
AAV-NDI1 Therapy Provides Significant Benefit to Murine and Cellular Models of Glaucoma

 , ,
, ,  , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
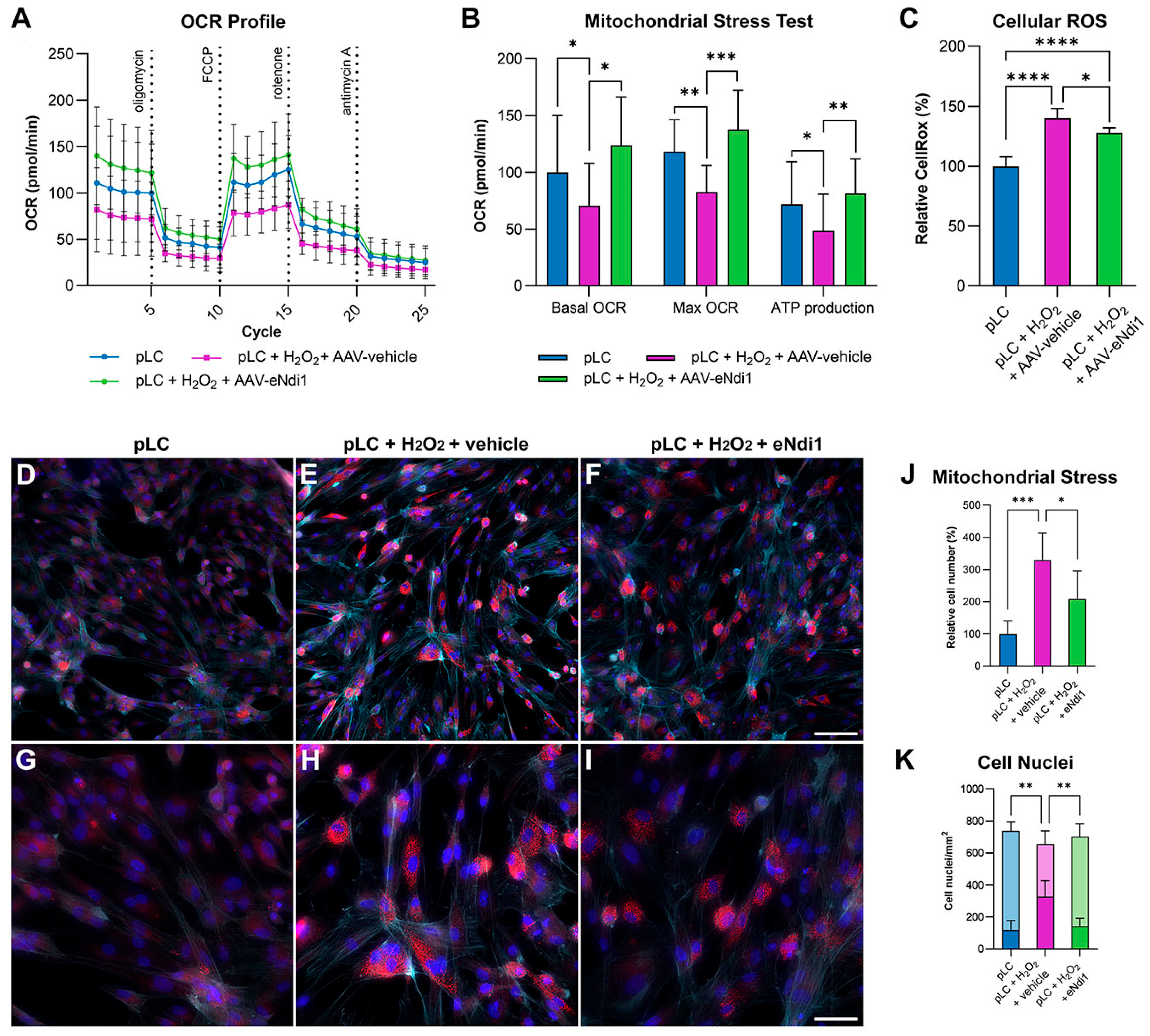
2.1. AAV-eNdi1 Provides Benefit in H2O2-Insulted Porcine LC Cells
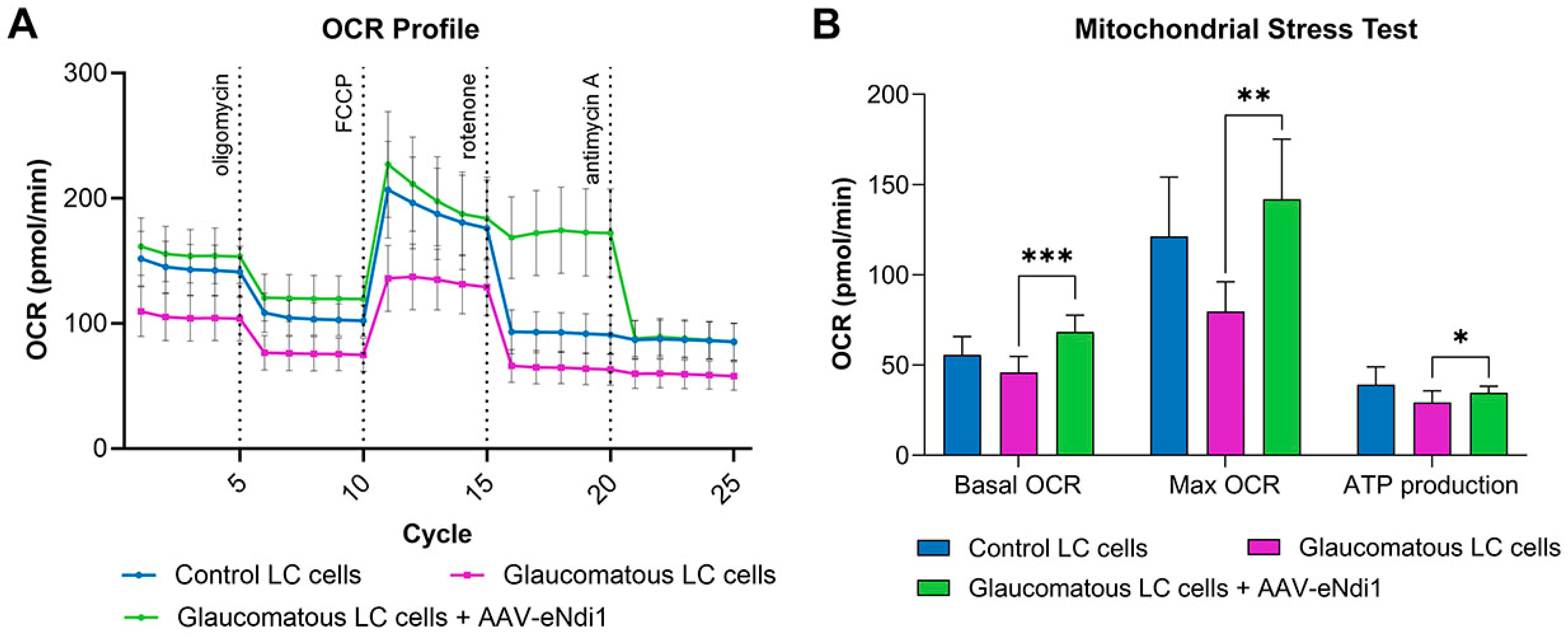
2.2. AAV-eNdi1 Provides Benefit in Human Glaucomatous LC Cells
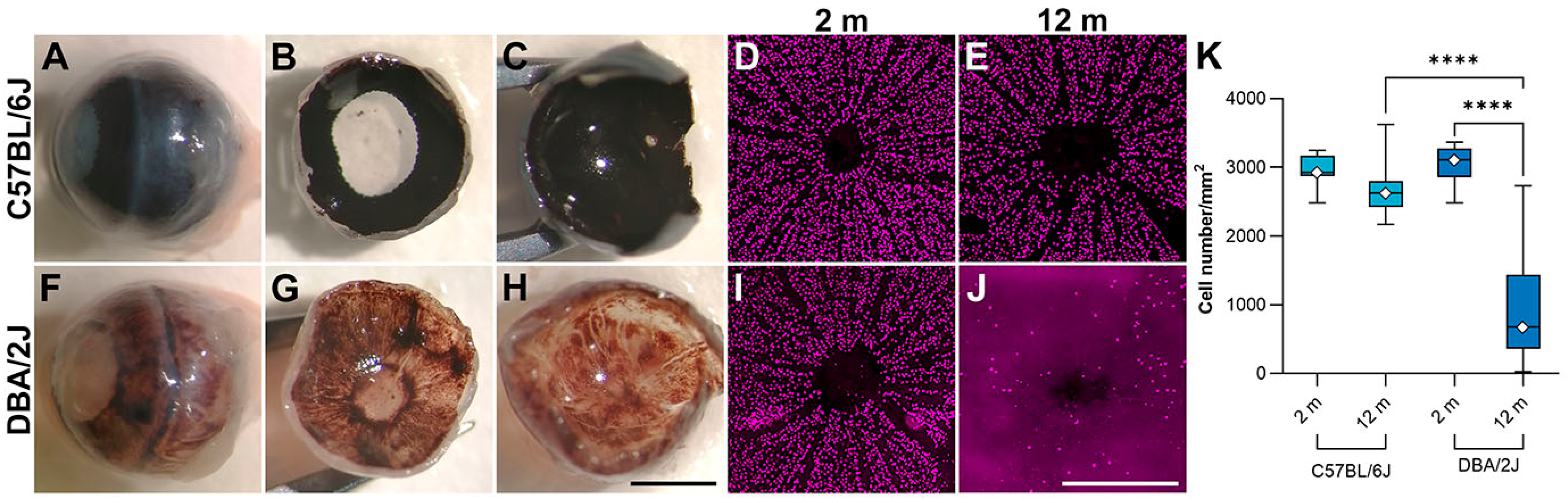
2.3. AAV-eNdi1 Provides Benefit in the DBA/2J Murine Model of Glaucoma
3. Discussion
4. Materials and Methods
4.1. Study Design
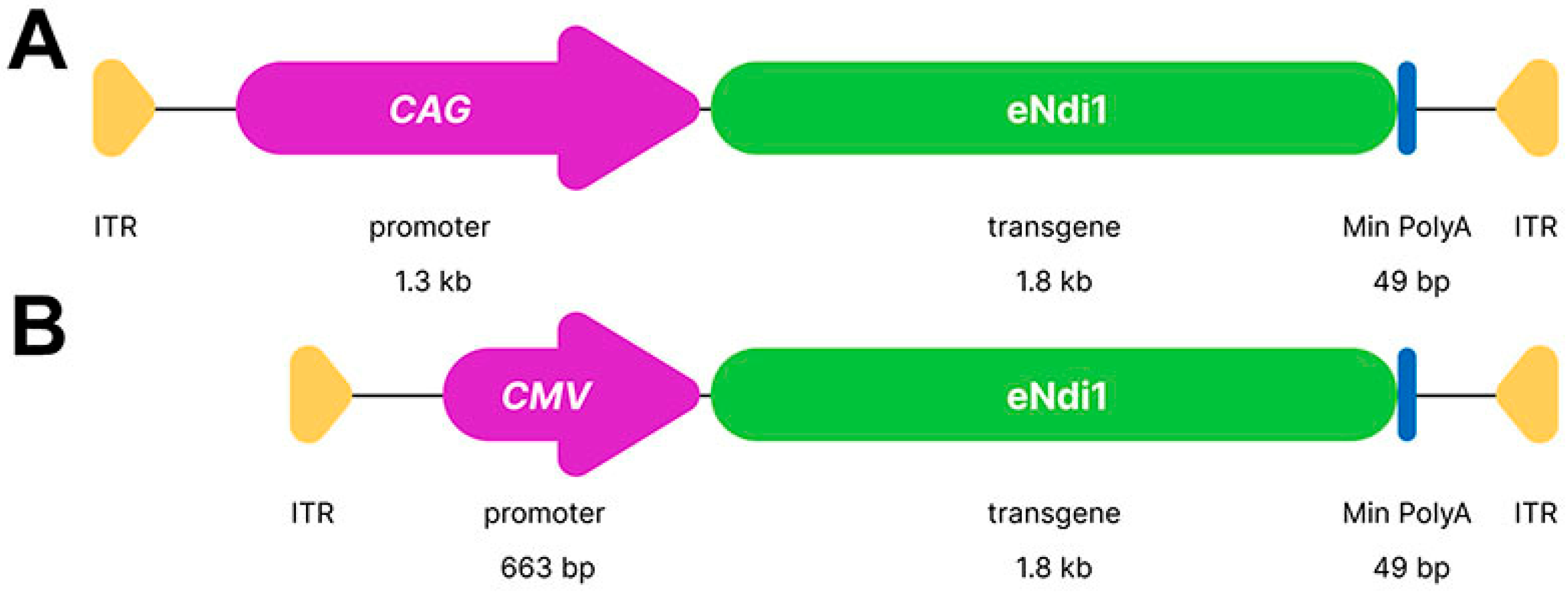
4.2. Plasmid Constructs and AAV Production
4.3. LC Cell Characterisation, Cell Culture and Mitochondrial Stress Test
4.4. Cellular ROS Assay
4.5. pLC Transfection Immunocytochemistry
4.6. Intravitreal Injection and Electroretinography in Mice
4.7. NADH Oxidation Rate Assay
4.8. Retinal Wholemounts and Immunohistochemistry
4.9. Fluorescence Microscopy
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Schuster, A.K.; Erb, C.; Hoffmann, E.M.; Dietlein, T.; Pfeiffer, N. The Diagnosis and Treatment of Glaucoma. Dtsch. Arztebl. Int. 2020, 117, 225–234. [Google Scholar] [CrossRef]
- Bourne, R.R.A.; Stevens, G.A.; White, R.A.; Smith, J.L.; Flaxman, S.R.; Price, H.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; et al. Causes of vision loss worldwide, 1990–2010: A systematic analysis. Lancet Glob. Health 2013, 6, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Killer, H.E.; Pircher, A. Normal tension glaucoma: Review of current understanding and mechanisms of the pathogenesis. Eye 2018, 32, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Tamm, E.R.; Ethier, C.R.; Lasker, I.R.R.F. initiative on astrocytes and glaucomatous neurodegeneration participants. Biological aspects of axonal damage in glaucoma: A brief review. Exp. Eye Res. 2017, 157, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kilpatrick, J.I.; Lukasz, B.; Jarvis, S.P.; McDonnell, F.; Wallace, D.M.; Clark, A.F.; O’Brien, C.J. Increased Substrate Stiffness Elicits a Myofibroblastic Phenotype in Human Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2018, 59, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Nickells, R.W. From ocular hypertension to ganglion cell death: A theoretical sequence of events leading to glaucoma. Can. J. Ophthalmol. 2007, 42, 278–287. [Google Scholar] [CrossRef]
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 2018, 4, 14. [Google Scholar] [CrossRef]
- Crish, S.D.; Sappington, R.M.; Inman, D.M.; Horner, P.J.; Calkins, D.J. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 5196–5201. [Google Scholar] [CrossRef]
- Rakic, P.; Riley, K.P. Overproduction and elimination of retinal axons in the fetal rhesus monkey. Science 1983, 219, 1441–1444. [Google Scholar] [CrossRef]
- Lu, L.J.; Tsai, J.C.; Liu, J. Novel Pharmacologic Candidates for Treatment of Primary Open-Angle Glaucoma. Yale J. Biol. Med. 2017, 90, 111–118. [Google Scholar]
- Catalani, E.; Brunetti, K.; Del Quondam, S.; Cervia, D. Targeting mitochondrial dysfunction and oxidative stress to prevent the neurodegeneration of retinal ganglion cells. Antioxidants 2023, 12, 2011. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, V.; Trounce, I.A.; Crowston, J.G. Mechanisms of retinal ganglion cell injury in aging and glaucoma. Ophthalmic Res. 2010, 44, 173–178. [Google Scholar] [CrossRef]
