
Metformin Lysosomal Targeting: A Novel Aspect to Be Investigated for Metformin Repurposing in Neurodegenerative Diseases?



Abstract
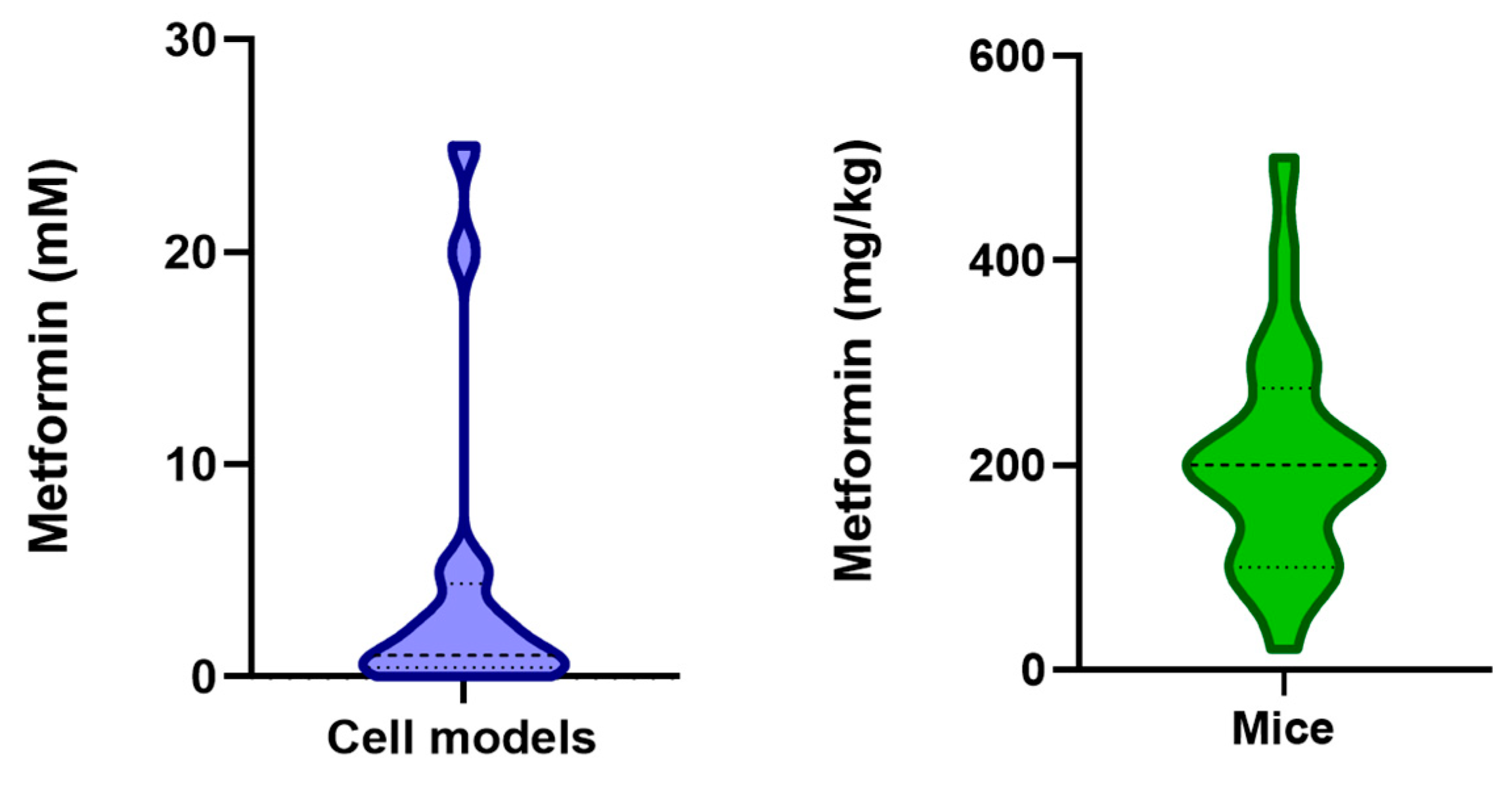
1. Introduction
2. Metformin Modulates Lysosomal-Dependent Processes
3. Lysosomal Targets Recognised by Metformin
4. Metformin and Neurodegenerative Diseases
5. Why Metformin Lysosomal Targeting Could Be Helpful in Neurodegenerative Diseases
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Schernthaner, G.; Schernthaner, G.H. The right place for metformin today. Diabetes Res. Clin. Pract. 2020, 159, 107946. [Google Scholar] [CrossRef] [PubMed]
- Kos, E.; Liszek, M.J.; Emanuele, M.A.; Durazo-Arvizu, R.; Camacho, P. Effect of metformin therapy on vitamin D and vitamin B₁₂ levels in patients with type 2 diabetes mellitus. Endocr. Pract. 2012, 18, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Sayedali, E.; Yalin, A.E.; Yalin, S. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes. World J. Diabetes 2023, 14, 585–593. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.M.H.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.Y.; Ahn, J.; Hyun, M.; Lee, I.; Min, K.J.; You, Y.J. NHX-5, an Endosomal Na+/H+ Exchanger, Is Associated with Metformin Action. J. Biol. Chem. 2016, 291, 18591–18599. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Bharath, L.P.; Nikolajczyk, B.S. The intersection of metformin and inflammation. Am. J. Physiol. Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Perera, R.M. Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology. Nat. Rev. Mol. Cell Biol. 2024, 25, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Tai, S.; Sun, J.; Zhou, Y.; Zhu, Z.; He, Y.; Chen, M.; Yang, H.; Xiao, Y.; Tu, T.; Tang, L.; et al. Metformin suppresses vascular smooth muscle cell senescence by promoting autophagic flux. J. Adv. Res. 2022, 41, 205–218. [Google Scholar] [CrossRef]
- Xu, Z.; Pan, Z.; Jin, Y.; Gao, Z.; Jiang, F.; Fu, H.; Chen, X.; Zhang, X.; Yan, H.; Yang, X.; et al. Inhibition of PRKAA/AMPK (Ser485/491) phosphorylation by crizotinib induces cardiotoxicity via perturbing autophagosome-lysosome fusion. Autophagy 2024, 20, 416–436. [Google Scholar] [CrossRef]
- Van, J.; Hahn, Y.; Silverstein, B.; Li, C.; Cai, F.; Wei, J.; Katiki, L.; Mehta, P.; Livatova, K.; DelPozzo, J.; et al. Metformin Inhibits Autophagy, Mitophagy and Antagonizes Doxorubicin-Induced Cardiomyocyte Death. Int. J. Drug Discov. Pharm. 2023, 2, 37–51. [Google Scholar] [CrossRef]
- Wu, H.; Feng, K.; Zhang, C.; Zhang, H.; Zhang, J.; Hua, Y.; Dong, Z.; Zhu, Y.; Yang, S.; Ma, C. Metformin attenuates atherosclerosis and plaque vulnerability by upregulating KLF2-mediated autophagy in apoE(-/-) mice. Biochem. Biophys. Res. Commun. 2021, 557, 334–341. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, S.; Fan, Z.; Li, Z.; Zhu, Y.; Shen, T.; Li, K.; Yan, Y.; Tian, J.; Liu, Z.; et al. Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice. Alzheimers Res. Ther. 2021, 13, 40. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Xiong, J.; He, Z.L.; Yuan, Y.; Wang, B.N.; Xu, J.Y.; Wu, M.; Zhang, S.S.; Cai, S.F.; Zhao, J.X.; et al. Metformin promotes microglial cells to facilitate myelin debris clearance and accelerate nerve repairment after spinal cord injury. Acta Pharmacol. Sin. 2022, 43, 1360–1371. [Google Scholar] [CrossRef]
