
Balanced Duality: H2O2-Based Therapy in Cancer and Its Protective Effects on Non-Malignant Tissues



Abstract
:1. Introduction
2. H2O2 as a Signaling Molecule
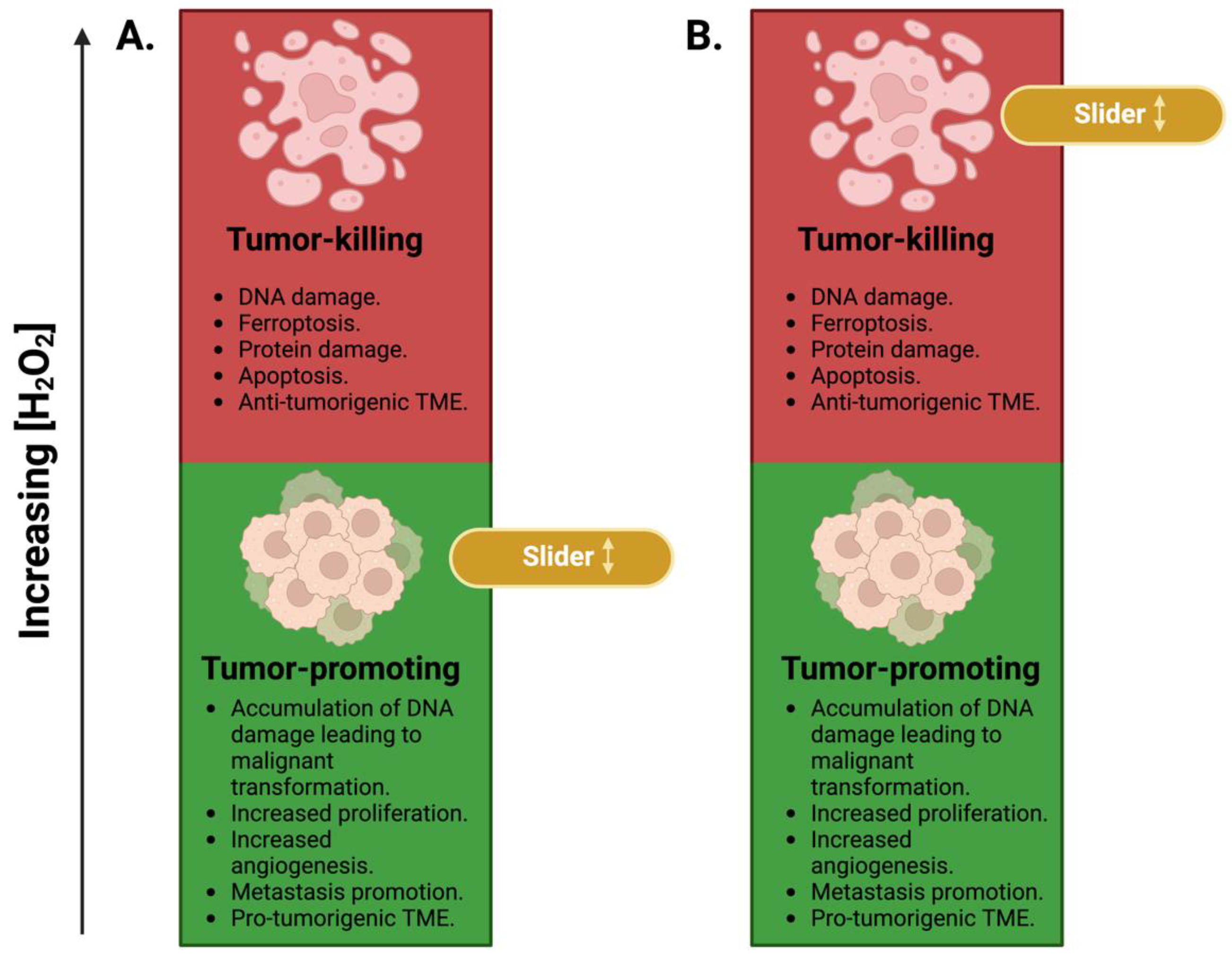
3. H2O2 in Cancer: A Sliding Scale
4. Targeting H2O2 in Cancer: Enhancing Conventional Therapy and Minimizing Its Normal Tissue Toxicities
4.1. Pharmacological Ascorbate

{kind=link}
| Trial | Disease | Therapy | Outcomes | Reference |
|---|---|---|---|---|
| NCT02344355 Phase 2 | GBM | P-AscH− with radiation and temozolomide. | Increased overall survival (19.6 months) compared to 14.6 months in historical controls. | [116] |
| NCT00228319 Phase 1/2a | Ovarian cancer | P-AscH− with carboplatin and paclitaxel. |
| [117] |
| NCT01049880 Phase 1 | Pancreatic cancer | P-AscH− with gemcitabine. |
| [118] |
| NCT00954525 Phase 1 | Pancreatic cancer | P-AscH− with gemcitabine and erlotinib. | P-AscH− was well tolerated. | [119] |
| NCT01364805 Phase 1/2a | Pancreatic cancer | P-AscH− with gemcitabine. | Trend toward increasing overall survival. | [120] |
| NCT02905578 Phase 2 | Pancreatic cancer | P-AscH− with gemcitabine. | Ongoing trial. | - |
| NCT02420314 Phase 2 | NSCLC | P-AscH− with carboplatin and paclitaxel. |
| [121] |
| NCT03146962 Phase 3 | Colorectal cancer | P-AscH− with FOLFOX ± Bevacizumab. | Increased progression-free survival to 9.2 months vs. 7.8 in the control group. This effect was limited to patients with RAS-mutant tumors. | [131] |
| NCT01080352 Phase 2 | Prostate cancer | P-AscH− | No benefits to the use of P-AscH- as a treatment. | [132] |
| NCT02516670 Phase 2 | Prostate cancer | P-AscH− with docetaxel. | No benefits from the addition of P-AscH- to the treatment regimen. | [133] |
| NCT03468075 Phase 2 | Soft tissue sarcoma | P-AscH− with gemcitabine. | Terminated. | - |
| NCT03508726 Phase 1b/2 | Soft tissue sarcoma | P-AscH− with radiation. | Completed. | - |
| NCT04900792 Phase 1 | GBM | P-AscH− with ferumoxytol, radiation, and temozolomide. | Ongoing trial. | - |
4.2. Superoxide Dismutase Mimetics
| Trial | Disease | Therapy | Outcomes | Reference |
|---|---|---|---|---|
| NCT02990468 Phase 1/2 | Head and neck cancer | MnBuOE with radiation and cisplatin | Ongoing trial. | - |
| NCT02655601 Phase 2 | High-grade glioma | MnBuOE with radiation and temozolomide | Ongoing trial. | - |
| NCT03386500 Phase 1 | Anal cancer | MnBuOE with radiation, 5FU, and mitomycin | Ongoing trial. | - |
| NCT05254327 Phase 2 | Rectal cancer | MnBuOE with radiation and chemotherapy (oxaliplatin, leucovorin, fluorouracil, capecitabine) | Ongoing trial. | - |
| NCT01921426 Phase 1b/2a | Head and neck cancer | AVA with radiation and cisplatin | Reduction in the incidence of severe radiation-induced oral mucositis by 50% (relative to historical controls). | [183] |
| NCT02508389 Phase 2b | Head and neck cancer | AVA with radiation and cisplatin |
| [180,182] |
| NCT03689712 Phase 3 | Head and neck cancer | AVA with radiation and cisplatin |
| [181] |
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thenard, L. Observations sur l’Influence de l’Eau dans la Formation des Acides Oxigénés. Ann. Chim. 1818, 9, 314–317. [Google Scholar]
- Thenard, L.J. Nouveaux résultats sur la combination de l’oxigène avec l’eau. Annales de Chimie et de Physique 1819, 10, 335–336. [Google Scholar]
- Thénard, L.J. Observations sur des nouvelles combinaisons entre l’oxigène et divers acides. Ann. Chim. Phys. 1818, 8, 306–312. [Google Scholar]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Henle, E.S.; Linn, S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J. Biol. Chem. 1997, 272, 19095–19098. [Google Scholar] [CrossRef]
- Gottfredsen, R.H.; Larsen, U.G.; Enghild, J.J.; Petersen, S.V. Hydrogen peroxide induce modifications of human extracellular superoxide dismutase that results in enzyme inhibition. Redox Biol. 2013, 1, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Grayson, C.; Koufos, O. Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation. Cells 2022, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Molavian, H.; Madani Tonekaboni, A.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, 13620. [Google Scholar] [CrossRef]
- Deisseroth, A.; Dounce, A.L. Catalase: Physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970, 50, 319–375. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef]
