrobial peptide prediction. BMC Res. Notes 2023, 16, 11. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef]
- Chung, C.-R.; Kuo, T.-R.; Wu, L.-C.; Lee, T.-Y.; Horng, J.-T. Characterization and identification of antimicrobial peptides with different functional activities. Briefings Bioinform. 2020, 21, 1098–1114. [Google Scholar] [CrossRef]
- Dong, G.; Zheng, L.; Huang, S.; Gao, J.; Zuo, Y. Amino acid reduction can help to improve the identification of antimicrobial peptides and their functional activities. Front. Genet. 2021, 12, 669328. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zhang, Y.; Li, W.; Chung, C.-R.; Guan, J.; Zhang, W.; Chiang, Y.-C.; Lee, T.-Y. D eepafp: An effective computational framework for identifying antifungal peptides based on deep learning. Protein Sci. 2023, 32, e4758. [Google Scholar] [CrossRef] [PubMed]
- Bajiya, N.; Choudhury, S.; Dhall, A.; Raghava, G.P. Antibp3: A method for predicting antibacterial peptides against gram-positive/negative/variable bacteria. Antibiotics 2024, 13, 168. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. Dbaasp v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- Grønning, A.G.; Kacprowski, T.; Scheele, C. Multipep: A hierarchical deep learning approach for multi-label classification of peptide bioactivities. Biol. Methods Protoc. 2021, 6, bpab021. [Google Scholar]
- Li, J.; Pu, Y.; Tang, J.; Zou, Q.; Guo, F. Deepavp: A dual-channel deep neural network for identifying variable-length antiviral peptides. IEEE J. Biomed. Health Inform. 2020, 24, 3012–3019. [Google Scholar] [CrossRef]
- Müller, A.T.; Gabernet, G.; Hiss, J.A.; Schneider, G. modlAMP: Python for antimicrobial peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef]
- Greener, J.G.; Moffat, L.; Jones, D.T. Design of metalloproteins and novel protein folds using variational autoencoders. Sci. Rep. 2018, 8, 16189. [Google Scholar] [CrossRef] [PubMed]
- Hawkins-Hooker, A.; Depardieu, F.; Baur, S.; Couairon, G.; Chen, A.; Bikard, D. Generating functional protein variants with variational autoencoders. PLoS Comput. Biol. 2021, 17, e1008736. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. Cd-hit: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Dallago, C.; Schütze, K.; Heinzinger, M.; Olenyi, T.; Littmann, M.; Lu, A.X.; Yang, K.K.; Min, S.; Yoon, S.; Morton, J.T.; et al. Learned embeddings from deep learning to visualize and predict protein sets. Curr. Protoc. 2021, 1, e113. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ortiz, D.; Contreras, S.; Quiroz, C.; Asenjo, J.A.; Olivera-Nappa, Á. Dmakit: A user-friendly web platform for bringing state-of-the-art data analysis techniques to non-specific users. Inf. Syst. 2020, 93, 101557. [Google Scholar] [CrossRef]
- Medina-Ortiz, D.; Contreras, S.; Amado-Hinojosa, J.; Torres-Almonacid, J.; Asenjo, J.A.; Navarrete, M.; Olivera-Nappa, Á. Generalized property-based encoders and digital signal processing facilitate predictive tasks in protein engineering. Front. Mol. Biosci. 2022, 9, 898627. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ortiz, D.; Contreras, S.; Amado-Hinojosa, J.; Torres-Almonacid, J.; Asenjo, J.A.; Navarrete, M.; Olivera-Nappa, Á. Combination of digital signal processing and assembled predictive models facilitates the rational design of proteins. arXiv 2020, arXiv:2010.03516. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 44, 7112–7127. [Google Scholar] [CrossRef]
