
Evaluation of the Antifibrotic Effects of Drugs Commonly Used in Inflammatory Intestinal Diseases on In Vitro Intestinal Cellular Models
 , ,
, ,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Selected Drugs on Cell Viability of CCD-18Co and IEC Caco-2
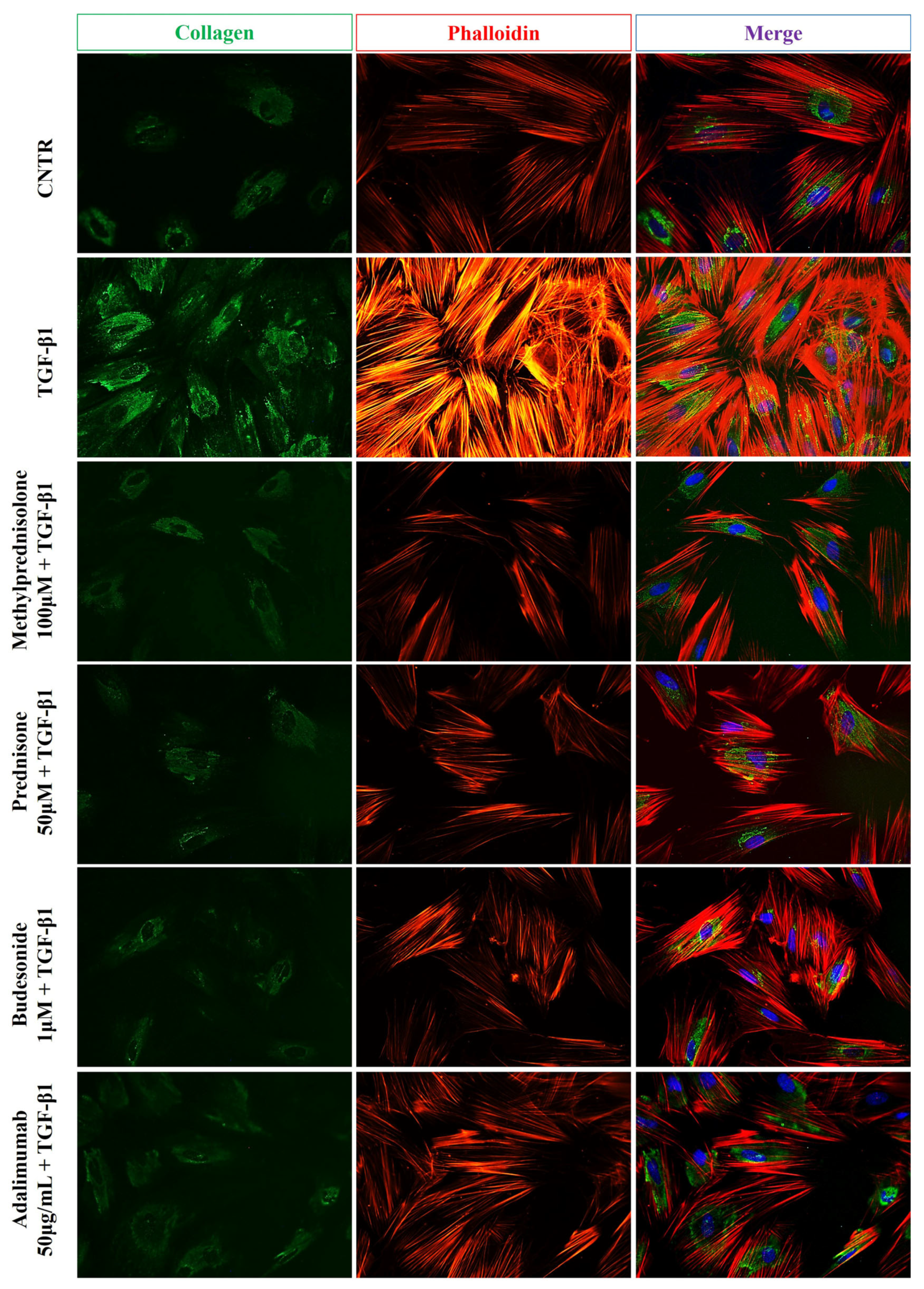
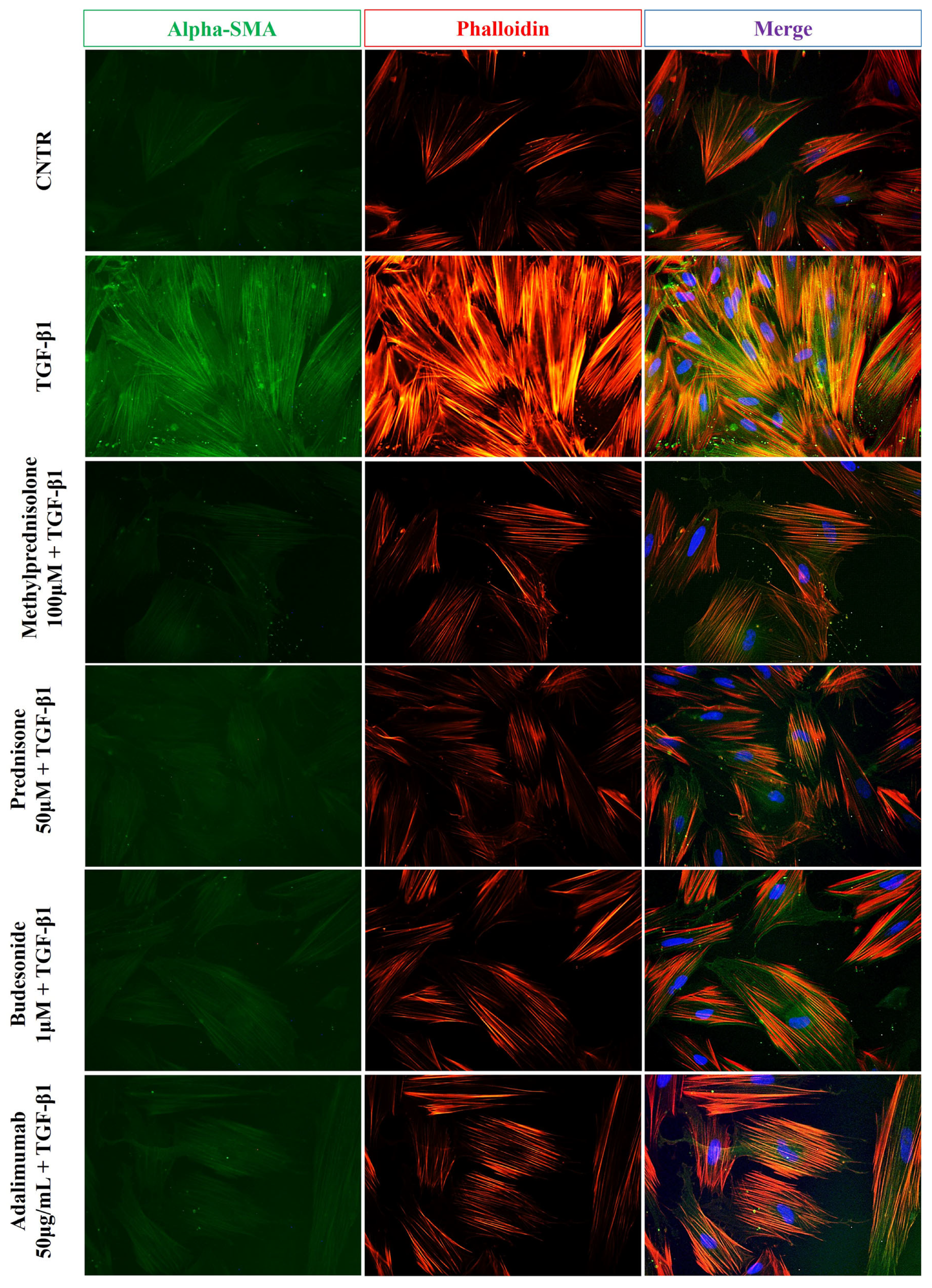
2.2. Effect of Selected Drugs on TGB-β1-Induced Collagen I and α-SMA Expression in CCD-18Co Cells
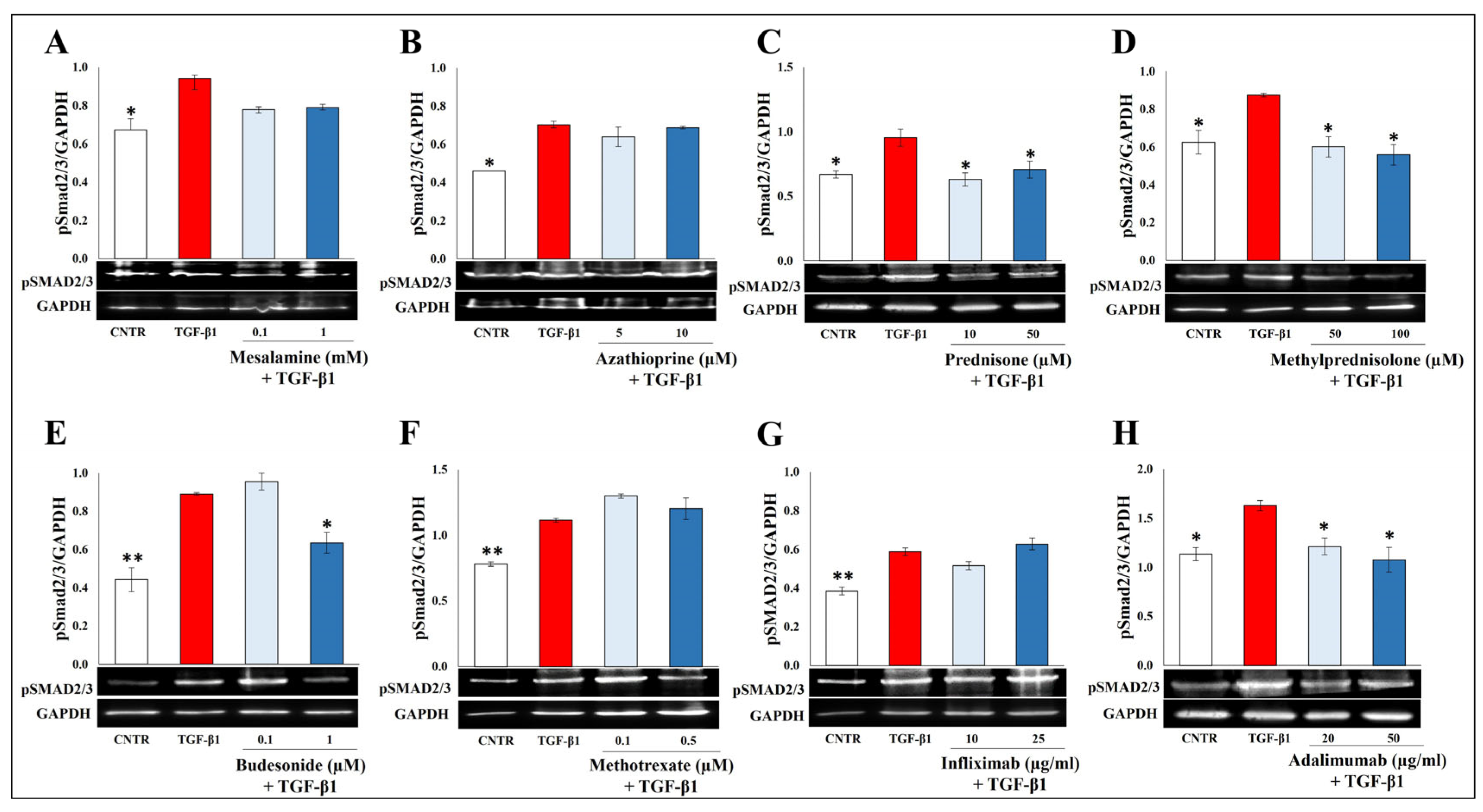
2.3. Effect of Selected Drugs on TGB-β1-Induced Smad Signaling
2.4. Effect of Selected Drugs on TGB-β1-Induced Epithelial to Mesenchymal Transition in Caco-2 IEC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cell Models and Treatments
4.3. Cell Viability Assay
4.4. Western Blot Analysis
4.5. Immunofluorescence Staining
4.6. TNF-α ELISA Assay
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Alessio, S.; Ungaro, F.; Noviello, D.; Lovisa, S.; Peyrin-Biroulet, L.; Danese, S. Revisiting fibrosis in inflammatory bowel disease: The gut thickens. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chang, E.B. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology 2021, 160, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Pizarro, T.T.; Cominelli, F. Immunological Regulation of Intestinal Fibrosis in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Latella, G.; Di Gregorio, J.; Flati, V.; Rieder, F.; Lawrance, I.C. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand. J. Gastroenterol. 2015, 50, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Speca, S.; Rousseaux, C.; Dubuquoy, C.; Rieder, F.; Vetuschi, A.; Sferra, R.; Giusti, I.; Bertin, B.; Dubuquoy, L.; Gaudio, E.; et al. Novel PPARgamma Modulator GED-0507-34 Levo Ameliorates Inflammation-driven Intestinal Fibrosis. Inflamm. Bowel Dis. 2016, 22, 279–292. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Xue, M.; Liu, C.; Xie, F.; Bai, L. Parathyroid Hormone-Like Hormone Induces Epithelial-to-Mesenchymal Transition of Intestinal Epithelial Cells by Activating the Runt-Related Transcription Factor 2. Am. J. Pathol. 2018, 188, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Seo, J.; Yoon, H.; Cho, S. The Post-Translational Regulation of Epithelial-Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. Int. J. Mol. Sci. 2021, 22, 3591. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Huber, N.; Lang, S.; Furst, A.; Jehle, E.; Rogler, G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin. Transl. Med. 2015, 4, e1. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Masia, D.; Salvador, P.; Macias-Ceja, D.C.; Gisbert-Ferrandiz, L.; Esplugues, J.V.; Manye, J.; Alos, R.; Navarro-Vicente, F.; Mamie, C.; Scharl, M.; et al. WNT2b Activates Epithelial-mesenchymal Transition Through FZD4: Relevance in Penetrating Crohn s Disease. J. Crohn’s Colitis 2020, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Mukherjee, P.K.; Massey, W.J.; Wang, Y.; Fiocchi, C. Fibrosis in IBD: From pathogenesis to therapeutic targets. Gut 2024, 73, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Lis-Lopez, L.; Bauset, C.; Seco-Cervera, M.; Macias-Ceja, D.; Navarro, F.; Alvarez, A.; Esplugues, J.V.; Calatayud, S.; Barrachina, M.D.; Ortiz-Masia, D.; et al. P2X7 Receptor Regulates Collagen Expression in Human Intestinal Fibroblasts: Relevance in Intestinal Fibrosis. Int. J. Mol. Sci. 2023, 24, 12936. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson, J.; Wick, M.J. Mechanism of fibrosis and stricture formation in Crohn’s disease. Scand. J. Immunol. 