- Shimokawa, H. Reactive oxygen species in cardiovascular health and disease: Special references to nitric oxide, hydrogen peroxide, and Rho-kinase. J. Clin. Biochem. Nutr. 2020, 66, 83–91. [Google Scholar] [CrossRef]
- Korac, B.; Kalezic, A.; Pekovic-Vaughan, V.; Korac, A.; Jankovic, A. Redox changes in obesity, metabolic syndrome, and diabetes. Redox Biol. 2021, 42, 101887. [Google Scholar] [CrossRef]
- Lee, Y.M.; He, W.; Liou, Y.C. The redox language in neurodegenerative diseases: Oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Devarie-Baez, N.O.; Silva Lopez, E.I.; Furdui, C.M. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 2016, 50, 172–194. [Google Scholar] [CrossRef]
- Garrido Ruiz, D.; Sandoval-Perez, A.; Rangarajan, A.V.; Gunderson, E.L.; Jacobson, M.P. Cysteine Oxidation in Proteins: Structure, Biophysics, and Simulation. Biochemistry 2022, 61, 2165–2176. [Google Scholar] [CrossRef]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Chen, C.Y.; Willard, D.; Rudolph, J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry 2009, 48, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Crump, K.E.; Juneau, D.G.; Poole, L.B.; Haas, K.M.; Grayson, J.M. The reversible formation of cysteine sulfenic acid promotes B-cell activation and proliferation. Eur. J. Immunol. 2012, 42, 2152–2164. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2012, 8, 57–64. [Google Scholar] [CrossRef]
- Wani, R.; Qian, J.; Yin, L.; Bechtold, E.; King, S.B.; Poole, L.B.; Paek, E.; Tsang, A.W.; Furdui, C.M. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc. Natl. Acad. Sci. USA 2011, 108, 10550–10555. [Google Scholar] [CrossRef] [PubMed]
- Stuhmer, W.; Ruppersberg, J.P.; Schroter, K.H.; Sakmann, B.; Stocker, M.; Giese, K.P.; Perschke, A.; Baumann, A.; Pongs, O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989, 8, 3235–3244. [Google Scholar] [CrossRef]
- Yuan, X.J.; Wang, J.; Juhaszova, M.; Golovina, V.A.; Rubin, L.J. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am. J. Physiol. 1998, 274, L621–L635. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, J.; Caballero, R.; Gomez, R.; Delpon, E. I(Kur)/Kv1.5 channel blockers for the treatment of atrial fibrillation. Expert Opin. Investig. Drugs 2009, 18, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Comes, N.; Bielanska, J.; Vallejo-Gracia, A.; Serrano-Albarras, A.; Marruecos, L.; Gomez, D.; Soler, C.; Condom, E.; Ramon, Y.C.S.; Hernandez-Losa, J.; et al. The voltage-dependent K(+) channels Kv1.3 and Kv1.5 in human cancer. Front. Physiol. 2013, 4, 283. [Google Scholar] [CrossRef]
- Wettwer, E.; Terlau, H. Pharmacology of voltage-gated potassium channel Kv1.5--impact on cardiac excitability. Curr. Opin. Pharmacol. 2014, 15, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O’Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012, 111, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H. Three functional facets of calbindin D-28k. Front. Mol. Neurosci. 2012, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.; Neuhauss, S.C.F. The Binding Properties and Physiological Functions of Recoverin. Front. Mol. Neurosci. 2018, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Berggard, T.; Borek, V.; Thulin, E.; Linse, S.; Akerfeldt, K.S. Redox sensitive cysteine residues in calbindin D28k are structurally and functionally important. Biochemistry 2005, 44, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-A.; Kwak, M.-K. The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef] [PubMed]
- Covas, G.; Marinho, H.S.; Cyrne, L.; Antunes, F. Chapter Nine—Activation of Nrf2 by H2O2: De Novo Synthesis Versus Nuclear Translocation. In Methods in Enzymology; Cadenas, E., Packer, L., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 528, pp. 157–171. [Google Scholar]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: Evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Pineda-Molina, E.; Klatt, P.; Vazquez, J.; Marina, A.; Garcia de Lacoba, M.; Perez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Morad, H.; Luqman, S.; Tan, C.H.; Swann, V.; McNaughton, P.A. TRPM2 ion channels steer neutrophils towards a source of hydrogen peroxide. Sci. Rep. 2021, 11, 9339. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Lorne, E.; Zhao, X.; Tsuruta, Y.; Sha, Y.; Liu, G.; Abraham, E. Antiinflammatory effects of hydrogen peroxide in neutrophil activation and acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 179, 694–704. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Chikuma, S.; Sugiyama, Y.; Kabashima, K.; Verkman, A.S.; Inoue, S.; Miyachi, Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J. Exp. Med. 2012, 209, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Guo, F. RhoA and Cdc42 in T cells: Are they targetable for T cell-mediated inflammatory diseases? Precis. Clin. Med. 2021, 4, 56–61. [Google Scholar] [CrossRef]
