
2,3-Diphosphoglyceric Acid Alleviating Hypoxic-Ischemic Brain Damage through p38 MAPK Modulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. 2,3-Diphosphoglyceric Acid Mitigates HI-Induced Brain Damage through Regulated MAPK Signaling
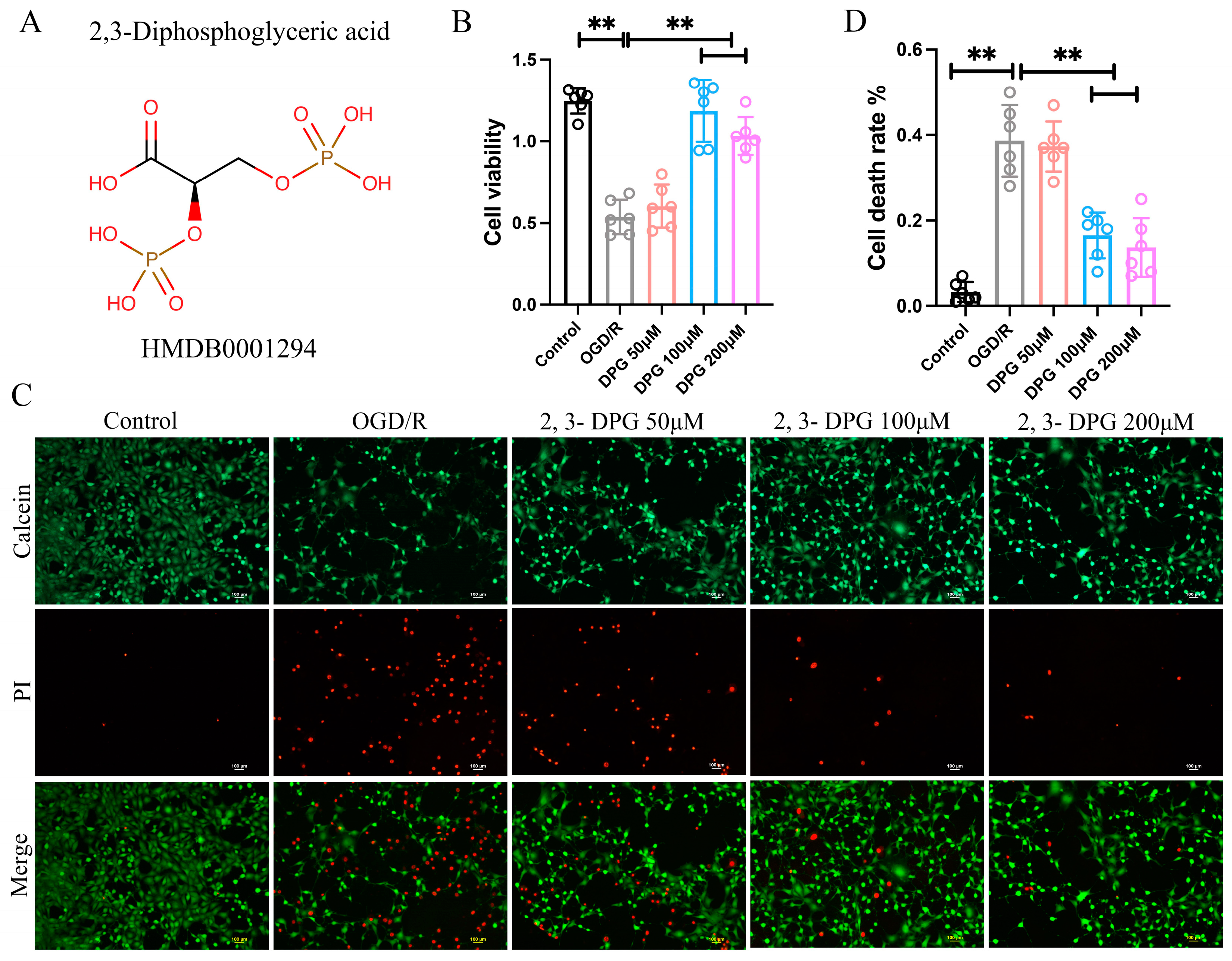
2.1.1. 2,3-Diphosphoglyceric Acid Reduced the Neuronal Cell Death Induce by OGD/R
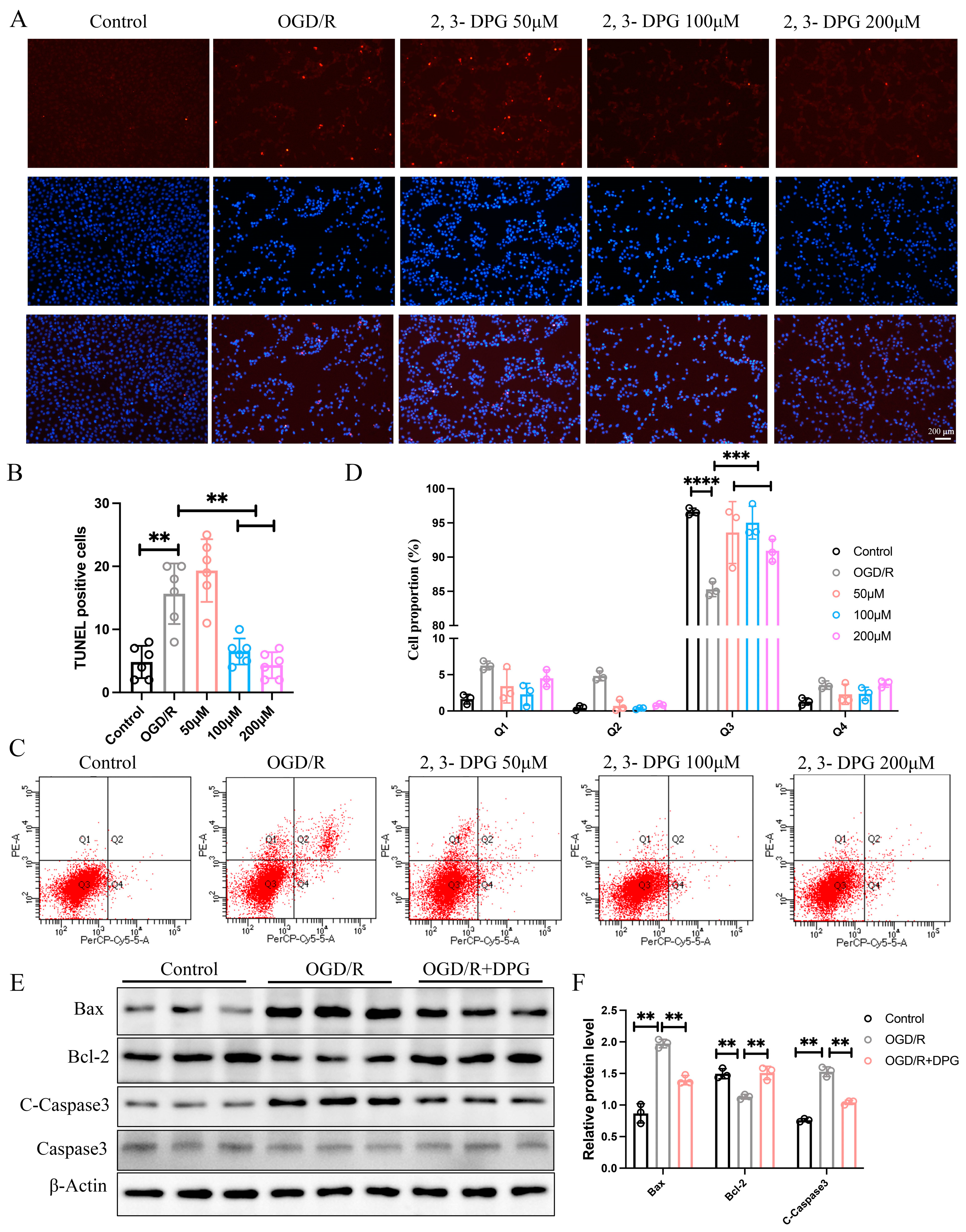
2.1.2. 2,3-Diphosphoglyceric Acid Inhibited HI-Induced Apoptosis In Vitro
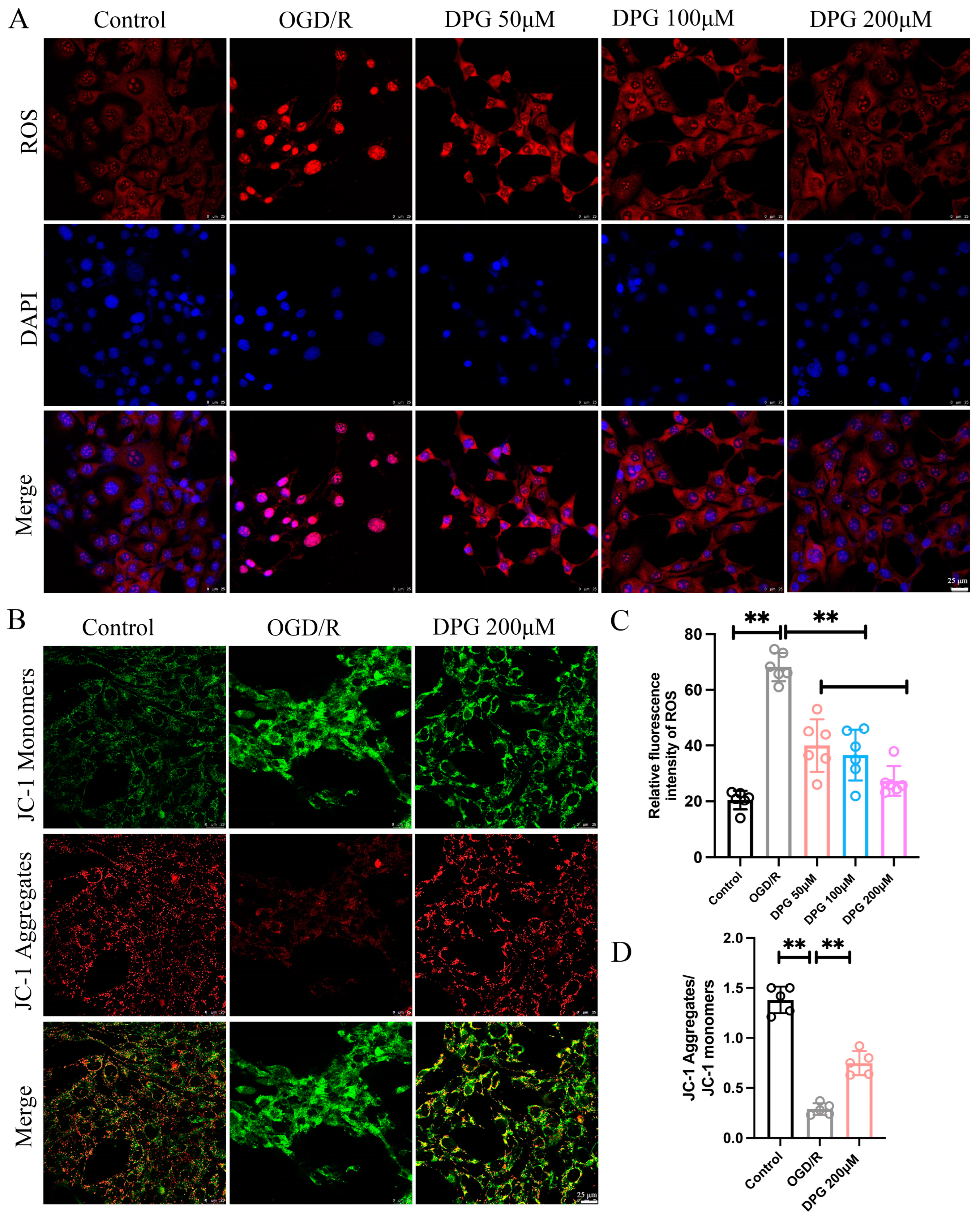
2.1.3. 2,3-Diphosphoglyceric Acid Suppressed the OGD/R-Induced ROS Generation and Alleviates OGD/R-Mediated Mitochondria Damage
2.1.4. 2,3-Diphosphoglyceric Acid Treatment Ameliorated HI-Induced Brain Injury in Neonatal HIBD Rats
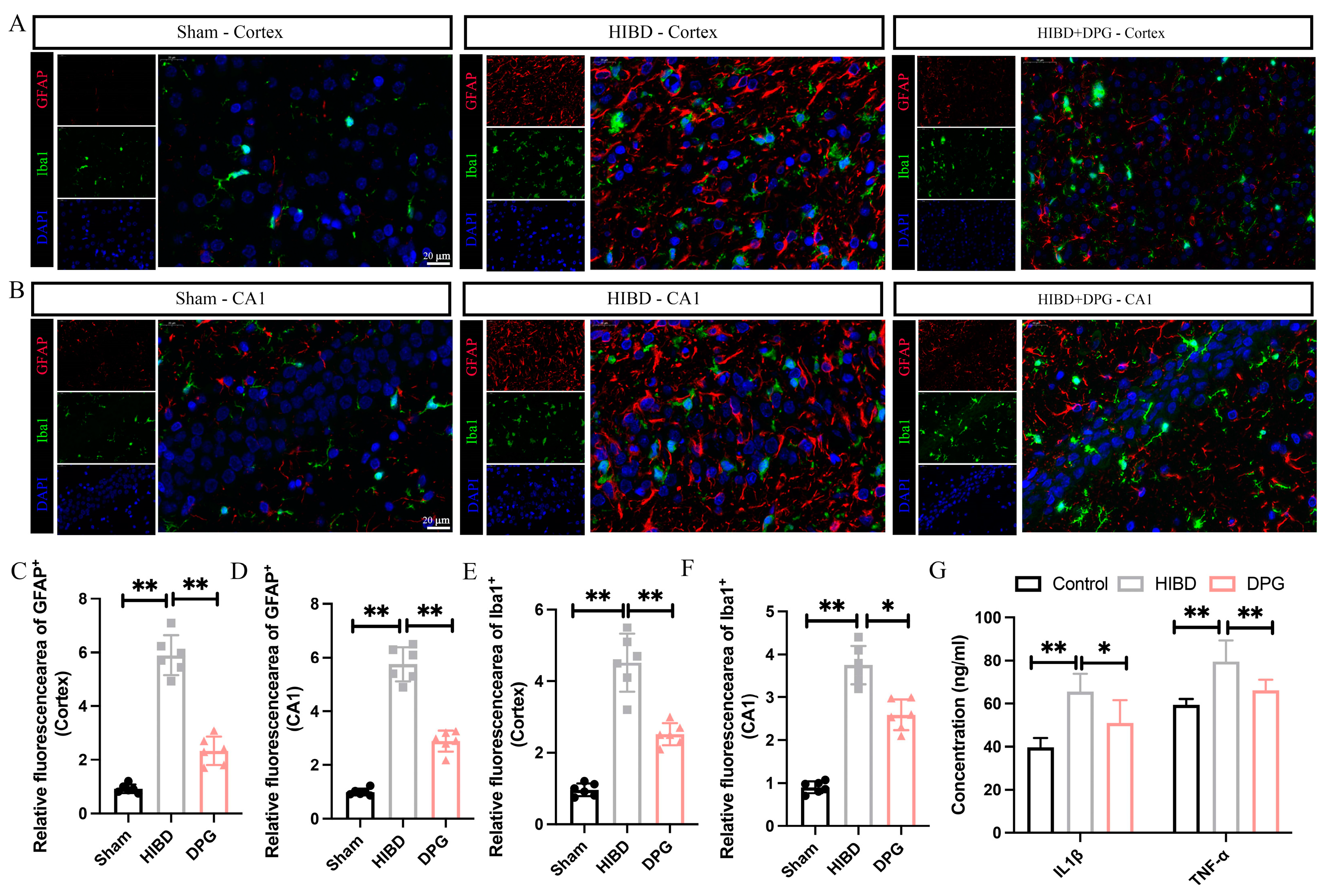
2.1.5. 2,3-Diphosphoglyceric Acid Inhibited HI-Induced Neuronal Apoptosis In Vivo
2.1.6. 2,3-Diphosphoglyceric Acid Effectively Mitigated Neuroinflammation in Rats with HIBD
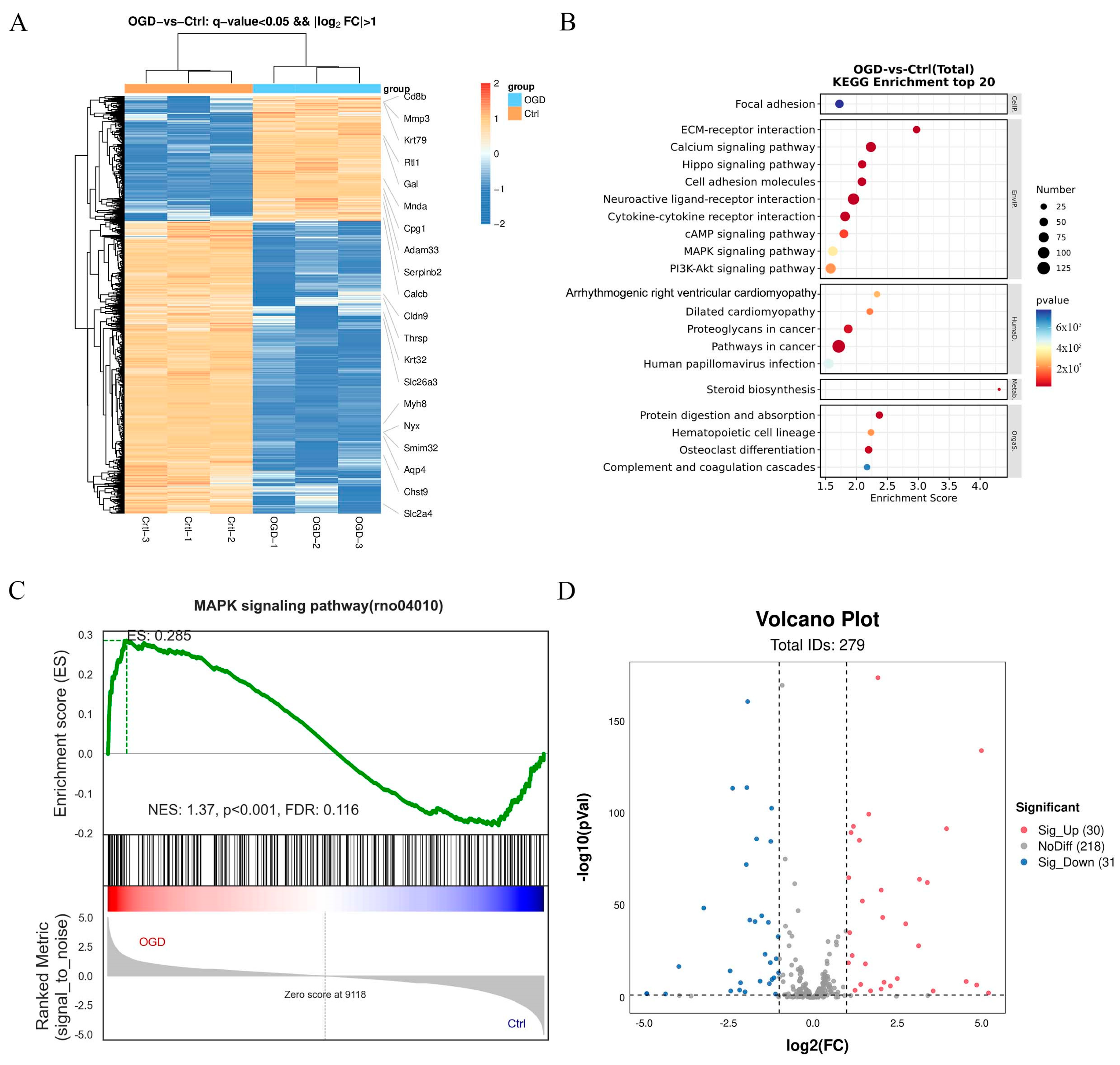
2.1.7. Differentially Expressed Signaling Pathways Involved in OGD/R-Induced Neuronal Injury
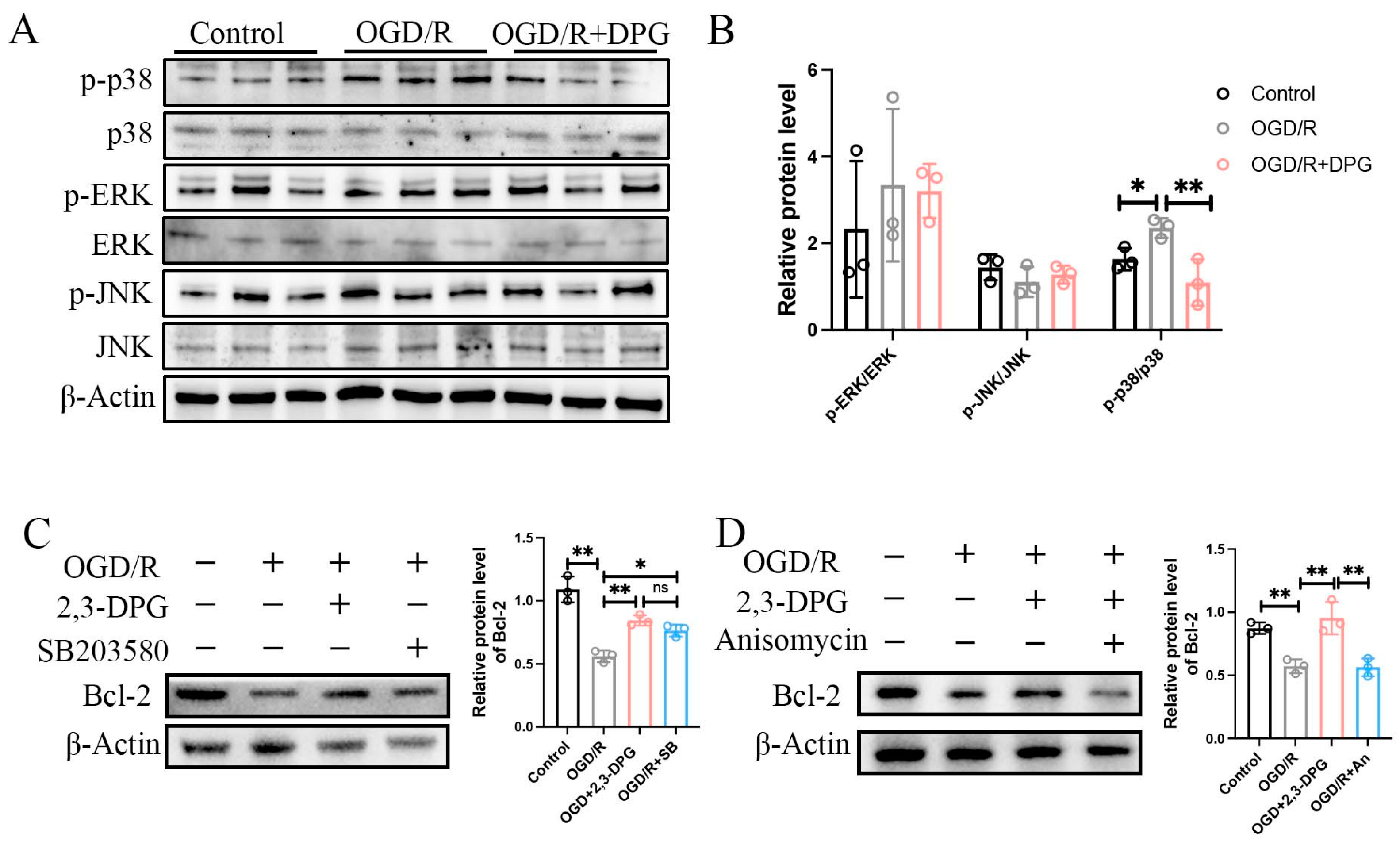
2.1.8. p38 MAPK Signaling Pathway Involved in the Neuroprotective Effect of 2,3-Diphosphoglyceric Acid
3. Discussion
4. Materials and Methods
4.1. Cell Culture and OGD Model
4.2. CCK8 Assay
4.3. Calcein-PI Staining
4.4. Flow Cytometry
4.5. TUNEL Assay
4.6. p38 MAPK Signaling Inhibition and Activation
4.7. ELISA Assay
4.8. Reactive Oxygen Species (ROS) Assay
4.9. Western Blot