- Son, S.M.; Shin, H.J.; Byun, J.; Kook, S.Y.; Moon, M.; Chang, Y.J.; Mook-Jung, I. Metformin Facilitates Amyloid-β Generation by β- and γ-Secretases via Autophagy Activation. J. Alzheimers Dis. 2016, 51, 1197–1208. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, X.; Yu, X.; Shang, R.; Lu, L.; Miao, J. Metformin protects fibroblasts from patients with GNE myopathy by restoring autophagic flux via an AMPK/mTOR-independent pathway. Biomed. Pharmacother. 2023, 164, 114958. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef]
- Watada, H.; Fujitani, Y. Minireview: Autophagy in pancreatic β-cells and its implication in diabetes. Mol. Endocrinol. 2015, 29, 338–348. [Google Scholar] [CrossRef]
- de Marañón, A.M.; Díaz-Pozo, P.; Canet, F.; Díaz-Morales, N.; Abad-Jiménez, Z.; López-Domènech, S.; Vezza, T.; Apostolova, N.; Morillas, C.; Rocha, M.; et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 2022, 53, 102342. [Google Scholar] [CrossRef]
- Guo, Y.; Jiang, H.; Wang, M.; Ma, Y.; Zhang, J.; Jing, L. Metformin alleviates cerebral ischemia/reperfusion injury aggravated by hyperglycemia via regulating AMPK/ULK1/PINK1/Parkin pathway-mediated mitophagy and apoptosis. Chem. Biol. Interact. 2023, 384, 110723. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Li, Z.; Zhao, M.; Wang, D.; Sun, Z.; Wen, P.; Dai, Y.; Gou, F.; Ji, Y.; et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res. Rev. 2023, 84, 101817. [Google Scholar] [CrossRef]
- Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.J.; Montes-de-Oca-Luna, R.; Rodriguez-Rocha, H.; Garcia-Garcia, A. Metformin and Trehalose-Modulated Autophagy Exerts a Neurotherapeutic Effect on Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 7253–7273. [Google Scholar] [CrossRef]
- Abdelaziz, D.H.; Thapa, S.; Abdulrahman, B.; Vankuppeveld, L.; Schatzl, H.M. Metformin reduces prion infection in neuronal cells by enhancing autophagy. Biochem. Biophys. Res. Commun. 2020, 523, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Sarkaki, A.; Farbood, Y.; Badavi, M.; Khalaj, L.; Khodagholi, F.; Ashabi, G. Metformin improves anxiety-like behaviors through AMPK-dependent regulation of autophagy following transient forebrain ischemia. Metab. Brain Dis. 2015, 30, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Benito-Cuesta, I.; Ordóñez-Gutiérrez, L.; Wandosell, F. AMPK activation does not enhance autophagy in neurons in contrast to MTORC1 inhibition: Different impact on β-amyloid clearance. Autophagy 2021, 17, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, L.; Ji, D.; Yu, W.; Zhang, Y.; Xiang, Y.; Zhou, C.; Deng, P.; Pi, H.; Lu, Y.; et al. Metformin attenuates cadmium-induced degeneration of spiral ganglion neuron via restoring autophagic flux in primary culture. J. Inorg. Biochem. 2022, 234, 111901. [Google Scholar] [CrossRef]
