

Evolution and Competitive Struggles of Lactiplantibacillus plantarum under Different Oxygen Contents


Abstract
:1. Introduction
2. Results and Discussion
2.1. Genome Summary and General Features
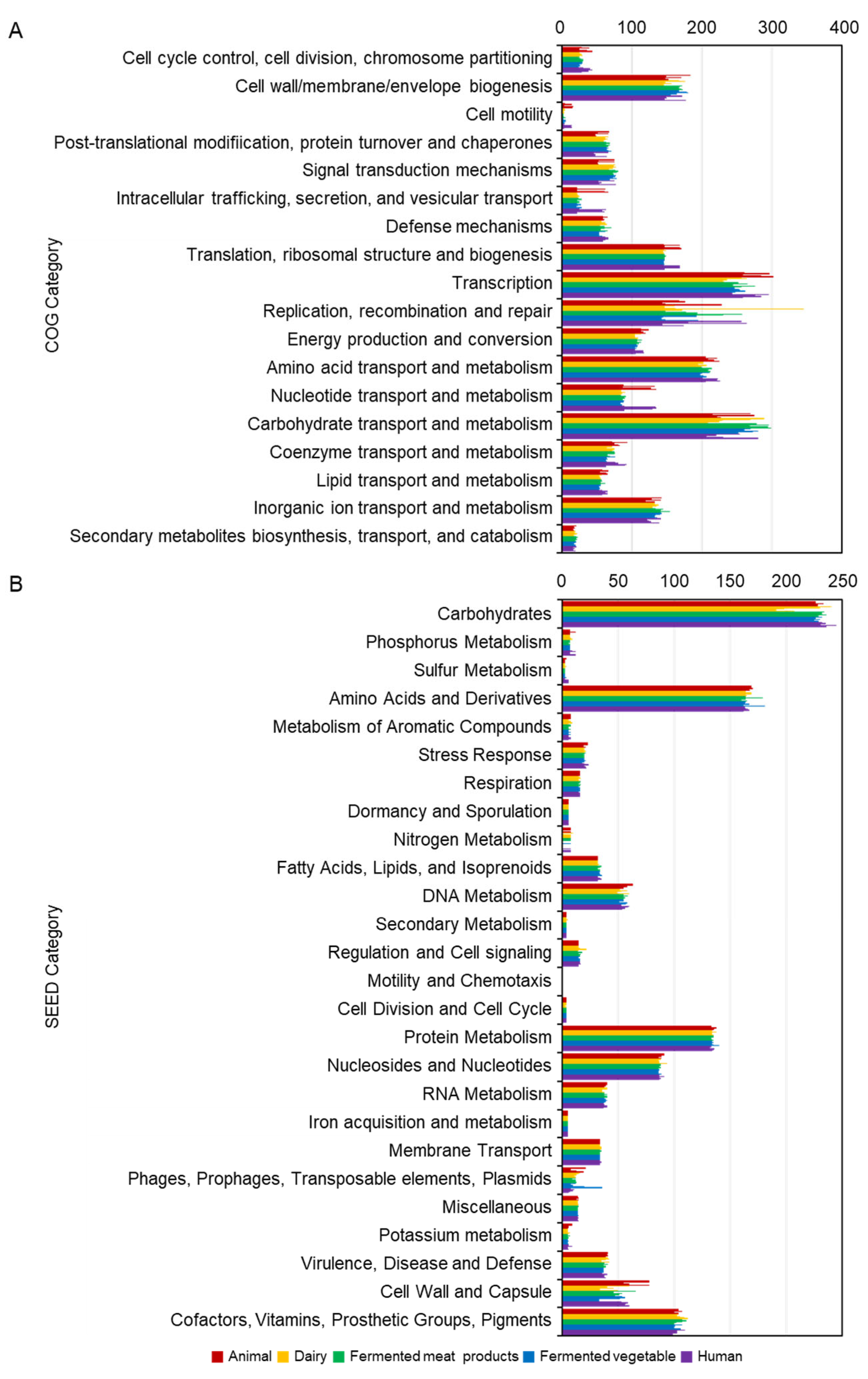
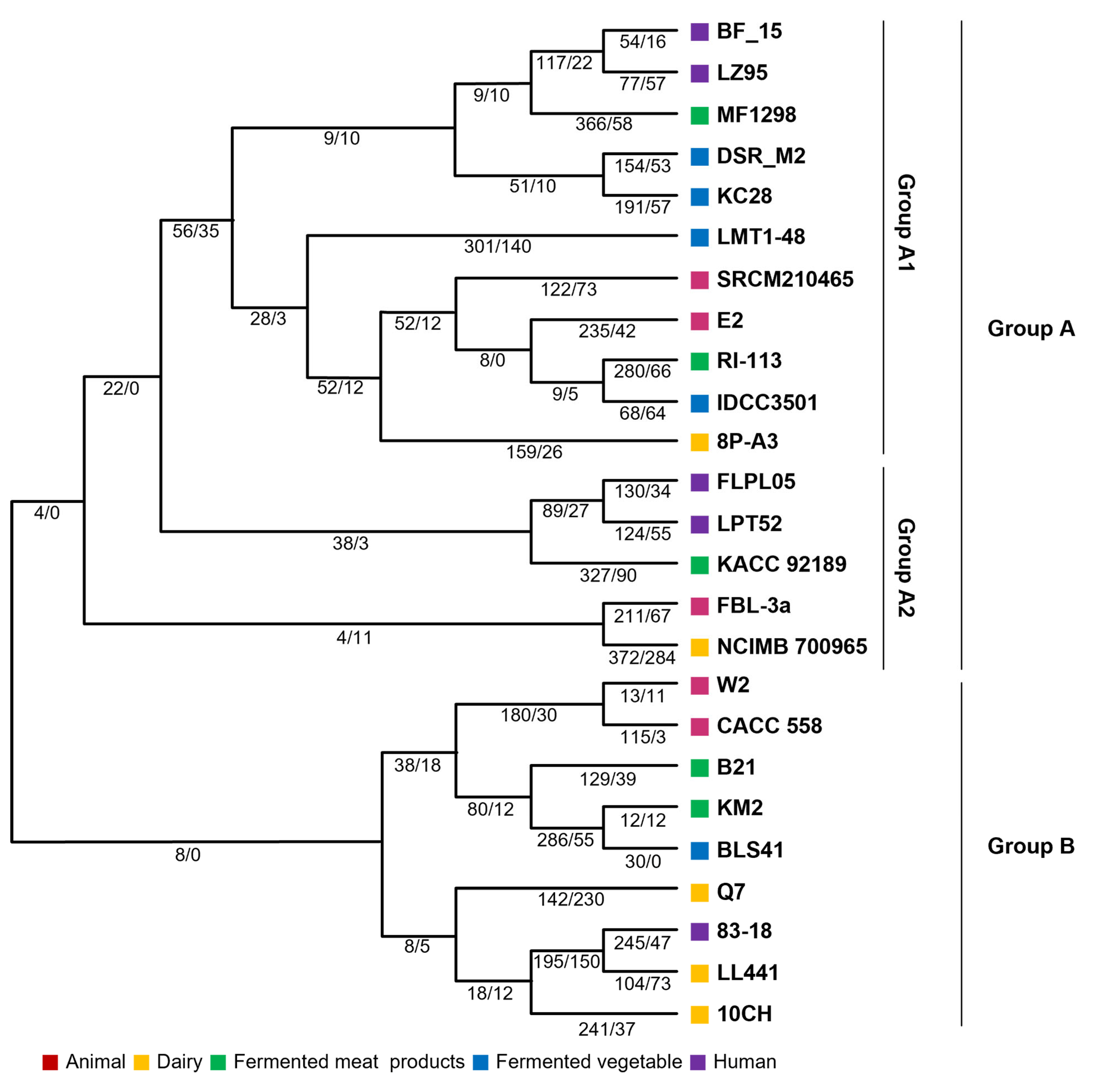
2.2. Comparative Analysis of Lb. plantarum
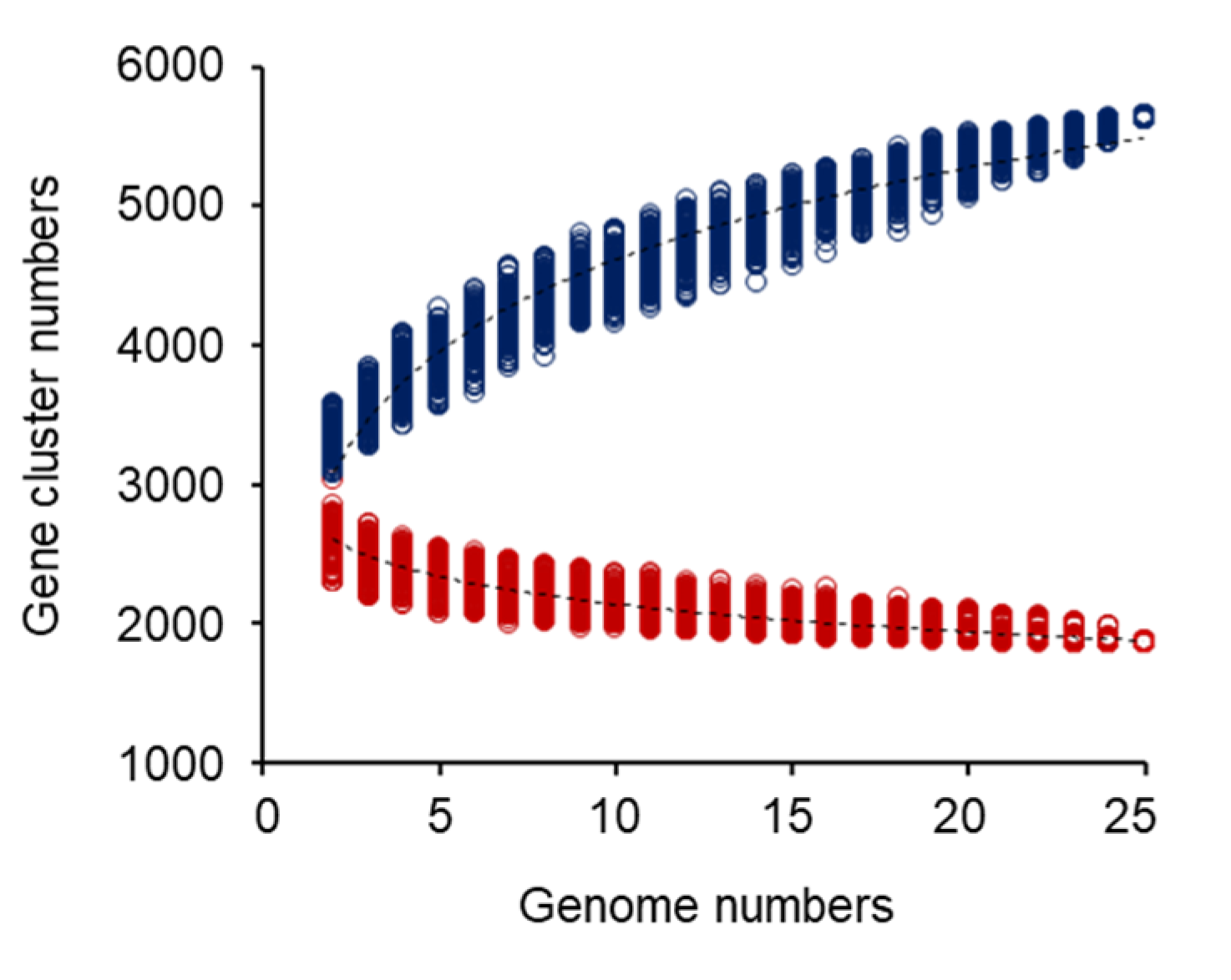
2.3. Pan-Genome and Core-Genome Analysis
2.4. Shared and Unique Gene Analysis of Lb. plantarum
2.5. Gene Gain and Loss Analysis—Evolutionary Events
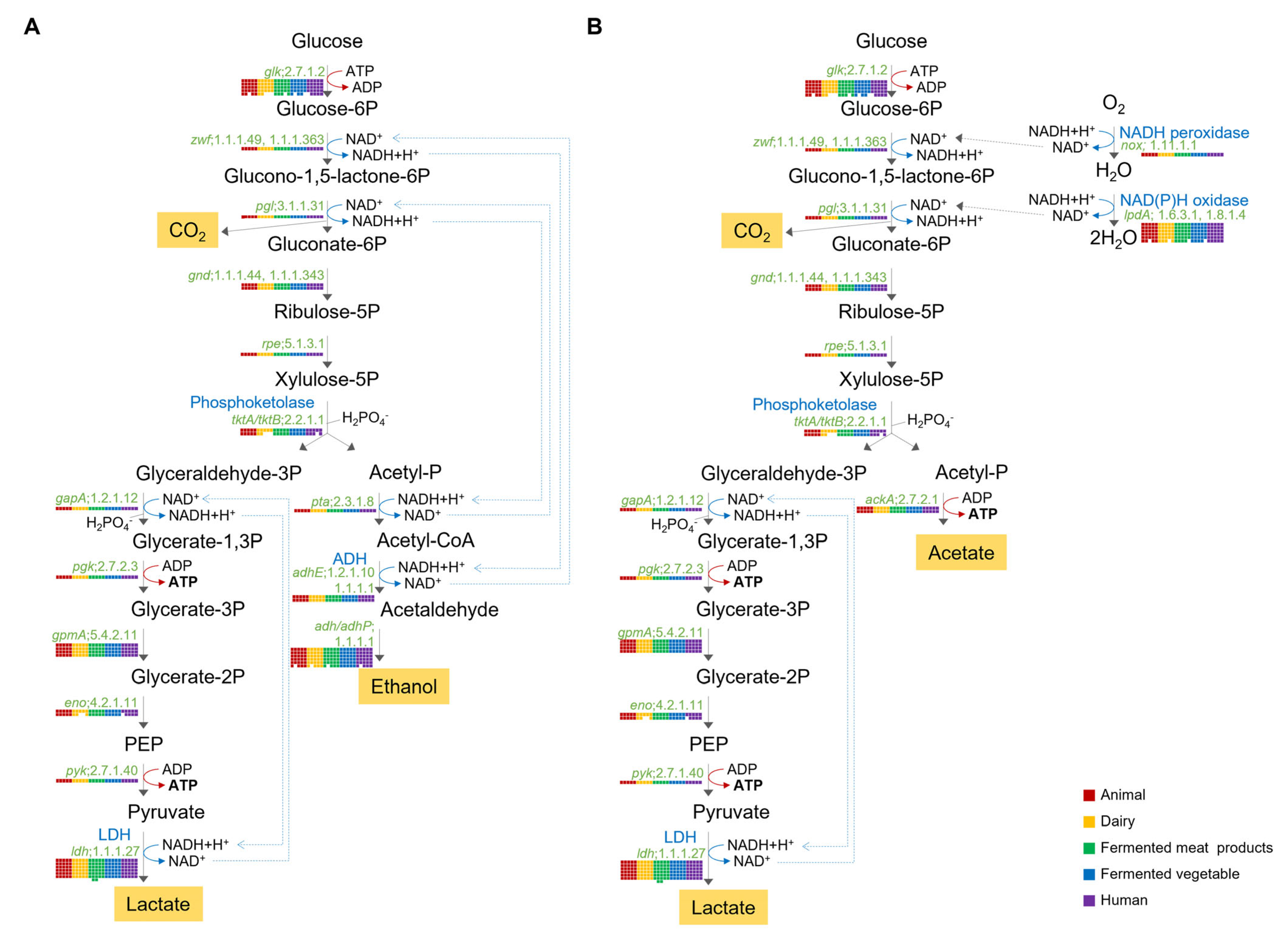
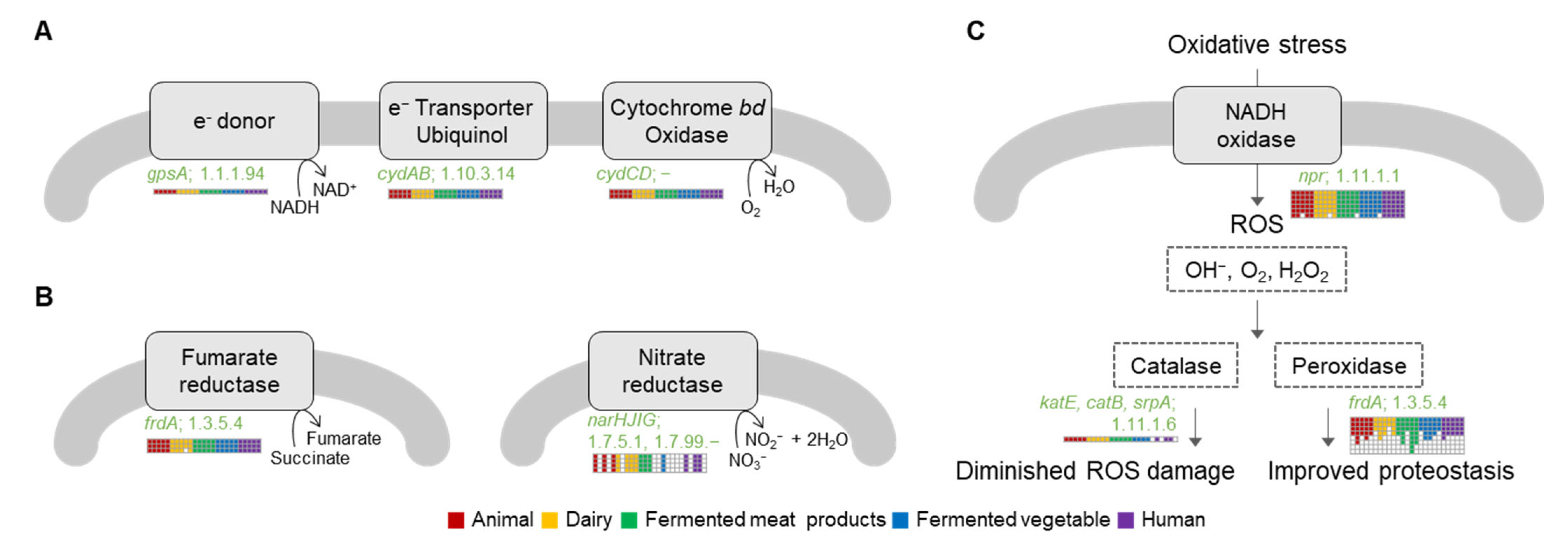
2.6. Genomic Insights for Respiration System
2.6.1. Hetero-Lactic Fermentation Pathway
2.6.2. Respiratory Chain
2.6.3. Nitrate Reductase
2.6.4. Reactive Oxidation Species (ROS) Persistence System
3. Materials and Methods
3.1. Datasets
3.2. Genome Similarity Measures
3.3. Comparative Genome
3.4. Construction of Phylogenetic Tree
3.5. Gene Gain and Loss
3.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Imlay, J.A. How oxygen damages microbes: Oxygen tolerance and obligate anaerobiosis. Adv. Microb. Physiol. 2002, 46, 111–153. [Google Scholar] [PubMed]
- Friedman, E.S.; Bittinger, K.; Esipova, T.V.; Hou, L.; Chau, L.; Jiang, J.; Mesaros, C.; Lund, P.J.; Liang, X.; FitzGerald, G.A.; et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc. Natl. Acad. Sci. USA 2018, 115, 4170–4175. [Google Scholar] [CrossRef]