2020, 92, e12990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, H.; Cheng, F.; Zhang, Z.; Long, S. MiR-21 regulates epithelial-mesenchymal transition in intestinal fibrosis of Crohn’s disease by targeting PTEN/mTOR. Dig. Liver Dis. 2022, 54, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Masia, D.; Gisbert-Ferrandiz, L.; Bauset, C.; Coll, S.; Mamie, C.; Scharl, M.; Esplugues, J.V.; Alos, R.; Navarro, F.; Cosin-Roger, J.; et al. Succinate Activates EMT in Intestinal Epithelial Cells through SUCNR1: A Novel Protagonist in Fistula Development. Cells 2020, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lin, X.; Tan, J.; Liu, Z.; He, J.; Hu, F.; Wang, Y.; Chen, M.; Liu, F.; Mao, R. Cellular and Molecular Mechanisms of Intestinal Fibrosis. Gut Liver 2023, 17, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-beta Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Chazotte, B. Labeling cytoskeletal F-actin with rhodamine phalloidin or fluorescein phalloidin for imaging. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot4947. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. α-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef]
- Gordon, H.; Minozzi, S.; Kopylov, U.; Verstockt, B.; Chaparro, M.; Buskens, C.; Warusavitarne, J.; Agrawal, M.; Allocca, M.; Atreya, R.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 14, 4–22. [CrossRef]
- Lemaitre, M.; Kirchgesner, J.; Rudnichi, A.; Carrat, F.; Zureik, M.; Carbonnel, F.; Dray-Spira, R. Association Between Use of Thiopurines or Tumor Necrosis Factor Antagonists Alone or in Combination and Risk of Lymphoma in Patients with Inflammatory Bowel Disease. JAMA 2017, 318, 1679–1686. [Google Scholar] [CrossRef]
- Quera, R.; Nunez, P.; Sicilia, B.; Flores, L.; Gomollon, F. Corticosteroids in inflammatory bowel disease: Are they still a therapeutic option? Gastroenterol. Hepatol. 2023, 46, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Palmisano, A.; Corradi, D.; Salvarani, C.; Buzio, C. Retroperitoneal fibrosis: Evolving concepts. Rheum. Dis. Clin. N. Am. 2007, 33, 803–817. [Google Scholar] [CrossRef]