- Chen, J.L.; Luo, C.; Pu, D.; Zhang, G.Q.; Zhao, Y.X.; Sun, Y.; Zhao, K.X.; Liao, Z.Y.; Lv, A.K.; Zhu, S.Y.; et al. Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp. Neurol. 2019, 311, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Escribano, A.P.; Bono-Yagüe, J.; García-Gimeno, M.A.; Sequedo, M.D.; Hervás, D.; Fornés-Ferrer, V.; Torres-Sánchez, S.C.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Synergistic activation of AMPK prevents from polyglutamine-induced toxicity in Caenorhabditis elegans. Pharmacol. Res. 2020, 161, 105105. [Google Scholar] [CrossRef]
- Chen, A.; Kristiansen, C.K.; Hong, Y.; Kianian, A.; Fang, E.F.; Sullivan, G.J.; Wang, J.; Li, X.; Bindoff, L.A.; Liang, K.X. Nicotinamide Riboside and Metformin Ameliorate Mitophagy Defect in Induced Pluripotent Stem Cell-Derived Astrocytes with POLG Mutations. Front. Cell Dev. Biol. 2021, 9, 737304. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Naruse, G.; Yoshida, A.; Minatoguchi, S.; Watanabe, T.; Kawaguchi, T.; Yamada, Y.; Mikami, A.; Kawasaki, M.; Takemura, G. Metformin Enhances Autophagy and Provides Cardioprotection in δ-Sarcoglycan Deficiency-Induced Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005418. [Google Scholar] [CrossRef]
- Mostafa, D.K.; Nayel, O.A.; Abdulmalek, S.; Abdelbary, A.A.; Ismail, C.A. Modulation of autophagy, apoptosis and oxidative stress: A clue for repurposing metformin in photoaging. Inflammopharmacology 2022, 30, 2521–2535. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; You, Y.J. Regulation of organelle function by metformin. IUBMB Life 2017, 69, 459–469. [Google Scholar] [CrossRef] [PubMed]
- van Ohgaki, R.; van IJzendoorn, S.C.D.; Matsushita, M.; Hoekstra, D.; Kanazawa, H. Organellar Na+/H+ exchangers: Novel players in organelle pH regulation and their emerging functions. Biochemistry 2011, 50, 443–450. [Google Scholar] [CrossRef]
- Pescosolido, M.F.; Ouyang, Q.; Liu, J.S.; Morrow, E.M. Loss of Christianson Syndrome Na(+)/H(+) Exchanger 6 (NHE6) Causes Abnormal Endosome Maturation and Trafficking Underlying Lysosome Dysfunction in Neurons. J. Neurosci. 2021, 41, 9235–9256. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.A.; Bah, F.; Ma, L.; Lee, Y.; Schmidt, M.; Welch, E.; Morrow, E.M.; Young-Pearse, T.L. Loss of endosomal exchanger NHE6 leads to pathological changes in tau in human neurons. Stem Cell Rep. 2022, 17, 2111–2126. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, K.; Takamatsu, H.; Kumanogoh, A. The Ragulator complex: Delving its multifunctional impact on metabolism and beyond. Inflamm. Regen. 2023, 43, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The γ-secretase complex: From structure to function. Front. Cell Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Patnana, P.K.; Nimmagadda, S.C. Low-dose metformin and PEN2-dependent lysosomal AMPK activation: Benefits outnumber side effects. Signal Transduct. Target. Ther. 2022, 7, 178. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Jessen, N. PEN2: Metformin’s new partner at lysosome. Cell Res. 2022, 32, 507–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, D.; Robinson, C.V.; Wu, H.; Fu, T.M. Structures of a Complete Human V-ATPase Reveal Mechanisms of Its Assembly. Mol. Cell 2020, 80, 501–511.e503. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, S.; Xue, J.; Zhang, H.; Yin, J. Evaluation of PEN2-ATP6AP1 axis as an antiparasitic target for metformin based on phylogeny analysis and molecular docking. Mol. Biochem. Parasitol. 2023, 255, 111580. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ding, J.; Li, S.; Lin, J.; Jiang, R.; Lin, C.; Dai, L.; Xie, C.; Lin, D.; Xu, H.; et al. Metformin Promotes the Survival of Random-Pattern Skin Flaps by Inducing Autophagy via the AMPK-mTOR-TFEB signaling pathway. Int. J. Biol. Sci. 2019, 15, 325–340. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Y.; Liu, J.; Deng, Y.; Zhou, B.; Wen, Y.; Li, M.; Wen, D.; Ying, Y.; Luo, S.; et al. Metformin Alleviates Hepatic Steatosis and Insulin Resistance in a Mouse Model of High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Promoting Transcription Factor EB-Dependent Autophagy. Front. Pharmacol. 2021, 12, 689111. [Google Scholar] [CrossRef]
- Park, J.; Rah, S.Y.; An, H.S.; Lee, J.Y.; Roh, G.S.; Ryter, S.W.; Park, J.W.; Yang, C.H.; Surh, Y.J.; Kim, U.H.; et al. Metformin-induced TTP mediates communication between Kupffer cells and hepatocytes to alleviate hepatic steatosis by regulating lipophagy and necroptosis. Metabolism 2023, 141, 155516. [Google Scholar] [CrossRef]
- Solberg, R.; Lunde, N.N.; Forbord, K.M.; Okla, M.; Kassem, M.; Jafari, A. The Mammalian Cysteine Protease Legumain in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15983. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Qin, M.; Li, Y.; Wu, W.; Tong, X. Metformin regulates autophagy via LGMN to inhibit choriocarcinoma. Gene 2023, 853, 147090. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Du, M.R.; Gao, Q.Y.; Liu, C.L.; Bai, L.Y.; Li, T.; Wei, F.L. Exploring the Pharmacological Potential of Metformin for Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 838173. [Google Scholar] [CrossRef]
- Cao, G.; Gong, T.; Du, Y.; Wang, Y.; Ge, T.; Liu, J. Mechanism of metformin regulation in central nervous system: Progression and future perspectives. Biomed. Pharmacother. 2022, 156, 113686. [Google Scholar] [CrossRef]
- Luo, A.; Ning, P.; Lu, H.; Huang, H.; Shen, Q.; Zhang, D.; Xu, F.; Yang, L.; Xu, Y. Association between Metformin and Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Clinical Observational Studies. J. Alzheimers Dis. 2022, 88, 1311–1323. [Google Scholar] [CrossRef]
- Malazy, O.T.; Bandarian, F.; Qorbani, M.; Mohseni, S.; Mirsadeghi, S.; Peimani, M.; Larijani, B. The effect of metformin on cognitive function: A systematic review and meta-analysis. J. Psychopharmacol. 2022, 36, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Jiménez, M.; Zaarkti, A.; García-Arnés, J.A.; García-Casares, N. Antidiabetic Drugs in Alzheimer’s Disease and Mild Cognitive Impairment: A Systematic Review. Dement. Geriatr. Cogn. Disord. 