- Andre, A.C.; Debande, L.; Marteyn, B.S. The selective advantage of facultative anaerobes relies on their unique ability to cope with changing oxygen levels during infection. Cell. Microbiol. 2021, 23, e13338. [Google Scholar] [CrossRef]
- Spees, A.M.; Wangdi, T.; Lopez, C.A.; Kingsbury, D.D.; Xavier, M.N.; Winter, S.E.; Tsolis, R.M.; Bäumler, A.J. Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. mBio 2013, 4, e00430-13. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; Winter, S.E.; Rivera-Chávez, F.; Xavier, M.N.; Poon, V.; Nuccio, S.P. Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. mBio 2012, 3, e00143-12. [Google Scholar] [CrossRef]
- Mokoena, M.P. Lactic acid bacteria and their bacteriocins: Classification, biosynthesis and applications against uropathogens: A mini-review. Molecules 2017, 22, 1255. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Tanizawa, Y.; Arita, M. Isolation and identification of lactic acid bacteria from environmental samples. Methods Mol. Biol. 2019, 1887, 3–13. [Google Scholar] [PubMed]
- Gorkiewicz, G.; Moschen, A. Gut microbiome: A new player in gastrointestinal disease. Virchows Arch. 2017, 472, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.B.; Gaudu, P.; Lechardeur, D.; Petit, M.A.; Gruss, A. Aerobic respiration metabolism in lactic acid bacteria and uses in biotechnology. Annu. Rev. Food Sci. Technol. 2012, 3, 37–58. [Google Scholar] [CrossRef]