- Kong, G.Y.; Van Bergen, N.J.; Trounce, I.A.; Crowston, J.G. Mitochondrial dysfunction and glaucoma. J. Glaucoma 2009, 18, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Munemasa, Y.; Kitaoka, Y.; Kuribayashi, J.; Ueno, S. Modulation of mitochondria in the axon and soma of retinal ganglion cells in a rat glaucoma model. J. Neurochem. 2010, 115, 1508–1519. [Google Scholar] [CrossRef]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. Smoking and Eye Pathologies. A systemic review. Part I. Anterior eye segment pathologies. Curr. Pharm. Des. 2017, 23, 629–638. [Google Scholar]
- Libby, R.T.; Anderson, M.G.; Pang, I.-H.; Robinson, Z.H.; Savinova, O.V.; Cosma, I.M.; Snow, A.; Wilson, L.A.; Smith, R.S.; Clar, A.F.; et al. Inherited glaucoma in DBA/2J mice: Pertinent disease features for studying the neurodegeneration. Vis. Neurosci. 2005, 22, 637–648. [Google Scholar] [CrossRef]
- Leadsham, J.E.; Sanders, G.; Giannaki, S.; Bastow, E.L.; Hutton, R.; Naeimi, W.R.; Breitenbach, M.; Gourlay, C.W. Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH oxidase, Yno1p, in yeast. Cell Metab. 2013, 18, 279–286. [Google Scholar] [CrossRef]
- Murphy, R.K.; Ketzler, L.; Rice, R.D.; Johnson, S.M.; Doss, M.S.; Jaccoma, E.H. Oral glucosamine supplements as a possible ocular hypertensive agent. JAMA Ophthalmol. 2013, 131, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic Retinopathy: The role of mitochondria in the neural retina and microvascular disease. Antioxidants 2020, 9, 905. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial dysfunction in Alzheimer’s disease: Opportunities for drug development. Curr. Neuropharmacol. 2022, 20, 675–692. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial dysfunction in Parkinson’s disease—A key disease hallmark with therapeutic potential. Mol. Neurodegener. 2023, 18, 83. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Xie, Z.; Chen, S.Y.; Zhang, X. Mitochondrial dysfunction in glaucomatous degeneration. Int. J. Ophthalmol. 2023, 16, 811–823. [Google Scholar] [CrossRef]
- Izzotti, A.; Sacca, S.C.; Longobardi, M.; Cartiglia, C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5251–5258. [Google Scholar] [CrossRef]
- Izzotti, A.; Sacca, S.C.; Longobardi, M.; Cartiglia, C. Mitochondrial damage in the trabecular meshwork of patients with glaucoma. Arch. Ophthalmol. 2010, 128, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Sacca, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA damage in the human trabecular meshwork: Clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Durango, R.; Fernandez-Martinez, A.; Garcia-Feijoo, J.; Castillo, A.; de la Casa, J.M.; Garcia-Bueno, B.; Perez-Nievas, B.G.; Fernandez-Cruz, A.; Leza, J.C. Expression of nitrotyrosine and oxidative consequences in the trabecular meshwork of patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2506–2511. [Google Scholar] [CrossRef] [PubMed]