4.10. Mitochondrial Membrane Potential (MMP) Measurement
4.11. Immunofluorescence (IF)
4.12. Neonatal Hypoxic-Ischemic Brain Damage (HIBD) Rat Model
4.13. TTC Staining
4.14. Hematoxylin-Eosin (HE) Staining
4.15. Nissl Staining
4.16. RNA-Sequencing Analysis (RNA-Seq)
4.17. Quantification and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef]
- Greco, P.; Nencini, G.; Piva, I.; Scioscia, M.; Volta, C.A.; Spadaro, S.; Neri, M.; Bonaccorsi, G.; Greco, F.; Cocco, I.; et al. Pathophysiology of hypoxic-ischemic encephalopathy: A review of the past and a view on the future. Acta Neurol. Belg. 2020, 120, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Verklan, M.T. The chilling details: Hypoxic-ischemic encephalopathy. J. Perinat. Neonatal Nurs. 2009, 23, 59–68; quiz 69–70. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef] [PubMed]
- Victor, S.; Rocha-Ferreira, E.; Rahim, A.; Hagberg, H.; Edwards, D. New possibilities for neuroprotection in neonatal hypoxic-ischemic encephalopathy. Eur. J. Pediatr. 2022, 181, 875–887. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, L. Neural stem cell therapies and hypoxic-ischemic brain injury. Prog. Neurobiol. 2019, 173, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Serrenho, I.; Rosado, M.; Dinis, A.; Cardoso, C.M.; Graos, M.; Manadas, B.; Baltazar, G. Stem Cell Therapy for Neonatal Hypoxic-Ischemic Encephalopathy: A Systematic Review of Preclinical Studies. Int. J. Mol. Sci. 2021, 22, 3142. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Chavez-Valdez, R.; Martin, L.J. Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 2011, 69, 743–758. [Google Scholar] [CrossRef]