- Kaul, N.; Forman, H.J. Activation of NFκB by the respiratory burst of macrophages. Free Radic. Biol. Med. 1996, 21, 401–405. [Google Scholar] [CrossRef]
- Lu, Y.; Wahl, L.M. Oxidative Stress Augments the Production of Matrix Metalloproteinase-1, Cyclooxygenase-2, and Prostaglandin E2 through Enhancement of NF-κB Activity in Lipopolysaccharide-Activated Human Primary Monocytes1. J. Immunol. 2005, 175, 5423–5429. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Bernardo, A.; Davies, K.J. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 2016, 603, 48–53. [Google Scholar] [CrossRef]
- Halliwell, B.; Clement, M.V.; Long, L.H. Hydrogen peroxide in the human body. FEBS Lett. 2000, 486, 10–13. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, T.; Qiu, Y.; Lin, Y.; Yao, Y.; Lian, W.; Lin, L.; Song, J.; Yang, H. An inorganic prodrug, tellurium nanowires with enhanced ROS generation and GSH depletion for selective cancer therapy. Chem. Sci. 2019, 10, 7068–7075. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Lopez-Lazaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Ali, T.; Li, D.; Ponnamperumage, T.N.F.; Peterson, A.K.; Pandey, J.; Fatima, K.; Brzezinski, J.; Jakusz, J.A.R.; Gao, H.; Koelsch, G.E.; et al. Generation of Hydrogen Peroxide in Cancer Cells: Advancing Therapeutic Approaches for Cancer Treatment. Cancers 2024, 16, 2171. [Google Scholar] [CrossRef]
- Okamoto, M.; Kawai, K.; Reznikoff, C.A.; Oyasu, R. Transformation in vitro of a nontumorigenic rat urothelial cell line by hydrogen peroxide. Cancer Res. 1996, 56, 4649–4653. [Google Scholar]
- Arnold, R.S.; Shi, J.; Murad, E.; Whalen, A.M.; Sun, C.Q.; Polavarapu, R.; Parthasarathy, S.; Petros, J.A.; Lambeth, J.D. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. USA 2001, 98, 5550–5555. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Erudaitius, D.; Huang, A.; Kazmi, S.; Buettner, G.R.; Rodgers, V.G. Peroxiporin Expression Is an Important Factor for Cancer Cell Susceptibility to Therapeutic H2O2: Implications for Pharmacological Ascorbate Therapy. PLoS ONE 2017, 12, e0170442. [Google Scholar] [CrossRef]
- Erudaitius, D.; Mantooth, J.; Huang, A.; Soliman, J.; Doskey, C.M.; Buettner, G.R.; Rodgers, V.G.J. Calculated cell-specific intracellular hydrogen peroxide concentration: Relevance in cancer cell susceptibility during ascorbate therapy. Free Radic. Biol. Med. 2018, 120, 356–367. [Google Scholar] [CrossRef]
- Blanchetot, C.; Boonstra, J. The ROS-NOX connection in cancer and angiogenesis. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Konate, M.M.; Doroshow, J.H. Hydrogen peroxide-producing NADPH oxidases and the promotion of migratory phenotypes in cancer. Arch. Biochem. Biophys. 2019, 675, 108076. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef]
- Liu, X.; Pei, C.; Yan, S.; Liu, G.; Liu, G.; Chen, W.; Cui, Y.; Liu, Y. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumour Biol. 2015, 36, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shyu, W.C.; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011, 6, e23945. [Google Scholar] [CrossRef]
- Garcia-Gomez, P.; Golan, I.; Dadras, M.S.; Mezheyeuski, A.; Bellomo, C.; Tzavlaki, K.; Moren, A.; Carreras-Puigvert, J.; Caja, L. NOX4 regulates TGFbeta-induced proliferation and self-renewal in glioblastoma stem cells. Mol. Oncol. 2022, 16, 1891–1912. [Google Scholar] [CrossRef]
- Lin, X.L.; Yang, L.; Fu, S.W.; Lin, W.F.; Gao, Y.J.; Chen, H.Y.; Ge, Z.Z. Overexpression of NOX4 predicts poor prognosis and promotes tumor progression in human colorectal cancer. Oncotarget 2017, 8, 33586–33600. [Google Scholar] [CrossRef]
- Laurent, A.; Nicco, C.; Chereau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Levy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar] [CrossRef]