- Martino, M.E.; Bayjanov, J.R.; Caffrey, B.E.; Wels, M.; Joncour, P.; Hughes, S.; Gillet, B.; Kleerebezem, M.; van Hijum, S.A.F.T.; Leulier, F. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 2016, 18, 4974–4989. [Google Scholar] [CrossRef]
- Heo, S.; Kim, J.-H.; Kwak, M.-S.; Jeong, D.-W.; Sung, M.-H. Complete genome sequence of Lactiplantibacillus plantarum KM2 from low-temperature aging beef. Korean J. Microbiol. 2021, 57, 303–306. [Google Scholar]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.A.; Richardson, D.J. Respiration of nitrate and nitrite. EcoSal Plus. 2008, 3, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.G.; O’Connor, L.; Walsh, D.; Condon, S. Oxygen dependent lactate utilization by Lactobacillus plantarum. Arch. Microbiol. 1985, 141, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Goffin, P.; Lorquet, F.; Kleerebezem, M.; Hols, P. Major role of NAD-dependent lactate dehydrogenases in aerobic lactate utilization in Lactobacillus plantarum during early stationary phase. J. Bacteriol. 2004, 186, 6661–6666. [Google Scholar] [CrossRef] [PubMed]
- Zharova, T.V.; Grivennikova, V.G.; Borisov, V.B. F1·Fo ATP synthase/ATPase: Contemporary view on unidirectional catalysis. Int. J. Mol. Sci. 2023, 24, 5417. [Google Scholar] [CrossRef] [PubMed]
- Parshina, S.N.; Sipma, J.; Henstra, A.M.; Stams, A.J. Carbon monoxide as an electron donor for the biological reduction of sulphate. Int. J. Microbiol. 2010, 2010, 319527. [Google Scholar] [CrossRef] [PubMed]