- Muenster, S.; Lieb, W.S.; Fabry, G.; Allen, K.N.; Kamat, S.S.; Guy, A.H.; Dordea, A.C.; Teixeira, L.; Tainsh, R.E.; Yu, B.; et al. The ability of nitric oxide to lower intraocular pressure is dependent on guanylyl cyclase. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4826–4835. [Google Scholar] [CrossRef] [PubMed]
- Ammar, D.A.; Hamweyah, K.M.; Kahook, M.Y. Antioxidants protect trabecular meshwork cells from hydrogen peroxide-induced cell death. Transl. Vis. Sci. Technol. 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Sacca, S.C.; Izzotti, A.; Rossi, P.; Traverso, C. Glaucomatous outflow pathway and oxidative stress. Exp. Eye Res. 2007, 84, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Luna, C.; Li, G.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Role of miR-29b on the regulation of the extracellular matrix in human trabecular meshwork cells under chronic oxidative stress. Mol. Vis. 2009, 15, 2488–2497. [Google Scholar]
- Park, C.H.; Kim, J.W. Effect of advanced glycation end products on oxidative stress and senescence of trabecular meshwork cells. Korean J. Ophthalmol. 2012, 26, 123–131. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ge, J.; Tombran-Tink, J. Mitochondrial defects and dysfunction in calcium regulation in glaucomatous trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4912–4922. [Google Scholar] [CrossRef]
- He, Y.; Leung, K.W.; Zhang, Y.H.; Duan, S.; Zhong, X.F.; Jiang, R.Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458. [Google Scholar] [CrossRef]
- McElnea, E.M.; Quill, B.; Docherty, N.G.; Irnaten, M.; Siah, W.F.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182–1191. [Google Scholar] [PubMed]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into hypoxia and oxidative stress at the optic nerve head in a rat model of glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Peng, Y.; Sun, S.L.; Sun, D. Mechanisms of immune system activation in glaucoma: Oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Investig. Ophthalmol. Vis. Sci. 2007, 48, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Feilchenfeld, Z.; Yucel, Y.H.; Gupta, N. Oxidative injury to blood vessels and glia of the pre-laminar optic nerve head in human glaucoma. Exp. Eye Res. 2008, 87, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Malone, P.E.; Hernandez, M.R. 4-Hydroxynonenal, a product of oxidative stress, leads to an antioxidant response in optic nerve head astrocytes. Exp. Eye Res. 2007, 84, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Kim, K.Y.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.S.; Kim, K.Y.; Bu, J.H.; Nam, H.S.; Jeong, S.W.; Park, T.L.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. Elevated intracellular cAMP exacerbates vulnerability to oxidative stress in optic nerve head astrocytes. Cell Death Dis. 2018, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Tribble, J.R.; Vasalauskaite, A.; Redmond, T.; Young, R.D.; Hassan, S.; Fautsch, M.P.; Sengpiel, F.; Williams, P.A.; Morgan, J.E. Midget retinal ganglion cell dendritic and mitochondrial degeneration is an early feature of human glaucoma. Brain Commun. 2019, 1, fcz035. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Yoshitomi, T.; Zorumski, C.F.; Izumi, y. Experimentally induced mammalian models of glaucoma. Biomed. Res. Int. 2015, 2015, 281214. [Google Scholar] [CrossRef]