2020, 49, 423–434. [Google Scholar] [CrossRef]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, J.; Jiang, J.; Liu, F.; Zhang, Y. Do oral antidiabetic medications alter the risk of Parkinson’s disease? An updated systematic review and meta-analysis. Neurol. Sci. 2023, 44, 4193–4203. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, X.; Li, P.; Wang, M.; Yan, L.; Bao, Z.; Liu, Q. Association between Diabetes Medications and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 678649. [Google Scholar] [CrossRef]
- van Gool, R.; Tucker-Bartley, A.; Yang, E.; Todd, N.; Guenther, F.; Goodlett, B.; Al-Hertani, W.; Bodamer, O.A.; Upadhyay, J. Targeting neurological abnormalities in lysosomal storage diseases. Trends Pharmacol. Sci. 2022, 43, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal dysfunction in neurodegeneration: Emerging concepts and methods. Trends Neurosci. 2022, 45, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Malnar, M.; Hecimovic, S.; Mattsson, N.; Zetterberg, H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 2014, 72, 37–47. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Cui, M.; Yoshimori, T.; Nakamura, S. Autophagy system as a potential therapeutic target for neurodegenerative diseases. Neurochem. Int. 2022, 155, 105308. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Bustos, V.; Flajolet, M.; Greengard, P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011, 25, 1934–1942. [Google Scholar] [CrossRef]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef]
- Atya, H.B.; Sharaf, N.M.; Abdelghany, R.M.; El-Helaly, S.N.; Taha, H. Autophagy and exosomes; inter-connected maestros in Alzheimer’s disease. Inflammopharmacology 2024, 32, 2061–2073. [Google Scholar] [CrossRef]
- Roth, J.R.; de Moraes, R.C.M.; Xu, B.P.; Crawley, S.R.; Khan, M.A.; Melkani, G.C. Rapamycin reduces neuronal mutant huntingtin aggregation and ameliorates locomotor performance in Drosophila. Front. Aging Neurosci. 2023, 15, 1223911. [Google Scholar] [CrossRef]
- Song, Q.; Meng, B.; Xu, H.; Mao, Z. The emerging roles of vacuolar-type ATPase-dependent Lysosomal acidification in neurodegenerative diseases. Transl. Neurodegener. 2020, 9, 17. [Google Scholar] [CrossRef]
- Wang, B.; Martini-Stoica, H.; Qi, C.; Lu, T.C.; Wang, S.; Xiong, W.; Qi, Y.; Xu, Y.; Sardiello, M.; Li, H.; et al. TFEB-vacuolar ATPase signaling regulates lysosomal function and microglial activation in tauopathy. Nat. Neurosci. 2024, 27, 48–62. [Google Scholar] [CrossRef]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Nixon, Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Tusi, S.K.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef]
- Bi, X.; Liao, G. Cholesterol in Niemann-Pick Type C disease. Subcell. Biochem. 2010, 51, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liu, X.; Zhang, Y.; Han, X.; Ma, C.; Liu, Y.; Guan, L.; Qiao, L.; Lin, J. The Effects of Combined Therapy with Metformin and Hydroxypropyl-β-Cyclodextrin in a Mouse Model of Niemann-Pick Disease Type C1. Front. Pharmacol. 2021, 12, 825425. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Álvarez, N.T.; Bautista-Niño, P.K.; Trejos-Suárez, J.; Serrano-Díaz, N.C. A model of metformin mitochondrial metabolism in metachromatic leukodystrophy: First description of human Schwann cells transfected with CRISPR-Cas9. Open Biol. 2022, 12, 210371. [Google Scholar] [CrossRef]