- Rothery, R.A.; Bertero, M.G.; Spreter, T.; Bouromand, N.; Strynadka, N.C.; Weiner, J.H. Protein crystallography reveals a role for the FS0 cluster of Escherichia coli nitrate reductase A (NarGHI) in enzyme maturation. J. Biol. Chem. 2010, 285, 8801–8807. [Google Scholar] [CrossRef]
- Rothery, R.A.; Blasco, F.; Magalon, A.; Weiner, J.H. The diheme cytochrome b subunit (Narl) of Escherichia coli nitrate reductase A (NarGHI): Structure, function, and interaction with quinols. J. Mol. Microbiol. Biotechnol. 2001, 3, 273–283. [Google Scholar] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Goker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic. Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.; Albaum, S.P.; Doppmeier, D.; Pühler, A.; Vorhölter, F.J.; Zakrzewski, M.; Goesmann, A. EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 2009, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An enhanced software platform for comparative gene content analyses. Nucleic. Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef]
- Lerat, E.; Daubin, V.; Moran, N.A. From gene trees to organismal phylogeny in prokaryotes: The case of the gamma-Proteobacteria. PLoS Biol. 2003, 1, E19. [Google Scholar] [CrossRef]
- Ogilvie, H.A.; Bouckaert, R.R.; Drummond, A.J. StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol. Biol. Evol. 2017, 34, 2101–2114. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Librado, P.; Vieira, F.G.; Rozas, J. BadiRate: Estimating family turnover rates by likelihood-based methods. Bioinformatics 2012, 28, 279–281. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | No. of Genome | Average Size of Genome (Mb) ns | Average G + C Content (%) ns | Average Number of CDSs ns |
|---|---|---|---|---|
| Animal | 30 | 3.34 ± 0.14 | 44.44 ± 0.16 | 3061 ± 151 |
| Dairy products | 25 | 3.27 ± 0.14 | 44.50 ± 0.14 | 2982 ± 127 |
| Fermented alcohol | 4 | 3.33 ± 0.16 | 44.46 ± 0.19 | 3043 ± 169 |
| Fermented fish | 1 | 3.20 ± 0.00 | 44.59 ± 0.00 | 2858 ± 0 |
| Fermented meat | 10 | 3.39 ± 0.10 | 44.35 ± 0.11 | 3084 ± 92 |
| Fermented soybean | 7 | 3.33 ± 0.07 | 44.48 ± 0.14 | 3028 ± 60 |
| Fermented vegetable | 65 | 3.31 ± 0.13 | 44.46 ± 0.19 | 3025 ± 143 |
| Human | 38 | 3.31 ± 0.07 | 44.48 ± 0.11 | 3015 ± 77 |
| Plant | 9 | 3.30 ± 0.07 | 44.45 ± 0.08 | 2976 ± 95 |
| Others | 39 | 3.31 ± 0.10 | 44.46 ± 0.14 | 3013 ± 100 |
| Strain | Genome Assembly | Country | Origin | Genome Size (Mb) | G + C Content (%) | No. of CDSs |
|---|---|---|---|---|---|---|
| CACC 558 | GCA_010092485.1 | Korea | Animal; Canine | 3.35 | 44.43 | 3056 |
| E2 | GCA_026013765.1 | China | Animal; Larimichthys crocea | 3.48 | 44.19 | 3165 |
| FBL-3a | GCA_003999275.1 | China | Animal; Beef cattle | 3.23 | 44.60 | 2918 |
| SRCM210465 | GCA_024969905.1 | Korea | Animal; Sus scrofa domesticus | 3.26 | 44.50 | 2994 |
| W2 | GCA_021560135.1 | China | Animal; Penaeus vannamei | 3.26 | 44.60 | 2944 |