- Nicco, C.; Laurent, A.; Chereau, C.; Weill, B.; Batteux, F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed. Pharmacother. 2005, 59, 169–174. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, D.; Liu, Y.; Zhang, Y.; Duan, C.; Otkur, W.; Chen, H.; Liu, X.; Xia, T.; Qi, H.; et al. AQP3-mediated H(2) O(2) uptake inhibits LUAD autophagy by inactivating PTEN. Cancer Sci. 2021, 112, 3278–3292. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, W.; Tao, G.Q.; Sun, J.; Shi, L.P. Study on the proliferation of human gastric cancer cell AGS by activation of EGFR in H2O2. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1006–1012. [Google Scholar] [PubMed]
- Polytarchou, C.; Hatziapostolou, M.; Papadimitriou, E. Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J. Biol. Chem. 2005, 280, 40428–40435. [Google Scholar] [CrossRef]
- Galli, S.; Labato, M.I.; Bal de Kier Joffé, E.; Carreras, M.a.C.; Poderoso, J.J. Decreased Mitochondrial Nitric Oxide Synthase Activity and Hydrogen Peroxide Relate Persistent Tumoral Proliferation to Embryonic Behavior1. Cancer Res. 2003, 63, 6370–6377. [Google Scholar]
- Jeronimo, A.; Rodrigues, G.; Vilas-Boas, F.; Martins, G.G.; Bagulho, A.; Real, C. Hydrogen peroxide regulates angiogenesis-related factors in tumor cells. Biochem. Cell Biol. 2017, 95, 679–685. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Li, W.; Ma, Q.; Li, J.; Guo, K.; Liu, H.; Han, L.; Ma, G. Hyperglycemia enhances the invasive and migratory activity of pancreatic cancer cells via hydrogen peroxide. Oncol. Rep. 2011, 25, 1279–1287. [Google Scholar] [CrossRef]
- Li, W.; Ma, Z.; Ma, J.; Li, X.; Xu, Q.; Duan, W.; Chen, X.; Lv, Y.; Zhou, S.; Wu, E.; et al. Hydrogen peroxide mediates hyperglycemia-induced invasive activity via ERK and p38 MAPK in human pancreatic cancer. Oncotarget 2015, 6, 31119–31133. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, L.; Chen, X.; Jiang, Z.; Zong, L.; Ma, Q. Hyperglycemia Promotes the Epithelial-Mesenchymal Transition of Pancreatic Cancer via Hydrogen Peroxide. Oxidative Med. Cell. Longev. 2016, 2016, 5190314. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Tamada, A.; Hyoudou, K.; Umeyama, Y.; Takahashi, Y.; Kobayashi, Y.; Kumai, H.; Ishida, E.; Staud, F.; Yabe, Y.; et al. Inhibition of experimental hepatic metastasis by targeted delivery of catalase in mice. Clin. Exp. Metastasis 2004, 21, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.K.; Ranganathan, A.C.; Mansouri, J.; Rodriguez, A.M.; Providence, K.M.; Rutter, J.L.; Pumiglia, K.; Bennett, J.A.; Melendez, J.A. Elevated Sod2 Activity Augments Matrix Metalloproteinase Expression: Evidence for the Involvement of Endogenous Hydrogen Peroxide in Regulating Metastasis1. Clin. Cancer Res. 2003, 9, 424–432. [Google Scholar] [PubMed]
- Stemberger, M.B.; Ju, J.A.; Thompson, K.N.; Mathias, T.J.; Jerrett, A.E.; Chang, K.T.; Ory, E.C.; Annis, D.A.; Mull, M.L.; Gilchrist, D.E.; et al. Hydrogen Peroxide Induces alpha-Tubulin Detyrosination and Acetylation and Impacts Breast Cancer Metastatic Phenotypes. Cells 2023, 12, 1266. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, J.; Guo, G.; Zhou, J. Norepinephrine-induced myeloid-derived suppressor cells block T-cell responses via generation of reactive oxygen species. Immunopharmacol. Immunotoxicol. 2015, 37, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Kraaij, M.D.; Savage, N.D.; van der Kooij, S.W.; Koekkoek, K.; Wang, J.; van den Berg, J.M.; Ottenhoff, T.H.; Kuijpers, T.W.; Holmdahl, R.; van Kooten, C. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.; Goodell, S.; Opipari, A.W.; Hao, L.Y.; Franchi, L.; Glick, G.D.; Ferrara, J.L.; Byersdorfer, C.A. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J. Immunol. 2015, 194, 5789–5800. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Spitz, D.R.; Buettner, G.R.; Allen, B.G. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers 2019, 11, 1077. [Google Scholar] [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H(2)O(2): Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef]