- Solini, A.; Tricò, D. Clinical efficacy and cost-effectiveness of metformin in different patient populations: A narrative review of real-world evidence. Diabetes Obes. Metab. 2024, 26 (Suppl. 3), 20–30. [Google Scholar] [CrossRef]
- Romero, R.; Erez, O.; Hüttemann, M.; Maymon, E.; Panaitescu, B.; Conde-Agudelo, A.; Pacora, P.; Yoon, B.H.; Grossman, L.I. Metformin, the aspirin of the 21st century: Its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Am. J. Obstet. Gynecol. 2017, 217, 282–302. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Green, R.; Miller, J.W. Vitamin B12 deficiency. Vitam. Horm. 2022, 119, 405–439. [Google Scholar] [CrossRef]
- Abdi, M.; Pasbakhsh, P.; Shabani, M.; Nekoonam, S.; Sadeghi, A.; Fathi, F.; Abouzaripour, M.; Mohamed, W.; Zibara, K.; Kashani, I.R.; et al. Metformin Therapy Attenuates Pro-inflammatory Microglia by Inhibiting NF-κB in Cuprizone Demyelinating Mouse Model of Multiple Sclerosis. Neurotox. Res. 2021, 39, 1732–1746. [Google Scholar] [CrossRef]
- Binyamin, O.; Frid, K.; Keller, G.; Saada, A.; Gabizon, R. Comparing anti-aging hallmark activities of Metformin and Nano-PSO in a mouse model of genetic Creutzfeldt-Jakob Disease. Neurobiol. Aging 2022, 110, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, W.; Gong, B.; Hou, L.; Dong, X.; Xu, C.; Zhao, R.; Yu, Q.; Zhou, Z.; Huang, S.; et al. Metformin attenuates cadmium-induced neuronal apoptosis in vitro via blocking ROS-dependent PP5/AMPK-JNK signaling pathway. Neuropharmacology 2020, 175, 108065. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Kim, E.W.; Park, S.J.; Phillips, B.U.; Jeong, J.; Kim, H.; Heath, C.J.; Kim, D.; Jang, Y.; López-Cruz, L.; et al. Reconsidering repurposing: Long-term metformin treatment impairs cognition in Alzheimer’s model mice. Transl. Psychiatry 2024, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Hoang, N.M.H.; Jo, W.H.; Ham, J.R.; Lee, M.K.; Kim, M.S. Associations among the TREM-1 Pathway, Tau Hyperphosphorylation, Prolactin Expression, and Metformin in Diabetes Mice. Neuroimmunomodulation 2022, 29, 359–368. [Google Scholar] [CrossRef]
- Esmaeilnejad, S.; Semnanian, S.; Javan, M. Metformin Protects Myelin from Degeneration in A Mouse Model of Iysophosphatidylcholine-Induced Demyelination in The Optic Chiasm. Cell J. 2021, 23, 119–128. [Google Scholar] [CrossRef] [PubMed]
- García-Juan, M.; Ordóñez-Gutiérrez, L.; Wandosell, F. Clearance of β-amyloid mediated by autophagy is enhanced by MTORC1 inhibition but not AMPK activation in APP/PSEN1 astrocytes. Glia 2024, 72, 588–606. [Google Scholar] [CrossRef]