- Ranjan, A.K.; Gulati, A. Advances in Therapies to Treat Neonatal Hypoxic-Ischemic Encephalopathy. J. Clin. Med. 2023, 12, 6653. [Google Scholar] [CrossRef]
- Zhao, J.; Le, M.; Li, J.; Huang, Q.; Chen, H.; Zhang, W.; Mao, H.; Sun, Q.; Li, A.; Zhao, Y.; et al. LINC00938 alleviates hypoxia ischemia encephalopathy induced neonatal brain injury by regulating oxidative stress and inhibiting JNK/p38 MAPK signaling pathway. Exp. Neurol. 2023, 367, 114449. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lin, Z.; Yuan, J.; Li, P.; Wang, Q.; Cho, N.; Wang, Y.; Lin, Z. The neuroprotective mechanisms of naringenin: Inhibition of apoptosis through the PI3K/AKT pathway after hypoxic-ischemic brain damage. J. Ethnopharmacol. 2024, 318, 116941. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, W.; Doycheva, D.M.; Gamdzyk, M.; Lu, W.; Tang, J.; Zhang, J.H. Ghrelin attenuates oxidative stress and neuronal apoptosis via GHSR-1alpha/AMPK/Sirt1/PGC-1alpha/UCP2 pathway in a rat model of neonatal HIE. Free. Radic. Biol. Med. 2019, 141, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R.U.S. FDA Approved Drugs from 2015-June 2020: A Perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Vogelzang, A.; Miyajima, M.; Sugiura, Y.; Wu, Y.; Chamoto, K.; Nakano, R.; Hatae, R.; Menzies, R.J.; Sonomura, K.; et al. B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature 2021, 599, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Serger, E.; Luengo-Gutierrez, L.; Chadwick, J.S.; Kong, G.; Zhou, L.; Crawford, G.; Danzi, M.C.; Myridakis, A.; Brandis, A.; Bello, A.T.; et al. The gut metabolite indole-3 propionate promotes nerve regeneration and repair. Nature 2022, 607, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, C.; Liu, X.; Jiang, C.X.; Cao, Q.; Yu, B.; Ni, Y.; Mao, S. The metabolomic profiling identifies N,N-dimethylglycine as a facilitator of dorsal root ganglia neuron axon regeneration after injury. FASEB J. 2022, 36, e22305. [Google Scholar] [CrossRef]
- Cade, R.; Conte, M.; Zauner, C.; Mars, D.; Peterson, J.; Lunne, D.; Hommen, N.; Packer, D. Effects of phosphate loading on 2,3-diphosphoglycerate and maximal oxygen uptake. Med. Sci. Sports Exerc. 1984, 16, 263–268. [Google Scholar] [CrossRef]
- Oslund, R.C.; Su, X.; Haugbro, M.; Kee, J.M.; Esposito, M.; David, Y.; Wang, B.; Ge, E.; Perlman, D.H.; Kang, Y.; et al. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat. Chem. Biol. 2017, 13, 1081–1087. [Google Scholar] [CrossRef]
- Poillon, W.N.; Kim, B.C. 2,3-Diphosphoglycerate and intracellular pH as interdependent determinants of the physiologic solubility of deoxyhemoglobin S. Blood 1990, 76, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.M. The role of hemoglobin oxygen affinity in oxygen transport at high altitude. Respir. Physiol. Neurobiol. 2007, 158, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.C.; Zaroulis, C.; Goetz, W.; Howland, W.S. Hemodynamic oxygen transport and 2,3-diphosphoglycerate changes after transfusion of patients in acute respiratory failure. Intensive Care Med. 1986, 12, 22–25. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Wu, H.; D'Alessandro, A.; Yegutkin, G.G.; Song, A.; Sun, K.; Li, J.; Cheng, N.Y.; Huang, A.; et al. Beneficial Role of Erythrocyte Adenosine A2B Receptor-Mediated AMP-Activated Protein Kinase Activation in High-Altitude Hypoxia. Circulation 2016, 134, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.K.; Korman, B.; Bassingthwaighte, J.B. Simple accurate mathematical models of blood HbO2 and HbCO2 dissociation curves at varied physiological conditions: Evaluation and comparison with other models. Eur. J. Appl. Physiol. 2016, 116, 97–113. [Google Scholar] [CrossRef]
- Ploszczyca, K.; Czuba, M.; Chalimoniuk, M.; Gajda, R.; Baranowski, M. Red Blood Cell 2,3-Diphosphoglycerate Decreases in Response to a 30 km Time Trial Under Hypoxia in Cyclists. Front. Physiol. 2021, 12, 670977. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Zhang, X.X.; Zhong, Z.Q.; Luo, X.X.; Wang, Y.F.; Xiao, X.P.; Huang, Z.Q.; Yu, S.Y.; Sun, J.Y.; Liu, M.J.; et al. Metabolomics Analysis of Electroacupuncture Pretreatment Induced Neuroprotection on Mice with Ischemic Stroke. Am. J. Chin. Med. 2023, 51, 1127–1151. [Google Scholar] [CrossRef]
- Baranovicova, E.; Grendar, M.; Kalenska, D.; Tomascova, A.; Cierny, D.; Lehotsky, J. NMR metabolomic study of blood plasma in ischemic and ischemically preconditioned rats: An increased level of ketone bodies and decreased content of glycolytic products 24 h after global cerebral ischemia. J. Physiol. Biochem. 2018, 74, 417–429. [Google Scholar] [CrossRef]