- Wagner, B.A.; Witmer, J.R.; van ‘t Erve, T.J.; Buettner, G.R. An Assay for the Rate of Removal of Extracellular Hydrogen Peroxide by Cells. Redox Biol. 2013, 1, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, M.A. Excretion of Ascorbic Acid in Relation to Saturation and Utilization. Arch. Intern. Med. 1939, 63, 1095–1116. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Levine, M. Absorption, transport, and disposition of ascorbic acid in humans. J. Nutr. Biochem. 1998, 9, 116–130. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Sun, A.Y.; Drisko, J.; Krishna, M.C.; Pooput, C.; Kirk, K.; Levine, M. Ascorbic acid as a pharmacologic agent decreases tumor growth by generating hydrogen peroxide in vivo. In Proceedings of the 14th Biennial Meeting of the Society for Free Radical Research International, Beijing, China, 18–22 October 2008. [Google Scholar]
- Pollard, H.B.; Levine, M.A.; Eidelman, O.; Pollard, M. Pharmacological ascorbic acid suppresses syngeneic tumor growth and metastases in hormone-refractory prostate cancer. In Vivo 2010, 24, 249–255. [Google Scholar]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Bradley, M.D.; Wagner, B.A.; Buettner, G.R.; Monga, V.; Milhem, M.; Spitz, D.R.; Allen, B.G. Redox active metals and H(2)O(2) mediate the increased efficacy of pharmacological ascorbate in combination with gemcitabine or radiation in pre-clinical sarcoma models. Redox Biol. 2018, 14, 417–422. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Hejleh, T.A. O2•− and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 2017, 31, 487–500.e8. [Google Scholar] [CrossRef] [PubMed]
- Fromberg, A.; Gutsch, D.; Schulze, D.; Vollbracht, C.; Weiss, G.; Czubayko, F.; Aigner, A. Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer Chemother. Pharmacol. 2011, 67, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yu, J.; Chalmers, B.; Drisko, J.; Yang, J.; Li, B.; Chen, Q. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anticancer Drugs 2012, 23, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Mamede, A.C.; Pires, A.S.; Abrantes, A.M.; Tavares, S.D.; Goncalves, A.C.; Casalta-Lopes, J.E.; Sarmento-Ribeiro, A.B.; Maia, J.M.; Botelho, M.F. Cytotoxicity of ascorbic acid in a human colorectal adenocarcinoma cell line (WiDr): In vitro and in vivo studies. Nutr. Cancer 2012, 64, 1049–1057. [Google Scholar] [CrossRef]
- Herst, P.M.; Broadley, K.W.; Harper, J.L.; McConnell, M.J. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic. Biol. Med. 2012, 52, 1486–1493. [Google Scholar] [CrossRef]
- Ma, E.; Chen, P.; Wilkins, H.M.; Wang, T.; Swerdlow, R.H.; Chen, Q. Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free Radic. Biol. Med. 2017, 113, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Wu, Q.N.; Chen, D.L.; Chen, L.Z.; Wang, Z.X.; Ren, C.; Mo, H.Y.; Chen, Y.; Sheng, H.; Wang, Y.N.; et al. Pharmacological Ascorbate Suppresses Growth of Gastric Cancer Cells with GLUT1 Overexpression and Enhances the Efficacy of Oxaliplatin Through Redox Modulation. Theranostics 2018, 8, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Calderon, P.B. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic. Biol. Med. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- O’Leary, B.R.; Houwen, F.K.; Johnson, C.L.; Allen, B.G.; Mezhir, J.J.; Berg, D.J.; Cullen, J.J.; Spitz, D.R. Pharmacological Ascorbate as an Adjuvant for Enhancing Radiation-Chemotherapy Responses in Gastric Adenocarcinoma. Radiat. Res. 2018, 189, 456–465. [Google Scholar] [CrossRef]
- Du, J.; Cieslak, J.A., 3rd; Welsh, J.L.; Sibenaller, Z.A.; Allen, B.G.; Wagner, B.A.; Kalen, A.L.; Doskey, C.M.; Strother, R.K.; Button, A.M.; et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015, 75, 3314–3326. [Google Scholar] [CrossRef]
- O’Leary, B.R.; Ruppenkamp, E.K.; Steers, G.J.; Du, J.; Carroll, R.S.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Pharmacological Ascorbate Enhances Chemotherapies in Pancreatic Ductal Adenocarcinoma. Pancreas 2022, 51, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, P.; Drisko, J.A.; Khabele, D.; Godwin, A.K.; Chen, Q. Pharmacological ascorbate induces ‘BRCAness’ and enhances the effects of Poly(ADP-Ribose) polymerase inhibitors against BRCA1/2 wild-type ovarian cancer. Oncol. Lett. 2020, 19, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, C.M.; Abukhiran, I.M.; Masaadeh, A.; Van Rheeden, R.V.; Kalen, A.L.; Rodman, S.N., 3rd; Petronek, M.S.; Mapuskar, K.A.; George, B.N.; Coleman, M.C.; et al. Manipulation of Redox Metabolism Using Pharmacologic Ascorbate Opens a Therapeutic Window for Radio-Sensitization by ATM Inhibitors in Colorectal Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2023, 115, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Monga, V.; Bodeker, K.L.; Kwofie, M.; Lee, C.Y.; Mapuskar, K.A.; Stolwijk, J.M.; Zaher, A.; Wagner, B.A.; Smith, M.C.; et al. Magnetic Resonance Imaging of Iron Metabolism with T2* Mapping Predicts an Enhanced Clinical Response to Pharmacologic Ascorbate in Patients with GBM. Clin. Cancer Res. 2024, 30, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J., 2nd; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef] [PubMed]