- Kang, K.; Xu, P.; Wang, M.; Chunyu, J.; Sun, X.; Ren, G.; Xiao, W.; Li, D. FGF21 attenuates neurodegeneration through modulating neuroinflammation and oxidant-stress. Biomed. Pharmacother. 2020, 129, 110439. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Park, P.H.; Hong, J.T.; Choi, D.Y. Activation of AMPK/aPKCζ/CREB pathway by metformin is associated with upregulation of GDNF and dopamine. Biochem. Pharmacol. 2020, 180, 114193. [Google Scholar] [CrossRef]
- Lu, X.Y.; Huang, S.; Chen, Q.B.; Zhang, D.; Li, W.; Ao, R.; Leung, F.C.; Zhang, Z.; Huang, J.; Tang, Y.; et al. Metformin Ameliorates Aβ Pathology by Insulin-Degrading Enzyme in a Transgenic Mouse Model of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2020, 2020, 2315106. [Google Scholar] [CrossRef]
- Oliveira, W.H.; Braga, C.F.; Lós, D.B.; Araújo, S.M.R.; França, M.R.; Duarte-Silva, E.; Rodrigues, G.B.; Rocha, S.W.S.; Peixoto, C.A. Metformin prevents p-tau and amyloid plaque deposition and memory impairment in diabetic mice. Exp. Brain Res. 2021, 239, 2821–2839. [Google Scholar] [CrossRef]
- Ozbey, G.; Nemutlu-Samur, D.; Parlak, H.; Yildirim, S.; Aslan, M.; Tanriover, G.; Agar, A. Metformin protects rotenone-induced dopaminergic neurodegeneration by reducing lipid peroxidation. Pharmacol. Rep. 2020, 72, 1397–1406. [Google Scholar] [CrossRef]
- Peralta, S.; Pinto, M.; Arguello, T.; Garcia, S.; Diaz, F.; Moraes, C.T. Metformin delays neurological symptom onset in a mouse model of neuronal complex I deficiency. JCI Insight 2020, 5, e141183. [Google Scholar] [CrossRef] [PubMed]
- Rabieipoor, S.; Zare, M.; Ettcheto, M.; Camins, A.; Javan, M. Metformin restores cognitive dysfunction and histopathological deficits in an animal model of sporadic Alzheimer’s disease. Heliyon 2023, 9, e17873. [Google Scholar] [CrossRef]
- Ryu, Y.K.; Go, J.; Park, H.Y.; Choi, Y.K.; Seo, Y.J.; Choi, J.H.; Rhee, M.; Lee, T.G.; Lee, C.H.; Kim, K.S. Metformin regulates astrocyte reactivity in Parkinson’s disease and normal aging. Neuropharmacology 2020, 175, 108173. [Google Scholar] [CrossRef] [PubMed]
- Sanz, F.J.; Solana-Manrique, C.; Lilao-Garzón, J.; Brito-Casillas, Y.; Muñoz-Descalzo, S.; Paricio, N. Exploring the link between Parkinson’s disease and type 2 diabetes mellitus in Drosophila. FASEB J. 2022, 36, e22432. [Google Scholar] [CrossRef] [PubMed]
- Syal, C.; Kosaraju, J.; Hamilton, L.; Aumont, A.; Chu, A.; Sarma, S.N.; Thomas, J.; Seegobin, M.; Dilworth, F.J.; He, L.; et al. Dysregulated expression of monoacylglycerol lipase is a marker for anti-diabetic drug metformin-targeted therapy to correct impaired neurogenesis and spatial memory in Alzheimer’s disease. Theranostics 2020, 10, 6337–6360. [Google Scholar] [CrossRef]
- Thapa, S.; Abdelaziz, D.H.; Abdulrahman, B.A.; Schatzl, H.M. Sephin1 Reduces Prion Infection in Prion-Infected Cells and Animal Model. Mol. Neurobiol. 2020, 57, 2206–2219. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, T.; Chen, S.; Wu, M.; Han, J.; Li, Z. Design and synthesis of 3-(4-pyridyl)-5-(4-sulfamido-phenyl)-1,2,4-oxadiazole derivatives as novel GSK-3β inhibitors and evaluation of their potential as multifunctional anti-Alzheimer agents. Eur. J. Med. Chem. 2021, 209, 112874. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tian, T.; Zhou, H.; Jiang, S.Y.; Jiao, Y.Y.; Zhu, Z.; Xia, J.; Ma, J.H.; Du, R.H. Metformin normalizes mitochondrial function to delay astrocyte senescence in a mouse model of Parkinson’s disease through Mfn2-cGAS signaling. J. Neuroinflamm. 2024, 21, 81. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z.; Mao, Y.; Li, B.; Zhu, Y.; Zhang, S.; Wang, S.; Jiang, Y.; Xu, N.; Xie, Y.; et al. NEAT1 regulates microtubule stabilization via FZD3/GSK3β/P-tau pathway in SH-SY5Y cells and APP/PS1 mice. Aging 2020, 12, 23233–23250. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, F.; Zheng, L.; Pan, Y.; Ni, L.; Fu, Z.; Shi, J.; Ni, Y. DPP-4 inhibition by linagliptin ameliorates age-related mild cognitive impairment by regulating microglia polarization in mice. Exp. Neurol. 2024, 373, 114689. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cell/Animal Model | Disease | Autophagy Induction | Signalling Pathway Involved | References |
|---|---|---|---|---|
| 293THK cells and male transgenic APP/PS1 (C57BL/6) mice | Alzheimer’s disease | ↑ | TGF-β activated kinase 1 (TAK1)-Ikappa B kinase (IKK)α/β-heat shock cognate protein (Hsc) 70 | [27] |
| Paraquat-treated SH-SY5Y cells and C57BL6 mice | Parkinson’s disease | ↑ | AMPK | [37] |
| RML prion-infected ScCAD5 cells | Prion disease | ↑ | Not reported | [38] |
| Adult male Wistar rats subjected to transient forebrain global ischaemia | Cerebral ischaemia | ↑ | AMPK | [39] |
| Primary cerebellar granule neurons and SH-SY-5Y cells | Alzheimer’s disease | = or ↓ | AMPK | [40] |
| Organotypic cultures of spiral ganglion neurons isolated from cadmium-treated C57BL/6 mice | Cadmium-induced spiral ganglion neuron degeneration | ↑ | Not reported | [41] |
| db/db mice and high glucose-cultured HT22 cells | Diabetes-induced tau hyperphosphorylation | ↑ | AMPK | [42] |
| Caenorhabditis elegans | Huntington’s disease | ↑ | AMPK | [43] |
| Induced pluripotent stem cell-derived astrocytes with POLG mutations | Mitochondrial diseases | ↑ | AMPK | [44] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papini, N.; Giussani, P.; Tringali, C. Metformin Lysosomal Targeting: A Novel Aspect to Be Investigated for Metformin Repurposing in Neurodegenerative Diseases? Int. J. Mol. Sci. 2024, 25, 8884. https://doi.org/10.3390/ijms25168884
Papini N, Giussani P, Tringali C. Metformin Lysosomal Targeting: A Novel Aspect to Be Investigated for Metformin Repurposing in Neurodegenerative Diseases? International Journal of Molecular Sciences. 2024; 25(16):8884. https://doi.org/10.3390/ijms25168884
Chicago/Turabian StylePapini, Nadia, Paola Giussani, and Cristina Tringali. 2024. "Metformin Lysosomal Targeting: A Novel Aspect to Be Investigated for Metformin Repurposing in Neurodegenerative Diseases?" International Journal of Molecular Sciences 25, no. 16: 8884. https://doi.org/10.3390/ijms25168884
APA StylePapini, N., Giussani, P., & Tringali, C. (2024). Metformin Lysosomal Targeting: A Novel Aspect to Be Investigated for Metformin Repurposing in Neurodegenerative Diseases? International Journal of Molecular Sciences, 25(16), 8884. https://doi.org/10.3390/ijms25168884