- Amato, R.; Cammalleri, M.; Melecchi, A.; Bagnoli, P.; Porciatti, V. Natural history of glaucoma progression in the DBA/2J model: Early contribution of müller cell gliosis. Cells 2023, 12, 1272. [Google Scholar] [CrossRef]
- Mahesh, N.; Maher, S.; Vittorio, P. IOP-dependent retinal ganglion cell dysfunction in glaucomatous DBA/2J mice. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4573–4579. [Google Scholar]
- John, S.W.; Smith, R.S.; Savinova, O.V.; Hawes, N.L.; Chang, B.; Turnbull, D.; Davisson, M.; Roderick, T.H.; Heckenlively, J.R. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Investig. Ophthalmol. Vis. Sci. 1998, 39, 951–962. [Google Scholar]
- Baltan, S.; Inman, D.M.; Danilov, C.A.; Morrison, R.S.; Calkins, D.J.; Horner, P.J. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J. Neurosci. 2010, 30, 5644–5652. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, S.H.; Nickerson, J.M.; Gao, F.J.; Sun, Z.; Chen, X.Y.; Zhang, S.J.; Gao, F.; Chen, J.Y.; Luo, Y.; et al. Cumulative mtDNA damage and mutations contribute to the progressive loss of RGCs in a rat model of glaucoma. Neurobiol. Dis. 2015, 74, 167–179. [Google Scholar] [CrossRef]
- Inman, D.M.; Sappington, R.M.; Horner, P.J.; Calkins, D.J. Quantitative correlation of optic Nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 986–996. [Google Scholar] [CrossRef]
- Dengler-Crish, C.M.; Smith, M.A.; Inman, D.M.; Wilson, G.N.; Young, J.W.; Crish, S.D. Anterograde transport blockade precedes deficits in retrograde transport in the visual projection of the DBA/2J mouse model of glaucoma. Front. Neurosci. 2014, 8, 290. [Google Scholar] [CrossRef]
- Bayer, A.U.; Neuhardt, T.; May, A.C.; Martus, P.; Maag, K.P.; Brodie, S.; Lütjen-Drecoll, E.; Podos, S.M.; Mittag, T. Retinal morphology and ERG response in the DBA/2NNia mouse model of angle-closure glaucoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1258–1265. [Google Scholar]
- Harazny, J.; Scholz, M.; Buder, T.; Lausen, B.; Kremers, J. Electrophysiological deficits in the retina of the DBA/2J mouse. Doc. Ophthalmol. 2009, 119, 181–197. [Google Scholar] [CrossRef]
- Cwerman-Thibault, H.; Lechauve, C.; Augustin, S.; Roussel, D.; Reboussin, É.; Mohammad, A.; Degardin-Chicaud, J.; Simonutti, M.; Liang, H.; Brignole-Baudouin, F.; et al. Neuroglobin can prevent or reverse glaucomatous progression in DBA/2J mice. Mol. Ther. Methods Clin. Dev. 2017, 5, 200–220. [Google Scholar] [CrossRef] [PubMed]
- Wax, M.B.; Tezel, G.; Yang, J.; Peng, G.; Patil, R.V.; Agarwal, N.; Sappington, R.M.; Calkins, D.J. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J. Neurosci. 2008, 28, 12085–12096. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Romero, C.O.; Breen, K.T.; Chagovetz, A.A.; Steele, M.R.; Ambati, B.K.; Vetter, M.L. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis. Model. Mech. 2015, 8, 443–455. [Google Scholar] [CrossRef]
- Wilson, G.N.; Inman, D.M.; Dengler Crish, C.M.; Smith, M.A.; Crish, S.D. Early pro-inflammatory cytokine elevations in the DBA/2J mouse model of glaucoma. J. Neuroinflam. 2015, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; O’Brien, C.J.; Zhdanov, A.V.; Papkovsky, D.B.; Clark, A.F.; Stamer, W.D.; Irnaten, M. Reduced Oxidative Phosphorylation and Increased Glycolysis in Human Glaucoma Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef] [PubMed]