- Medina-Ortiz, D.; Contreras, S.; Quiroz, C.; Olivera-Nappa, Á. Development of supervised learning predictive models for highly non-linear biological, biomedical, and general datasets. Front. Mol. Biosci. 2020, 7, 13. [Google Scholar] [CrossRef]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A next-generation hyperparameter optimization framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar]
- Zhang, J.; Yang, L.; Tian, Z.; Zhao, W.; Sun, C.; Zhu, L.; Huang, M.; Guo, G.; Liang, G. Large-scale screening of antifungal peptides based on quantitative structure–activity relationship. ACS Med. Chem. Lett. 2021, 13, 99–104. [Google Scholar] [CrossRef]
- Youmans, M.; Spainhour, J.C.G.; Qiu, P. Classification of antibacterial peptides using long short-term memory recurrent neural networks. IEEE/Acm Trans. Comput. Biol. Bioinform. 2020, 17, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Guan, J.; Xie, P.; Chung, C.-R.; Deng, J.; Huang, Y.; Chiang, Y.-C.; Lee, T.-Y. Ampactipred: A three-stage framework for predicting antibacterial peptides and activity levels with deep forest. Protein Sci. 2024, 33, e5006. [Google Scholar] [CrossRef]
- Yan, W.; Tang, W.; Wang, L.; Bin, Y.; Xia, J. Prmftp: Multi-functional therapeutic peptides prediction based on multi-head self-attention mechanism and class weight optimization. PLoS Comput. Biol. 2022, 18, e1010511. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W. Deep-ampep30: Improve short antimicrobial peptides prediction with deep learning. Mol. Ther. Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Zhang, J.; Li, C.; Huo, Y.; Dong, G. iamp-attenpred: A novel antimicrobial peptide predictor based on bert feature extraction method and cnn-bilstm-attention combination model. Briefings Bioinform. 2024, 25, bbad443. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, P.; Lin, W.-Z.; Jia, J.-H.; Chou, K.-C. iamp-2l: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shao, Y.-T.; Cheng, X.; Stamatovic, B. iamp-ca2l: A new cnn-bilstm-svm classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Briefings Bioinform. 2021, 22, bbab209. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C. E-cleap: An ensemble learning model for efficient and accurate identification of antimicrobial peptides. PLoS ONE 2024, 19, e0300125. [Google Scholar] [CrossRef]
- Wang, R.; Wang, T.; Zhuo, L.; Wei, J.; Fu, X.; Zou, Q.; Yao, X. Diff-amp: Tailored designed antimicrobial peptide framework with all-in-one generation, identification, prediction and optimization. Briefings Bioinform. 2024, 25, bbae078. [Google Scholar] [CrossRef]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS ONE 2011, 6, e18476. [Google Scholar] [CrossRef]
- Torrent, M.; Di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. Ampa: An automated web server for prediction of protein antimicrobial regions. Bioinformatics 2012, 28, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Qureshi, A.; Kumar, M. Avppred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S. Stable-abppred: A stacked ensemble predictor based on bilstm and attention mechanism for accelerated discovery of antibacterial peptides. Briefings Bioinform. 2022, 23, bbab439. [Google Scholar] [CrossRef] [PubMed]