| 10CH | GCA_002005385.2 | UK | Dairy products; Cheese | 3.31 | 44.50 | 2993 |
| 8P-A3 | GCA_009762745.1 | Russia | Dairy products; Probiotic preparation “Lactobacterinum” | 3.33 | 44.38 | 3030 |
| LL441 | GCA_027474465.1 | Spain | Dairy products; Traditional cheese | 3.20 | 44.50 | 2913 |
| NCIMB 700965 | GCA_003611015.1 | New Zealand | Dairy products; Cheese | 3.22 | 44.54 | 2978 |
| Q7 | GCA_003999605.1 | China | Dairy products; Yak fermented milk | 2.98 | 44.84 | 2730 |
| B21 | GCA_000931425.2 | Australia | Fermented meat; Vietnamese fermented sausage (nem chua) | 3.31 | 44.45 | 3011 |
| KACC 92189 | GCA_003692595.1 | Korea | Fermented meat; Starter | 3.46 | 44.30 | 3133 |
| KM2 | GCA_016838645.1 | Korea | Fermented meat; Ripening beef | 3.42 | 44.28 | 3111 |
| MF1298 | GCA_001880185.2 | Norway | Fermented meat; Fermented sausage | 3.58 | 44.22 | 3243 |
| RI-113 | GCA_001990145.1 | Switzerland | Fermented meat; Fermented salami | 3.46 | 44.34 | 3188 |
| BLS41 | GCA_002116955.1 | Korea | Fermented vegetable; Kimchi | 3.48 | 44.15 | 3195 |
| DSR_M2 | GCA_003286955.1 | Korea | Fermented vegetable; Kimchi | 3.25 | 44.59 | 2971 |
| IDCC3501 | GCA_003428355.1 | Korea | Fermented vegetable; Kimchi | 3.24 | 44.50 | 2957 |
| KC28 | GCA_002948215.1 | Korea | Fermented vegetable; Kimchi | 3.29 | 44.50 | 3042 |
| LMT1-48 | GCA_003813125.1 | Korea | Fermented vegetable; Kimchi | 3.28 | 44.50 | 3032 |