- Irnaten, M.; Zhdanov, A.; Brennan, D.; Crotty, T.; Clark, A.; Papkovsky, D.; O’Brien, C. Activation of the NFAT-calcium signaling pathway in human lamina cribrosa cells in glaucoma. Investig. Ophthalmol. Vis. Sci. 2018, 59, 831–842. [Google Scholar] [CrossRef] [PubMed]
- McElnea, E.M.; Hughes, E.; McGoldrick, A.; McCann, A.; Quill, B.; Docherty, N.; Irnaten, M.; Farrell, M.; Clark, A.F.; O’Brien, C.J.; et al. Lipofuscin accumulation and autophagy in glaucomatous human lamina cribrosa cells. BMC Ophthalmol. 2014, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Finnegan, L.K.; Post, I.J.M.; Carrigan, M.; Gardiner, T.; Peixoto, E.; Maloney, D.; Humphries, M.M.; Stitt, A.; et al. AAV-mediated gene therapy improving mitochondrial function provides benefit in age-related macular degeneration models. Clin. Transl. Med. 2022, 12, e952. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Finnegan, L.K.; Post, I.J.M.; Carrigan, M.; Nixon, R.; Humphries, M.M.; Humphries, P.; Kenna, P.F.; Palfi, A.; et al. RPE-directed gene therapy improves mitochondrial function in murine dry AMD models. Int. J. Mol. Sci. 2023, 24, 3847. [Google Scholar] [CrossRef] [PubMed]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef]
- Chadderton, N.; Palfi, A.; Maloney, D.M.; Carrigan, M.; Finnegan, L.K.; Hanlon, K.S.; Shortall, C.; O’Reilly, M.; Humphries, P.; Cassidy, L.; et al. Optimisation of AAV-NDI1 significantly enhances its therapeutic value for correcting retinal mitochondrial dysfunction. Pharmaceutics 2023, 15, 322. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Muthuswamy, A.; Chen, H.; Hu, Y.; Turner, O.C.; Aina, O.H. Mammalian Retinal Cell Quantification. Toxicol. Pathol. 2021, 49, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Lo Faro, V.; Bhattacharya, A.; Zhou, W.; Zhou, D.; Wang, Y.; Läll, K.; Kanai, M.; Lopera-Maya, E.; Straub, P.; Pawar, P.; et al. Novel ancestry-specific primary open-angle glaucoma loci and shared biology with vascular mechanisms and cell proliferation. Cell Rep. Med. 2024, 5, 101430. [Google Scholar] [CrossRef] [PubMed]
- NEIGHBORHOOD Consortium; ANZRAG Consortium; Biobank Japan Project; FinnGen Study; UK Biobank Eye and Vision Consortium; GIGA Study Group; 23 and Me Research Team. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 2021, 12, 1258. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Bhardwaj, A.; Yadav, A.; Dada, R.; Tanwar, M. Molecular genetics of primary open-angle glaucoma. Indian. J. Ophthalmol. 2023, 71, 1739–1756. [Google Scholar] [CrossRef] [PubMed]
- Doucette, L.P.; Rasnitsyn, A.; Seifi, M.; Walter, M.A. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv. Ophthalmol. 2015, 60, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Sulak, R.; Liu, X.; Smedowski, A. The concept of gene therapy for glaucoma: The dream that has not come true yet. Neural Regen. Res. 2024, 19, 92–99. [Google Scholar] [CrossRef]
- Pietrucha-Dutczak, M.; Amadio, M.; Govoni, S.; Lewin-Kowalik, J.; Smedowski, A. The Role of Endogenous Neuroprotective Mechanisms in the Prevention of Retinal Ganglion Cells Degeneration. Front. Neurosci. 2018, 12, 834. [Google Scholar] [CrossRef]