- Sola, A.; Berrios, M.; Sheldon, R.A.; Ferriero, D.M.; Gregory, G.A. Fructose-1,6-bisphosphate after hypoxic ischemic injury is protective to the neonatal rat brain. Brain Res. 1996, 741, 294–299. [Google Scholar] [CrossRef]
- Chen, H.; Wusiman, Y.; Zhao, J.; Zhang, W.; Liu, W.; Wang, S.; Qian, G.; Zhang, G.; Le, M.; Dong, X. Metabolomics analysis revealed the neuroprotective role of 2-phosphoglyceric acid in hypoxic-ischemic brain damage through GPX4/ACSL4 axis regulation. Eur. J. Pharmacol. 2024, 971, 176539. [Google Scholar] [CrossRef]
- Duhm, J.; Gerlach, E. On the mechanisms of the hypoxia-induced increase of 2,3-diphosphoglycerate in erythrocytes. Studies on rat erythrocytes in vivo and on human erythrocytes in vitro. Pflug. Arch. 1971, 326, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Spasojevic, I.; Zakrzewska, J.; Bacic, G.G. 31P NMR spectroscopy and polarographic combined study of erythrocytes treated with 5-fluorouracil: Cardiotoxicity-related changes in ATP, 2,3-BPG, and O2 metabolism. Ann. N. Y. Acad. Sci. 2005, 1048, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Koh, N.; Sakakibara, F.; Hamada, Y.; Wakao, T.; Hara, T.; Mori, K.; Nakashima, E.; Naruse, K.; Hotta, N. Polyol pathway, 2,3-diphosphoglycerate in erythrocytes and diabetic neuropathy in rats. Eur. J. Pharmacol. 