- Badea, I.; Taylor, M.; Rosenberg, A.; Foldvari, M. Pathogenesis and therapeutic approaches for improved topical treatment in localized scleroderma and systemic sclerosis. Rheumatology 2009, 48, 213–221. [Google Scholar] [CrossRef]
- Baiula, M.; Spampinato, S.; Gentilucci, L.; Tolomelli, A. Novel Ligands Targeting alpha(4)beta(1) Integrin: Therapeutic Applications and Perspectives. Front. Chem. 2019, 7, 489. [Google Scholar] [CrossRef]
- Ohmatsu, H.; Kadono, T.; Sugaya, M.; Tomita, M.; Kai, H.; Miyagaki, T.; Saeki, H.; Tamaki, K.; Steeber, D.A.; Tedder, T.F.; et al. alpha4beta7 Integrin is essential for contact hypersensitivity by regulating migration of T cells to skin. J. Allergy Clin. Immunol. 2010, 126, 1267–1276. [Google Scholar] [CrossRef]
- Petrovic, A.; Alpdogan, O.; Willis, L.M.; Eng, J.M.; Greenberg, A.S.; Kappel, B.J.; Liu, C.; Murphy, G.J.; Heller, G.; van den Brink, M.R. LPAM (alpha 4 beta 7 integrin) is an important homing integrin on alloreactive T cells in the development of intestinal graft-versus-host disease. Blood 2004, 103, 1542–1547. [Google Scholar] [CrossRef]
- Aggeletopoulou, I.; Assimakopoulos, S.F.; Konstantakis, C.; Triantos, C. Interleukin 12/interleukin 23 pathway: Biological basis and therapeutic effect in patients with Crohn’s disease. World J. Gastroenterol. 2018, 24, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- Allocca, M.; Bonifacio, C.; Fiorino, G.; Spinelli, A.; Furfaro, F.; Balzarini, L.; Bonovas, S.; Danese, S. Efficacy of tumour necrosis factor antagonists in stricturing Crohn’s disease: A tertiary center real-life experience. Dig. Liver Dis. 2017, 49, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Bouhnik, Y.; Carbonnel, F.; Laharie, D.; Stefanescu, C.; Hebuterne, X.; Abitbol, V.; Nachury, M.; Brixi, H.; Bourreille, A.; Picon, L.; et al. Efficacy of adalimumab in patients with Crohn’s