- O’Callaghan, J.; Delaney, C.; O’Connor, M.; van Batenburg-Sherwood, J.; Schicht, M.; Lütjen-Drecoll, E.; Hudson, N.; Ni Dhubhghaill, S.; Humphries, P.; Stanley, C.; et al. Matrix metalloproteinase-3 (MMP-3)-mediated gene therapy for glaucoma. Sci. Adv. 2023, 9, eadf6537. [Google Scholar] [CrossRef]
- Zuo, L.; Khan, R.S.; Lee, V.; Dine, K.; Wu, W.; Shindler, K.S. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5097–5102. [Google Scholar] [CrossRef]
- Ross, A.G.; McDougald, D.S.; Khan, R.S.; Duong, T.T.; Dine, K.E.; Aravand, P.; Bennett, J.; Chavali, V.R.M.; Shindler, K.S. Rescue of retinal ganglion cells in optic nerve injury using cell-selective AAV mediated delivery of SIRT1. Gene Ther. 2021, 28, 256–264. [Google Scholar] [CrossRef]
- Yue, J.; Khan, R.S.; Duong, T.T.; Dine, K.E.; Cui, Q.N.; O’Neill, N.; Aravand, P.; Liu, T.; Chaqour, B.; Shindler, K.S.; et al. Cell-specific expression of human SIRT1 by gene therapy reduces retinal ganglion cell loss induced by elevated intraocular pressure. Neurotherapeutics 2023, 20, 896–907. [Google Scholar] [CrossRef]
- McKinnon, S.J.; Lehman, D.M.; Tahzib, N.G.; Ransom, N.L.; Reitsamer, H.A.; Liston, P.; LaCasse, E.; Li, Q.; Korneluk, R.G.; Hauswirth, W.W. Baculoviral IAP repeat-containing-4 protects optic nerve axons in a rat glaucoma model. Mol. Ther. 2002, 5, 780–787. [Google Scholar] [CrossRef]
- Visuvanathan, S.; Baker, A.N.; Lagali, P.S.; Coupland, S.G.; Miller, G.; Hauswirth, W.W.; Tsilfidis, C. XIAP gene therapy effects on retinal ganglion cell structure and function in a mouse model of glaucoma. Gene Ther. 2022, 29, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Shi, S.; Palacios, E.; Liu, W.; Buscho, S.E.; Li, J.; Huang, S.; Vizzeri, G.; Dong, X.C.; Motamedi, M.; et al. Sirt6 protects retinal ganglion cells and optic nerve from degeneration during aging and glaucoma. Mol. Ther. 2024, 32, 1760–1778. [Google Scholar] [CrossRef]
- Thananthirige, K.P.M.; Chitranshi, N.; Basavarajappa, D.; Rajput, R.; Abbasi, M.; Palanivel, V.; Gupta, V.B.; Paulo, J.A.; Koronyo-Hamaoui, M.; Mirzaei, M.; et al. Tau modulation through AAV9 therapy augments Akt/Erk survival signalling in glaucoma mitigating the retinal degenerative phenotype. Acta Neuropathol. Commun. 2024, 12, 89. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.K.S.; Ren, S.T.; Chan, P.P.M.; Wan, K.H.N.; Kam, A.K.W.; Lai, G.W.K.; Chiu, V.S.M.; Ko, M.W.L.; You, C.K.F.; Yu, M.C.Y. Nicotinamide riboside as a neuroprotective therapy for glaucoma: Study protocol for a randomized, double-blind, placebo-control trial. Trials 2022, 23, 45. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, C.G.; John, S.W.M.; Williams, P.A.; Blumberg, D.M.; Cioffi, G.A.; Liebmann, J.M. Nicotinamide and pyruvate for neuroenhancement in open-angle glaucoma: A phase 2 randomized clinical trial. JAMA Ophthalmol. 2022, 140, 11–18. [Google Scholar] [CrossRef]
- Garg Shukla, A.; Cioffi, G.A.; Liebmann, J.M. Drug-induced liver injury during a glaucoma neuroprotection clinical trial. J. Glaucoma. 2024, 33, e58–e59. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative stress in optic neuropathies. Antioxidants 2021, 10, 1538. [Google Scholar] [CrossRef]