| 83-18 | GCA_009759825.1 | Russia | Human; Human feces (woman, 60 years old) | 3.36 | 44.39 | 3107 |
| BF_15 | GCA_025723165.1 | China | Human; The feces of breast-fed infants | 3.31 | 44.50 | 3016 |
| FLPL05 | GCA_027557615.1 | China | Human; Homo sapiens-faces | 3.30 | 44.60 | 2992 |
| LPT52 | GCA_023348525.1 | Ireland | Human; Homo sapiens-faces | 3.27 | 44.60 | 2958 |
| LZ95 | GCA_001484005.1 | China | Human; Homo sapiens | 3.32 | 44.49 | 3005 |
| Query Genome | Strain | ANI Value | dDDH Value | Distance | Probability that dDDH > 70% | G + C Difference |
|---|---|---|---|---|---|---|
| KM2 | CACC 558 | 99.26 | 93.7 | 0.0081 | 96.88 | 0.19 |
| E2 | 98.96 | 91.8 | 0.0102 | 96.35 | 86.50 | |
| FBL-3a | 99.12 | 93.1 | 0.0087 | 96.74 | 0.36 | |
| SRCM210465 | 99.06 | 93.4 | 0.0084 | 96.82 | 86.90 | |
| W2 | 99.24 | 94.5 | 0.0071 | 97.09 | 90.30 | |
| 10CH | 99.15 | 93.2 | 0.0086 | 96.76 | 0.04 | |
| 8P-A3 | 99.11 | 92.6 | 0.0092 | 96.60 | 0.11 | |
| LL441 | 98.80 | 89.2 | 0.0130 | 95.52 | 86.10 | |
| NCIMB 700965 | 98.59 | 88.4 | 0.0138 | 95.23 | 0.23 | |
| Q7 | 99.07 | 92.5 | 0.0094 | 96.58 | 85.70 | |
| B21 | 99.41 | 95.9 | 0.0056 | 97.41 | 0.09 | |
| KACC 92189 | 99.17 | 93.9 | 0.0079 | 96.93 | 0.09 | |
| MF1298 | 99.06 | 93.5 | 0.0083 | 96.83 | 0.04 | |
| RI-113 | 98.97 | 92.6 | 0.0093 | 96.59 | 0.01 | |
| BLS41 | 99.96 | 100 | 0.0000 | 98.30 | 0.01 | |
| DSR_M2 | 99.10 | 93.1 | 0.0087 | 96.74 | 0.09 | |
| IDCC3501 | 99.11 | 93.0 | 0.0089 | 96.70 | 88.60 | |