- Polireddy, K.; Dong, R.; Reed, G.; Yu, J.; Chen, P.; Williamson, S.; Violet, P.C.; Pessetto, Z.; Godwin, A.K.; Fan, F.; et al. High Dose Parenteral Ascorbate Inhibited Pancreatic Cancer Growth and Metastasis: Mechanisms and a Phase I/IIa study. Sci. Rep. 2017, 7, 17188. [Google Scholar] [CrossRef]
- Furqan, M.; Abu-Hejleh, T.; Stephens, L.M.; Hartwig, S.M.; Mott, S.L.; Pulliam, C.F.; Petronek, M.; Henrich, J.B.; Fath, M.A.; Houtman, J.C.; et al. Pharmacological ascorbate improves the response to platinum-based chemotherapy in advanced stage non-small cell lung cancer. Redox Biol. 2022, 53, 102318. [Google Scholar] [CrossRef]
- Magri, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chila, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R., Jr.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef] [PubMed]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Kanter, M.; Akpolat, M. Vitamin C protects against ionizing radiation damage to goblet cells of the ileum in rats. Acta Histochem. 2008, 110, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Faridi, U.; Alatawi, F.; Mostafa, M. Protective role of tocopherol and ascorbic acid in taxol-treated human erythrocytes in vitro. Toxicol. Res. Appl. 2017, 1, 2397847317705813. [Google Scholar] [CrossRef]
- Liu, Q.; Shi, Y.; Chong, Y.; Ge, C. Pharmacological Ascorbate Promotes the Tumor Radiosensitization of Au@Pd Nanoparticles with Simultaneous Protection of Normal Tissues. ACS Appl. Bio Mater. 2021, 4, 1843–1851. [Google Scholar] [CrossRef]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M.; et al. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; O’Leary, B.R.; Du, J.; Carroll, R.S.; Steers, G.J.; Buettner, G.R.; Cullen, J.J. Pharmacologic Ascorbate Radiosensitizes Pancreatic Cancer but Radioprotects Normal Tissue: The Role of Oxidative Stress-Induced Lipid Peroxidation. Antioxidants 2024, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; He, M.M.; Xiao, J.; Zhang, Y.Q.; Yuan, X.L.; Fang, W.J.; Zhang, Y.; Wang, W.; Hu, X.H.; Ma, Z.G.; et al. A Randomized, Open-Label, Multicenter, Phase 3 Study of High-Dose Vitamin C Plus FOLFOX ± Bevacizumab versus FOLFOX ± Bevacizumab in Unresectable Untreated Metastatic Colorectal Cancer (VITALITY Study). Clin. Cancer Res. 2022, 28, 4232–4239. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Hojgaard, M.; Andersen, J.T.; Jorgensen, N.R.; Zerahn, B.; Kristensen, B.; Henriksen, T.; Lykkesfeldt, J.; Mikines, K.J.; Poulsen, H.E. Weekly ascorbic acid infusion in castration-resistant prostate cancer patients: A single-arm phase II trial. Transl. Androl. Urol. 2017, 6, 517–528. [Google Scholar] [CrossRef]
- Paller, C.J.; Zahurak, M.L.; Mandl, A.; Metri, N.A.; Lalji, A.; Heath, E.; Kelly, W.K.; Hoimes, C.; Barata, P.; Taksey, J.; et al. High-Dose Intravenous Vitamin C Combined with Docetaxel in Men with Metastatic Castration-Resistant Prostate Cancer: A Randomized Placebo-Controlled Phase 2 Trial. Cancer Res. Commun. 2024. [Google Scholar] [CrossRef]
- Cushing, C.M.; Petronek, M.S.; Bodeker, K.L.; Vollstedt, S.; Brown, H.A.; Opat, E.; Hollenbeck, N.J.; Shanks, T.; Berg, D.J.; Smith, B.J.; et al. Magnetic resonance imaging (MRI) of pharmacological ascorbate-induced iron redox state as a biomarker in subjects undergoing radio-chemotherapy. Redox Biol. 2021, 38, 101804. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Teferi, N.; Caster, J.M.; Stolwijk, J.M.; Zaher, A.; Buatti, J.M.; Hasan, D.; Wafa, E.I.; Salem, A.K.; Gillan, E.G.; et al. Magnetite nanoparticles as a kinetically favorable source of iron to enhance GBM response to chemoradiosensitization with pharmacological ascorbate. Redox Biol. 2023, 62, 102651. [Google Scholar] [CrossRef]