- Mirra, S.; Marfany, G. mitochondrial gymnastics in retinal cells: A resilience mechanism against oxidative stress and neurodegeneration. Adv. Exp. Med. Biol. 2019, 1185, 513–517. [Google Scholar] [PubMed]
- Burgoyne, C.F. The non-human primate experimental glaucoma model. Exp. Eye Res. 2015, 141, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, B.; Yang, Z.; Zhan, Z.; Li, J.; Lan, Y. Protocol for laser-induced chronic ocular hypertension and intracameral injection in nonhuman primates. STAR Protoc. 2022, 3, 101801. [Google Scholar] [CrossRef]
- Zhou, X.; Li, F.; Kong, L.; Tomita, H.; Li, C.; Cao, W. Involvement of inflammation, degradation, and apoptosis in a mouse model of glaucoma. J. Biol. Chem. 2005, 280, 31240–31248. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Perkins, G.A.; Kim, K.Y.; Kong, Y.; Lee, Y.; Choi, S.H.; Liu, Y.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Zangwill, L.; et al. Loss of AKAP1 triggers Drp1 dephosphorylation-mediated mitochondrial fission and loss in retinal ganglion cells. Cell Death Dis. 2020, 11, 254. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, K.Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.N.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.; et al. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol. 2020, 37, 101703. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk between dysfunctional mitochondria and inflammation in glaucomatous neurodegeneration. Front. Pharmacol. 2021, 12, 699623. [Google Scholar] [CrossRef]
- Johnson, T.V.; Tomarev, S.I. Rodent models of glaucoma. Brain Res. Bull. 2010, 81, 349–358. [Google Scholar] [CrossRef]
- Kimura, A.; Noro, T.; Harada, T. Role of animal models in glaucoma research. Neural Regen. Res. 2020, 15, 1257–1258. [Google Scholar] [CrossRef]
- Yu, A.L.; Fuchshofer, R.; Kampik, A.; Welge-Lüssen, U. Effects of oxidative stress in trabecular meshwork cells are reduced by prostaglandin analogues. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4872–4880. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Moriniere, J.; Welge-Lussen, U. TGF-β2- and H2O2-induced biological changes in optic nerve head astrocytes are reduced by the antioxidant alpha-lipoic acid. Ophthalmic Res. 2012, 48, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.M.; Clark, A.F.; Lipson, K.E.; Andrews, D.; Crean, J.K.; O’Brien, C.J. Anti-connective tissue growth factor antibody treatment reduces extracellular matrix production in trabecular meshwork and lamina cribrosa cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7836–7848. [Google Scholar] [CrossRef] [PubMed]
- Tektas, O.Y.; Lutjen-Drecoll, E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp. Eye Res. 2009, 88, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Browne, J.G.; Ho, S.L.; Kane, R. Connective tissue growth factor is increased in pseudoexfoliation glaucoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3660–3666. [Google Scholar] [CrossRef] [PubMed]
- Millington-Ward, S.; Chadderton, N.; Berkeley, M.; Finnegan, L.K.; Hanlon, K.S.; Carrigan, M.; Humphries, P.; Kenna, P.F.; Palfi, A.; Farrar, G.J. Novel 199 base pair NEFH promoter drives expression in retinal ganglion cells. Sci. Rep. 2020, 10, 16515. [Google Scholar] [CrossRef]