| KC28 | 99.09 | 92.5 | 0.0094 | 96.57 | 0.05 | |
| LMT1-48 | 98.98 | 91.0 | 0.0110 | 96.13 | 86.40 | |
| 83-18 | 98.86 | 91.4 | 0.0106 | 96.25 | 0.21 | |
| BF_15 | 99.07 | 92.1 | 0.0098 | 96.46 | 90.50 | |
| FLPL05 | 99.03 | 93.0 | 0.0089 | 96.69 | 86.90 | |
| LPT52 | 99.06 | 92.8 | 0.0091 | 96.65 | 90.30 | |
| LZ95 | 99.08 | 93.3 | 0.0085 | 96.77 | 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, S.; Jung, E.J.; Park, M.-K.; Sung, M.-H.; Jeong, D.-W. Evolution and Competitive Struggles of Lactiplantibacillus plantarum under Different Oxygen Contents. Int. J. Mol. Sci. 2024, 25, 8861. https://doi.org/10.3390/ijms25168861
Heo S, Jung EJ, Park M-K, Sung M-H, Jeong D-W. Evolution and Competitive Struggles of Lactiplantibacillus plantarum under Different Oxygen Contents. International Journal of Molecular Sciences. 2024; 25(16):8861. https://doi.org/10.3390/ijms25168861
Chicago/Turabian StyleHeo, Sojeong, Eun Jin Jung, Mi-Kyung Park, Moon-Hee Sung, and Do-Won Jeong. 2024. "Evolution and Competitive Struggles of Lactiplantibacillus plantarum under Different Oxygen Contents" International Journal of Molecular Sciences 25, no. 16: 8861. https://doi.org/10.3390/ijms25168861
APA StyleHeo, S., Jung, E. J., Park, M.-K., Sung, M.-H., & Jeong, D.-W. (2024). Evolution and Competitive Struggles of Lactiplantibacillus plantarum under Different Oxygen Contents. International Journal of Molecular Sciences, 25(16), 8861. https://doi.org/10.3390/ijms25168861