- Zaher, A.; Mapuskar, K.A.; Sarkaria, J.N.; Spitz, D.R.; Petronek, M.S.; Allen, B.G. Differential H2O2 Metabolism among Glioblastoma Subtypes Confers Variable Responses to Pharmacological Ascorbate Therapy Combined with Chemoradiation. Int. J. Mol. Sci. 2023, 24, 17158. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide dismutases. Annu. Rev. Biochem. 1975, 44, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.L.; Holme, E.; Hellner, L. Superoxide dismutase in extracellular fluids. Clin. Chim. Acta 1982, 126, 41–51. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Benov, L.; Spasojevic, I.; Fridovich, I. The ortho effect makes manganese(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin a powerful and potentially useful superoxide dismutase mimic. J. Biol. Chem. 1998, 273, 24521–24528. [Google Scholar] [CrossRef]
- Policar, C.; Bouvet, J.; Bertrand, H.C.; Delsuc, N. SOD mimics: From the tool box of the chemists to cellular studies. Curr. Opin. Chem. Biol. 2022, 67, 102109. [Google Scholar] [CrossRef]
- Borgstahl, G.E.O.; Oberley-Deegan, R.E. Superoxide Dismutases (SODs) and SOD Mimetics. Antioxidants 2018, 7, 156. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Anand, N.; Varma, S.R.; Ramamurthy, S.; Vichitra, C.; Sharma, A.; Mahalakshmi, A.M.; Essa, M.M. Superoxide dismutase and neurological disorders. IBRO Neurosci. Rep. 2024, 16, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar] [CrossRef] [PubMed]
- Friedel, F.C.; Lieb, D.; Ivanovic-Burmazovic, I. Comparative studies on manganese-based SOD mimetics, including the phosphate effect, by using global spectral analysis. J. Inorg. Biochem. 2012, 109, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.P. Functional mimics of superoxide dismutase enzymes as therapeutic agents. Chem. Rev. 1999, 99, 2573–2588. [Google Scholar] [CrossRef]
- Raber, J.; Davis, M.J.; Pfankuch, T.; Rosenthal, R.; Doctrow, S.R.; Moulder, J.E. Mitigating effect of EUK-207 on radiation-induced cognitive impairments. Behav. Brain Res. 2017, 320, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Langan, A.R.; Khan, M.A.; Yeung, I.W.; Van Dyk, J.; Hill, R.P. Partial volume rat lung irradiation: The protective/mitigating effects of Eukarion-189, a superoxide dismutase-catalase mimetic. Radiother. Oncol. 2006, 79, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, J.; Jelveh, S.; Calveley, V.; Zaidi, A.; Doctrow, S.R.; Hill, R.P. Mitigation of radiation-induced lung injury by genistein and EUK-207. Int. J. Radiat. Biol. 2011, 87, 889–901. [Google Scholar] [CrossRef]
- Mahmood, J.; Jelveh, S.; Zaidi, A.; Doctrow, S.R.; Hill, R.P. Mitigation of radiation-induced lung injury with EUK-207 and genistein: Effects in adolescent rats. Radiat. Res. 2013, 179, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Fish, B.L.; Szabo, A.; Doctrow, S.R.; Kma, L.; Molthen, R.C.; Moulder, J.E.; Jacobs, E.R.; Medhora, M. Short-term treatment with a SOD/catalase mimetic, EUK-207, mitigates pneumonitis and fibrosis after single-dose total-body or whole-thoracic irradiation. Radiat. Res. 2012, 178, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Doctrow, S.R.; Lopez, A.; Schock, A.M.; Duncan, N.E.; Jourdan, M.M.; Olasz, E.B.; Moulder, J.E.; Fish, B.L.; Mader, M.; Lazar, J.; et al. A synthetic superoxide dismutase/catalase mimetic EUK-207 mitigates radiation dermatitis and promotes wound healing in irradiated rat skin. J. Investig. Dermatol. 2013, 133, 1088–1096. [Google Scholar] [CrossRef]
- Vujaskovic, Z.; Batinic-Haberle, I.; Rabbani, Z.N.; Feng, Q.F.; Kang, S.K.; Spasojevic, I.; Samulski, T.V.; Fridovich, I.; Dewhirst, M.W.; Anscher, M.S. A small molecular weight catalytic metalloporphyrin antioxidant with superoxide dismutase (SOD) mimetic properties protects lungs from radiation-induced injury. Free Radic. Biol. Med. 2002, 33, 857–863. [Google Scholar] [CrossRef]
- Gauter-Fleckenstein, B.; Fleckenstein, K.; Owzar, K.; Jiang, C.; Batinic-Haberle, I.; Vujaskovic, Z. Comparison of two Mn porphyrin-based mimics of superoxide dismutase in pulmonary radioprotection. Free Radic. Biol. Med. 2008, 44, 982–989. [Google Scholar] [CrossRef]
- Spasojevic, I.; Chen, Y.; Noel, T.J.; Fan, P.; Zhang, L.; Reboucas, J.S.; St Clair, D.K.; Batinic-Haberle, I. Pharmacokinetics of the potent redox-modulating manganese porphyrin, MnTE-2-PyP(5+), in plasma and major organs of B6C3F1 mice. Free Radic. Biol. Med. 2008, 45, 943–949. [Google Scholar] [CrossRef]
- Gauter-Fleckenstein, B.; Fleckenstein, K.; Owzar, K.; Jiang, C.; Reboucas, J.S.; Batinic-Haberle, I.; Vujaskovic, Z. Early and late administration of MnTE-2-PyP5+ in mitigation and treatment of radiation-induced lung damage. Free Radic. Biol. Med. 2010, 48, 1034–1043. [Google Scholar] [CrossRef]