- Leroy, B.; Fischer, M.D.; Flannery, J.G.; MacLaren, R.E.; Dalkara, D.; Scholl, H.P.N.; Chung, D.C.; Spera, C.; Viriato, D.; Banhazi, J. Gene therapy for inherited retinal disease: Long-term durability of effect. Ophthalmic Res. 2023, 66, 179–196. [Google Scholar] [CrossRef]
- Palfi, A.; Yesmambetov, A.; Millington-Ward, S.; Shortall, C.; Humphries, P.; Kenna, P.F.; Chadderton, N.; Farrar, G.J. AAV-delivered Tulp1 supplementation therapy targeting photoreceptors provides minimal benefit in Tulp1-/- retinas. Front. Neurosci. 2020, 14, 891. [Google Scholar] [CrossRef]
- O’Reilly, M.; Palfi, A.; Chadderton, N.; Millington-Ward, S.; Ader, M.; Cronin, T.; Tuohy, T.; Auricchio, A.; Hildinger, M.; Tivnan, A.; et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am. J. Hum. Genet. 2007, 81, 127–135. [Google Scholar] [CrossRef]
- Rohr, U.P.; Wulf, M.A.; Stahn, S.; Steidl, U.; Haas, R.; Kronenwett, R. Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time PCR. J. Virol. Methods 2002, 106, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.N.; Clark, A.F.; Tovar-Vidales, T. Isolation and characterization of human optic nerve head astrocytes and lamina cribrosa cells. Exp. Eye Res. 2020, 197, 108103. [Google Scholar] [CrossRef] [PubMed]
- Irnaten, M.; Barry, R.C.; Wallace, D.M.; Docherty, N.G.; Quill, B.; Clark, A.F.; O’Brien, C.J. Elevated maxi-K+ ion channel current in glaucomatous lamina cribrosa cells. Exp. Eye Res. 2013, 115, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Lambert, W.; Agarwal, R.; Howe, W.; Clark, A.F.; Wordinger, R.J. Neurotrophin and neurotrophin receptor expression by cells of the human lamina cribrosa. Investig Ophthalmol. Vis. Sci. 2001, 42, 2315–2323. [Google Scholar]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef]
- Finnegan, L.K.; Chadderton, N.; Kenna, P.F.; Palfi, A.; Carty, M.; Bowie, A.G.; Millington-Ward, S.; Farrar, G.J. SARM1 ablation is protective and preserves spatial vision in an in vivo mouse model of retinal ganglion cell degeneration. Int. J. Mol. Sci. 2022, 23, 1606. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millington-Ward, S.; Palfi, A.; Shortall, C.; Finnegan, L.K.; Bargroff, E.; Post, I.J.M.; Maguire, J.; Irnaten, M.; O′Brien, C.; Kenna, P.F.; et al. AAV-NDI1 Therapy Provides Significant Benefit to Murine and Cellular Models of Glaucoma. Int. J. Mol. Sci. 2024, 25, 8876. https://doi.org/10.3390/ijms25168876
Millington-Ward S, Palfi A, Shortall C, Finnegan LK, Bargroff E, Post IJM, Maguire J, Irnaten M, O′Brien C, Kenna PF, et al. AAV-NDI1 Therapy Provides Significant Benefit to Murine and Cellular Models of Glaucoma. International Journal of Molecular Sciences. 2024; 25(16):8876. https://doi.org/10.3390/ijms25168876
Chicago/Turabian StyleMillington-Ward, Sophia, Arpad Palfi, Ciara Shortall, Laura K. Finnegan, Ethan Bargroff, Iris J. M. Post, John Maguire, Mustapha Irnaten, Colm O′Brien, Paul F. Kenna, and et al. 2024. "AAV-NDI1 Therapy Provides Significant Benefit to Murine and Cellular Models of Glaucoma" International Journal of Molecular Sciences 25, no. 16: 8876. https://doi.org/10.3390/ijms25168876