- Simeon, S.; Li, H.; Win, T.S.; Malik, A.A.; Kandhro, A.H.; Piacham, T.; Shoombuatong, W.; Nuchnoi, P.; Wikberg, J.E.; Gleeson, M.P.; et al. Pepbio: Predicting the bioactivity of host defense peptides. RSC Adv. 2017, 7, 35119–35134. [Google Scholar] [CrossRef]
- Sharma, R.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S.; Kumar Singh, R. Deep-afppred: Identifying novel antifungal peptides using pretrained embeddings from seq2vec with 1dcnn-bilstm. Briefings Bioinform. 2022, 23, bbab422. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S.; Kumar Singh, R. Deep-abppred: Identifying antibacterial peptides in protein sequences using bidirectional lstm with word2vec. Briefings Bioinform. 2021, 22, bbab065. [Google Scholar] [CrossRef]
- Randou, E.G.; Veltri, D.; Shehu, A. Binary response models for recognition of antimicrobial peptides. In Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics, Washington, DC, USA, 22–25 September 2013; pp. 76–85. [Google Scholar]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into chou’s general pseaac. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef]
- Lin, W.; Xu, D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016, 32, 3745–3752. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Lee, I.; Nam, H. Amp-bert: Prediction of antimicrobial peptide function based on a bert model. Protein Sci. 2023, 32, e4529. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, N.K.; Raghava, G.P. Antibp2: Improved version of antibacterial peptide prediction. BMC Bioinform. 2010, 11, S19. [Google Scholar] [CrossRef]
- Kavousi, K.; Bagheri, M.; Behrouzi, S.; Vafadar, S.; Atanaki, F.F.; Lotfabadi, B.T.; Ariaeenejad, S.; Shockravi, A.; Moosavi-Movahedi, A.A. Iampe: Nmr-assisted computational prediction of antimicrobial peptides. J. Chem. Inf. Model. 2020, 60, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Karnik, S.; Nilawe, P.; Jayaraman, V.K.; Idicula-Thomas, S. Classamp: A prediction tool for classification of antimicrobial peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Gull, S.; Shamim, N.; Minhas, F. Amap: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput. Biol. Med. 2019, 107, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Yao, L.; Xie, P.; Chung, C.-R.; Huang, Y.; Chiang, Y.-C.; Lee, T.-Y. A two-stage computational framework for identifying antiviral peptides and their functional types based on contrastive learning and multi-feature fusion strategy. Briefings Bioinform. 2024, 25, bbae208. [Google Scholar] [CrossRef] [PubMed]
- Fernández, D.; Olivera-Nappa, Á.; Uribe-Paredes, R.; Medina-Ortiz, D. Exploring machine learning algorithms and protein language models strategies to develop enzyme classification systems. In International Work-Conference on Bioinformatics and Biomedical Engineering; Springer: Cham, Switzerland, 2023; pp. 307–319. [Google Scholar]
- Feng, J.; Sun, M.; Liu, C.; Zhang, W.; Xu, C.; Wang, J.; Wang, G.; Wan, S. Samp: Identifying antimicrobial peptides by an ensemble learning model based on proportionalized split amino acid composition. bioRxiv 2024. [Google Scholar] [CrossRef]
- Du, Z.; Ding, X.; Xu, Y.; Li, Y. Unidl4biopep: A universal deep learning architecture for binary classification in peptide bioactivity. Briefings Bioinform. 2023, 24, bbad135. [Google Scholar] [CrossRef]