disease and symptomatic small bowel stricture: A multicentre, prospective, observational cohort (CREOLE) study. Gut 2018, 67, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Frei, R.; Fournier, N.; Zeitz, J.; Scharl, M.; Morell, B.; Greuter, T.; Schreiner, P.; Misselwitz, B.; Safroneeva, E.; Schoepfer, A.M.; et al. Early Initiation of Anti-TNF is Associated with Favourable Long-term Outcome in Crohn’s Disease: 10-Year-Follow-up Data from the Swiss IBD Cohort Study. J. Crohn’s Colitis 2019, 13, 1292–1301. [Google Scholar] [CrossRef]
- Valvano, M.; Vinci, A.; Cesaro, N.; Frassino, S.; Ingravalle, F.; Ameli, M.; Viscido, A.; Necozione, S.; Latella, G. The long-term effect on surgery-free survival of biological compared to conventional therapy in Crohn’s disease in real world-data: A retrospective study. BMC Gastroenterol. 2023, 23, 438. [Google Scholar] [CrossRef] [PubMed]
- Aratari, A.; Scribano, M.L.; Pugliese, D.; Baccolini, V.; De Biasio, F.; Verna, S.; Morretta, C.; Festa, S.; Armuzzi, A.; Papi, C. Crohn’s disease after surgery: Changes in post-operative management strategies over time and their impact on long-term re-operation rate-A retrospective multicentre real-world study. Aliment. Pharm. Ther. 2024, 59, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Theiss, A.L.; Simmons, J.G.; Jobin, C.; Lund, P.K. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J. Biol. Chem. 2005, 280, 36099–36109. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, I.; Carissimi, C.; Scafa, N.; Bastianelli, A.; Fulci, V.; Renzini, A.; Russo, G.; Oliva, S.; Vitali, R.; Palone, F.; et al. Characterization of patient-derived intestinal organoids for modelling fibrosis in Inflammatory Bowel Disease. Inflamm. Res. 2024, 73, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Cuomo, C.; Mignini, I.; Gasbarrini, A.; Papa, A. Focus on Anti-Tumour Necrosis Factor (TNF)-alpha-Related Autoimmune Diseases. Int. J. Mol. Sci. 2023, 24, 8187. [Google Scholar] [CrossRef] [PubMed]