1995, 294, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Sharifzadeh, M.; Rezayat, S.M.; Hassanzadeh, G.; Hassani, S.; Baeeri, M.; Shetab-Bushehri, V.; Kuznetsov, D.A.; Abdollahi, M. Benefit of magnesium-25 carrying porphyrin-fullerene nanoparticles in experimental diabetic neuropathy. Int. J. Nanomed. 2010, 5, 517–523. [Google Scholar]
- Graham, J.; Hobson, D.; Ponnampalam, A. High affinity hemoglobin and Parkinson’s disease. Med. Hypotheses 2014, 83, 819–821. [Google Scholar] [CrossRef]
- Yu, L.; Liu, S.; Zhou, R.; Sun, H.; Su, X.; Liu, Q.; Li, S.; Ying, J.; Zhao, F.; Mu, D.; et al. Atorvastatin inhibits neuronal apoptosis via activating cAMP/PKA/p-CREB/BDNF pathway in hypoxic-ischemic neonatal rats. FASEB J. 2022, 36, e22263. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Song, N.; Ma, K.; Tong, Y.; Yang, L. The effects of protein-rich extract from Rhizoma Gastrodiae against cerebral ischemia/reperfusion injury via regulating MAPK and PI3K/AKT signaling pathway. Brain Res. Bull. 2023, 203, 110772. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; King, J.S.; Qian, X.; Harwood, A.J.; Shears, S.B. Dephosphorylation of 2020, 23-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc. Natl. Acad. Sci. USA 2008, 105, 5998–6003. [Google Scholar] [CrossRef]
- Fothergill-Gilmore, L.A.; Watson, H.C. The phosphoglycerate mutases. Adv. Enzym. Relat. Areas Mol. Biol. 1989, 62, 227–313. [Google Scholar]
- Sun, Q.; Li, S.; Wang, Y.; Peng, H.; Zhang, X.; Zheng, Y.; Li, C.; Li, L.; Chen, R.; Chen, X.; et al. Phosphoglyceric acid mutase-1 contributes to oncogenic mTOR-mediated tumor growth and confers non-small cell lung cancer patients with poor prognosis. Cell Death Differ. 2018, 25, 1160–1173. [Google Scholar] [CrossRef]