- Chowdhury, A.S.; Reehl, S.M.; Kehn-Hall, K.; Bishop, B.; Webb-Robertson, B.-J.M. Better understanding and prediction of antiviral peptides through primary and secondary structure feature importance. Sci. Rep. 2020, 10, 19260. [Google Scholar] [CrossRef]
- Burdukiewicz, M.; Sidorczuk, K.; Rafacz, D.; Pietluch, F.; Chilimoniuk, J.; Rødiger, S.; Gagat, P. Proteomic screening for prediction and design of antimicrobial peptides with ampgram. Int. J. Mol. Sci. 2020, 21, 4310. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Chaudhary, K.; Kumar, R.; Sharma, M.; Raghava, G.P. In silico approach for prediction of antifungal peptides. Front. Microbiol. 2018, 9, 318353. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Algorithm | Encoder | Training Performance | Validation Performance | Testing Performance |
|---|---|---|---|---|---|
| Antiangiogenic | HistGradientBoosting | ProTrans t5 BDF | (0.75, 0.78) | (0.74, 0.76) | 0.82 |
| Antidiabetic | RandomForest | ProTrans t5 Uniref | (0.81, 0.83) | (0.81, 0.82) | 0.81 |
| Anti-Gram (−) | XGBClassifier | Esm1B | (0.89, 0.89) | (0.88, 0.89) | 0.88 |
| Anti-Gram (+) | ExtraTrees | Esm1B | (0.88, 0.88) | (0.88, 0.88) | 0.88 |
| Anti-inflammatory | Random Forest | ProTrans t5 xlu50 | (0.87, 0.88) | (0.87, 0.87) | 0.89 |
| Antimalarial | ExtraTrees | ProTrans t5 BERT | (0.82, 0.85) | (0.83, 0.84) | 0.78 |
| Antimammalian cell | ExtraTrees | ProTrans t5 Uniref | (0.90, 0.90) | (0.90, 0.90) | 0.90 |
| Anti-methicillin-resistant S. aureus | ExtraTrees | Esm1B | (0.87, 0.88) | (0.87, 0.87) | 0.90 |
| Antioxidative | RandomForest | ProTrans t5 xlu50 | (0.75, 0.77) | (0.75, 0.76) | 0.82 |
| Antibacterial | RandomForest | ProTrans t5 xlu50 | (0.89, 0.90) | (0.89, 0.89) | 0.92 |
| Antifungal | RandomForest | ProTrans t5 xlu50 | (0.83, 0.83) | (0.82, 0.82) | 0.84 |
| Antimicrobial | RandomForest | ProTrans t5 Uniref | (0.88, 0.88) | (0.87, 0.88) | 0.88 |
| Antiparasitic | RandomForest | Esm1b | (0.83, 0.84) | (0.83, 0.83) | 0.85 |
| Antiviral | RandomForest | ProTrans t5 Uniref | (0.81, 0.81) | (0.80, 0.81) | 0.79 |
| Anuran defense | Hist Gradient Boosting | ProTrans t5 xlu50 | (0.90, 0.91) | (0.90, 0.90) | 0.93 |
| Blood–brain barrier penetrating | ExtraTrees | ProTrans t5 Uniref | (0.77, 0.80) | (0.78, 0.79) | 0.85 |
| Cell–cell communication | Hist Gradient Boosting | ProTrans t5 xlu50 | (0.90, 0.91) | (0.90, 0.90) | 0.91 |
| Cell-penetrating | ExtraTrees | ProTrans t5 ALBERT | (0.87, 0.88) | (0.87, 0.87) | 0.86 |
| Neuropeptide | ExtraTrees | ProTrans t5 xlu50 | (0.86, 0.86) | (0.86, 0.86) | 0.87 |
| Quorum sensing | ExtraTrees | ProTrans t5 ALBERT | (0.83, 0.86) | (0.87, 0.88) | 0.87 |
| Drug delivery vehicle | ExtraTrees | Esm1B | (0.85, 0.85) | (0.84, 0.85) | 0.92 |
| Task | Method | Reference | Sensitivity | Specificity | F1 |
|---|---|---|---|---|---|
| Antimicrobial | AMP-Detector | — | 0.91 | 0.85 | 0.88 |
| AMP-discover | [31] | 0.66 | 0.78 | 0.74 | |
| amplify | [32] | 0.8 | 0.7 | 0.72 | |
| TPpred-LE | [30] | 0.62 | 0.71 | 0.71 | |
| AMPScanner | [33] | 0.59 | 0.55 | 0.55 | |
| Antifungal | AMP-Detector | — | 0.85 | 0.79 | 0.84 |
| AMPfun | [34] | 0.6 | 0.56 | 0.67 | |
| IAMP-RAAC | [35] | 0.59 | 0.54 | 0.67 | |
| AMP-discover | [31] | 0.53 | 0.38 | 0.61 | |
| DeepAFP | [36] | 0.55 | 0.44 | 0.52 | |