- Angelis, I.D.; Turco, L. Caco-2 cells as a model for intestinal absorption. Curr. Protoc. Toxicol. 2011, 47, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.M.; Hu, X.Y.; Chen, Y.; Xie, J.H.; Ying, M.X.; Wang, Y.D.; Yu, Q. Differentiated Caco-2 cell models in food-intestine interaction study: Current applications and future trends. Trends Food Sci. Technol. 2021, 107, 455–465. [Google Scholar] [CrossRef]
- Fedi, A.; Vitale, C.; Ponschin, G.; Ayehunie, S.; Fato, M.; Scaglione, S. In vitro models replicating the human intestinal epithelium for absorption and metabolism studies: A systematic review. J. Control. Release 2021, 335, 247–268. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artone, S.; Ciafarone, A.; Augello, F.R.; Lombardi, F.; Cifone, M.G.; Palumbo, P.; Cinque, B.; Latella, G. Evaluation of the Antifibrotic Effects of Drugs Commonly Used in Inflammatory Intestinal Diseases on In Vitro Intestinal Cellular Models. Int. J. Mol. Sci. 2024, 25, 8862. https://doi.org/10.3390/ijms25168862
Artone S, Ciafarone A, Augello FR, Lombardi F, Cifone MG, Palumbo P, Cinque B, Latella G. Evaluation of the Antifibrotic Effects of Drugs Commonly Used in Inflammatory Intestinal Diseases on In Vitro Intestinal Cellular Models. International Journal of Molecular Sciences. 2024; 25(16):8862. https://doi.org/10.3390/ijms25168862
Chicago/Turabian StyleArtone, Serena, Alessia Ciafarone, Francesca Rosaria Augello, Francesca Lombardi, Maria Grazia Cifone, Paola Palumbo, Benedetta Cinque, and Giovanni Latella. 2024. "Evaluation of the Antifibrotic Effects of Drugs Commonly Used in Inflammatory Intestinal Diseases on In Vitro Intestinal Cellular Models" International Journal of Molecular Sciences 25, no. 16: 8862. https://doi.org/10.3390/ijms25168862