- Dong, X.; Jiang, D.; Wang, L.; Zhao, J.; Yu, L.; Huang, Y.; Wu, X.; Zhu, Y.; Zhao, Y.; Zhao, Q.; et al. VPS28 regulates brain vasculature by controlling neuronal VEGF trafficking through extracellular vesicle secretion. iScience 2022, 25, 104042. [Google Scholar] [CrossRef]
- Rice, J.E., 3rd; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, J.; Zhao, J.; Chen, H.; Liu, W.; Le, M.; Guo, X.; Dong, X. 2,3-Diphosphoglyceric Acid Alleviating Hypoxic-Ischemic Brain Damage through p38 MAPK Modulation. Int. J. Mol. Sci. 2024, 25, 8877. https://doi.org/10.3390/ijms25168877
Ni J, Zhao J, Chen H, Liu W, Le M, Guo X, Dong X. 2,3-Diphosphoglyceric Acid Alleviating Hypoxic-Ischemic Brain Damage through p38 MAPK Modulation. International Journal of Molecular Sciences. 2024; 25(16):8877. https://doi.org/10.3390/ijms25168877
Chicago/Turabian StyleNi, Jiawei, Jing Zhao, Haocong Chen, Wenjuan Liu, Meini Le, Xirong Guo, and Xiaohua Dong. 2024. "2,3-Diphosphoglyceric Acid Alleviating Hypoxic-Ischemic Brain Damage through p38 MAPK Modulation" International Journal of Molecular Sciences 25, no. 16: 8877. https://doi.org/10.3390/ijms25168877
APA StyleNi, J., Zhao, J., Chen, H., Liu, W., Le, M., Guo, X., & Dong, X. (2024). 2,3-Diphosphoglyceric Acid Alleviating Hypoxic-Ischemic Brain Damage through p38 MAPK Modulation. International Journal of Molecular Sciences, 25(16), 8877. https://doi.org/10.3390/ijms25168877