| TPpred-LE | [30] | 0.75 | 0.46 | 0.14 | |
| Antibacterial | AMP-Detector | — | 0.95 | 0.81 | 0.96 |
| AMP-discover | [31] | 0.92 | 0.39 | 0.89 | |
| AntiBP3 Gram variable | [37] | 0.98 | 0.39 | 0.86 | |
| AntiBP3 Gram (−) | [37] | 0.94 | 0.3 | 0.82 | |
| AntiBP3 Gram + | [37] | 0.94 | 0.3 | 0.82 | |
| DBAASP E. coli | [38] | 0.98 | 0.21 | 0.59 | |
| DBAASP S. aureus | [38] | 0.98 | 0.19 | 0.49 | |
| AMPActiPred | [38] | 0.84 | 0.13 | 0.49 | |
| DBAASP Klebsiella | [38] | 0.99 | 0.18 | 0.45 | |
| TPpred-LE | [30] | 0.97 | 0.17 | 0.41 | |
| DBAASP Pseudomonas | [38] | 0.97 | 0.17 | 0.4 | |
| Antiparasitic | AMP-Detector | — | 0.89 | 0.8 | 0.84 |
| AMP-discover | [31] | 0.56 | 0.56 | 0.66 | |
| AMPfun | [34] | 0.67 | 0.48 | 0.2 | |
| IAMP-RAAC | [35] | 0.49 | 0.46 | 0.16 | |
| multipep_max | [39] | 0.71 | 0.47 | 0.07 | |
| TPpred-LE | [30] | 1.0 | 0.47 | 0.01 | |
| Antiviral | AMP-Detector | — | 0.76 | 0.77 | 0.78 |
| IAMP-RAAC | [35] | 0.72 | 0.65 | 0.68 | |
| TPpred-LE | [30] | 0.89 | 0.65 | 0.67 | |
| AMPfun | [34] | 0.75 | 0.63 | 0.66 | |
| AMP-discover | [31] | 0.56 | 0.57 | 0.66 | |
| DeepAVP | [40] | 0.52 | 0.46 | 0.57 | |
| AVP-IFT | [12] | 0.52 | 0.47 | 0.46 |
| Activity | # Discovered from Peptide Atlas | # Generated Using Trained VAE | # Generated Using Positive Examples | # Generated Used Negative Examples |
|---|---|---|---|---|
| Antibacterial | 403,367 | 336 | 63,709 | 58,826 |
| Anti-Gram (+) | 83,271 | 147 | 34,468 | 31,960 |
| Antifungal | 406,191 | 554 | 37,147 | 37,976 |
| Blood–brain barrier penetrating | 1,259,618 | 38 | 12,384 | 11,277 |
| Antiparasitic | 133,555 | 563 | 2887 | 2077 |
| Anti-inflamatory | 2,999,776 | 1 | 27 | 27 |
| Cell-penetrating | 698,536 | 58 | 12,468 | 14,066 |
| Anti mammalian cell | 81,701 | 93 | 16,360 | 14,611 |
| Anuran defense | 593,692 | 40 | 4729 | 4554 |
| Anti-methicillin-resistant S. aureus | 29,964 | 114 | 12,984 | 12,774 |
| Cell–cell communication | 129,651 | 2 | 13,237 | 13,475 |
| Antioxidative | 290,289 | 75 | 5307 | 4821 |
| Antiangiogenic | 292,815 | 34 | 37,954 | 33,980 |
| Antiviral | 2,582,176 | 29 | 24,987 | 24,178 |
| Quorum sensing | 2,088,671 | 17 | 16,477 | 15,624 |
| Antimicrobial | 305,496 | 640 | 49,497 | 45,325 |
| Antimalarial | 259,169 | 311 | 35,862 | 34,647 |
| Anti-Gram (−) | 152,568 | 232 | 37,009 | 33,168 |
| Drug delivery vehicle | 113,0481 | 60 | 14,147 | 15,857 |
| Antidiabetic | 3,344,580 | 1 | 4571 | 4518 |
| Neuropeptide | 2,499,312 | 1 | 20,188 | 19,898 |
| # | Pre-Trained Model | Description | Tensor Size | Reference |
|---|---|---|---|---|
| 1 | ProTrans t5 UniRef | The ProtTrans UniRef pre-trained model is a deep learning model specifically trained for protein sequence representation and understanding. It is trained on the UniRef50 database, which contains clustered protein sequences to reduce redundancy and improve diversity. | 1024 | [49] |
| 2 | ProTrans t5 xlu50 | ProtT5-XL-UniRef50 is based on the t5-3b model and was pre-trained on a large corpus of protein sequences in a self-supervised fashion. This means that it was pre-trained on the raw protein sequences only, with no humans labeling them in any way (which is why it can be used with a large amount of publicly available data), with an automatic process to generate inputs and labels from the protein sequences. | 1024 | [49] |
| 3 | ProTrans T5-BDF | ProtT5-XL-BFD is based on the t5-3b model and was trained on a large corpus of protein sequences in a self-supervised fashion. This means that it was trained on the raw protein sequences only, with no human labeling them in any way (which is why it can use many publicly available data), with an automatic process to generate inputs and labels from the protein sequences. | 1024 | [49] |
| 4 | Esm1b | The ESM-1b (Evolutionary Scale Modeling) pre-trained model is a variant of the ESM model, designed for protein sequence modeling. It is based on self-supervised learning techniques and utilizes a Transformer architecture, similar to those used in natural language processing tasks. | 1280 | [45] |
| 5 | ProTrans XLNet | The ProtTrans XLNet pre-trained model is a variant of the XLNet model customized for protein sequence analysis. XLNet is an extension of the Transformer-based architecture, which integrates bidirectional context learning with permutation-based training. Similarly, ProtTrans XLNet leverages these features to learn contextual representations of amino acids in protein sequences. | 1024 | [49] |
| 6 | ProTrans ALBERT | The ProtTrans ALBERT (A Lite BERT) pre-trained model is a variant of the ALBERT model specifically adapted for protein sequence analysis. ALBERT is a lightweight version of the BERT model, designed to reduce the computational resource usage while maintaining its performance. Similarly, ProtTrans ALBERT leverages this efficiency to provide effective representations of amino acids in protein sequences. | 4096 | [49] |
| 7 | ProTrans BERT | The ProtTrans BERT (Bidirectional Encoder Representations from Transformers) pre-trained model is a variant of the BERT model specifically tailored to protein sequence analysis. Like its counterpart in natural language processing, ProtTrans BERT utilizes a Transformer-based architecture to learn contextual representations of amino acids in protein sequences. | 1024 | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Ortiz, D.; Contreras, S.; Fernández, D.; Soto-García, N.; Moya, I.; Cabas-Mora, G.; Olivera-Nappa, Á. Protein Language Models and Machine Learning Facilitate the Identification of Antimicrobial Peptides. Int. J. Mol. Sci. 2024, 25, 8851. https://doi.org/10.3390/ijms25168851
Medina-Ortiz D, Contreras S, Fernández D, Soto-García N, Moya I, Cabas-Mora G, Olivera-Nappa Á. Protein Language Models and Machine Learning Facilitate the Identification of Antimicrobial Peptides. International Journal of Molecular Sciences. 2024; 25(16):8851. https://doi.org/10.3390/ijms25168851
Chicago/Turabian StyleMedina-Ortiz, David, Seba Contreras, Diego Fernández, Nicole Soto-García, Iván Moya, Gabriel Cabas-Mora, and Álvaro Olivera-Nappa. 2024. "Protein Language Models and Machine Learning Facilitate the Identification of Antimicrobial Peptides" International Journal of Molecular Sciences 25, no. 16: 8851. https://doi.org/10.3390/ijms25168851
APA StyleMedina-Ortiz, D., Contreras, S., Fernández, D., Soto-García, N., Moya, I., Cabas-Mora, G., & Olivera-Nappa, Á. (2024). Protein Language Models and Machine Learning Facilitate the Identification of Antimicrobial Peptides. International Journal of Molecular Sciences, 25(16), 8851. https://doi.org/10.3390/ijms25168851