- Archambeau, J.O.; Tovmasyan, A.; Pearlstein, R.D.; Crapo, J.D.; Batinic-Haberle, I. Superoxide dismutase mimic, MnTE-2-PyP(5+) ameliorates acute and chronic proctitis following focal proton irradiation of the rat rectum. Redox Biol. 2013, 1, 599–607. [Google Scholar] [CrossRef]
- Gauter-Fleckenstein, B.; Reboucas, J.S.; Fleckenstein, K.; Tovmasyan, A.; Owzar, K.; Jiang, C.; Batinic-Haberle, I.; Vujaskovic, Z. Robust rat pulmonary radioprotection by a lipophilic Mn N-alkylpyridylporphyrin, MnTnHex-2-PyP(5+). Redox Biol. 2014, 2, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Shrishrimal, S.; Kosmacek, E.A.; Chatterjee, A.; Tyson, M.J.; Oberley-Deegan, R.E. The SOD Mimic, MnTE-2-PyP, Protects from Chronic Fibrosis and Inflammation in Irradiated Normal Pelvic Tissues. Antioxidants 2017, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.; Sheng, H.; Weitner, T.; Arulpragasam, A.; Lu, M.; Warner, D.S.; Vujaskovic, Z.; Spasojevic, I.; Batinic-Haberle, I. Design, mechanism of action, bioavailability and therapeutic effects of mn porphyrin-based redox modulators. Med. Princ. Pract. 2013, 22, 103–130. [Google Scholar] [CrossRef]
- Weitner, T.; Kos, I.; Sheng, H.; Tovmasyan, A.; Reboucas, J.S.; Fan, P.; Warner, D.S.; Vujaskovic, Z.; Batinic-Haberle, I.; Spasojevic, I. Comprehensive pharmacokinetic studies and oral bioavailability of two Mn porphyrin-based SOD mimics, MnTE-2-PyP5+ and MnTnHex-2-PyP5+. Free Radic. Biol. Med. 2013, 58, 73–80. [Google Scholar] [CrossRef]
- Leu, D.; Spasojevic, I.; Nguyen, H.; Deng, B.; Tovmasyan, A.; Weitner, T.; Sampaio, R.S.; Batinic-Haberle, I.; Huang, T.-T. CNS bioavailability and radiation protection of normal hippocampal neurogenesis by a lipophilic Mn porphyrin-based superoxide dismutase mimic, MnTnBuOE-2-PyP5+. Redox Biol. 2017, 12, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, D.H.; Tovmasyan, A.; Ashcraft, K.A.; Rajic, Z.; Weitner, T.; Liu, C.; Li, W.; Buckley, A.F.; Prasad, M.R.; Young, K.H.; et al. Radioprotection of the brain white matter by Mn(III) n-Butoxyethylpyridylporphyrin-based superoxide dismutase mimic MnTnBuOE-2-PyP5+. Mol. Cancer Ther. 2015, 14, 70–79. [Google Scholar] [CrossRef]
- McElroy, T.; Brown, T.; Kiffer, F.; Wang, J.; Byrum, S.D.; Oberley-Deegan, R.E.; Allen, A.R. Assessing the Effects of Redox Modifier MnTnBuOE-2-PyP 5+ on Cognition and Hippocampal Physiology Following Doxorubicin, Cyclophosphamide, and Paclitaxel Treatment. Int. J. Mol. Sci. 2020, 21, 1867. [Google Scholar] [CrossRef] [PubMed]
- Yulyana, Y.; Tovmasyan, A.; Ho, I.A.; Sia, K.C.; Newman, J.P.; Ng, W.H.; Guo, C.M.; Hui, K.M.; Batinic-Haberle, I.; Lam, P.Y. Redox-Active Mn Porphyrin-based Potent SOD Mimic, MnTnBuOE-2-PyP(5+), Enhances Carbenoxolone-Mediated TRAIL-Induced Apoptosis in Glioblastoma Multiforme. Stem Cell Rev. Rep. 2016, 12, 140–155. [Google Scholar] [CrossRef]
- Ashcraft, K.A.; Boss, M.K.; Tovmasyan, A.; Roy Choudhury, K.; Fontanella, A.N.; Young, K.H.; Palmer, G.M.; Birer, S.R.; Landon, C.D.; Park, W.; et al. Novel Manganese-Porphyrin Superoxide Dismutase-Mimetic Widens the Therapeutic Margin in a Preclinical Head and Neck Cancer Model. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 892–900. [Google Scholar] [CrossRef]
- Birer, S.R.; Lee, C.T.; Choudhury, K.R.; Young, K.H.; Spasojevic, I.; Batinic-Haberle, I.; Crapo, J.D.; Dewhirst, M.W.; Ashcraft, K.A. Inhibition of the Continuum of Radiation-Induced Normal Tissue Injury by a Redox-Active Mn Porphyrin. Radiat. Res. 2017, 188, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Choi, C.; Lee, G.H.; Son, A.; Kim, S.H.; Park, H.C.; Batinic-Haberle, I.; Park, W. Mechanism of the Antitumor and Radiosensitizing Effects of a Manganese Porphyrin, MnHex-2-PyP. Antioxid. Redox Signal. 2017, 27, 1067–1082. [Google Scholar] [CrossRef]
- Chaiswing, L.; Yarana, C.; St Clair, W.; Tovmasyan, A.; Batinic-Haberle, I.; Spasojevic, I.; St Clair, D. A Redox-active Mn Porphyrin, MnTnBuOE-2-PyP(5+), Synergizes with Carboplatin in Treatment of Chemoresistant Ovarian Cell Line. Oxidative Med. Cell. Longev. 2022, 2022, 9664636. [Google Scholar] [CrossRef]
- Soares, R.B.; Manguinhas, R.; Costa, J.G.; Saraiva, N.; Gil, N.; Rosell, R.; Camões, S.P.; Batinic-Haberle, I.; Spasojevic, I.; Castro, M.; et al. The Redox-Active Manganese(III) Porphyrin, MnTnBuOE-2-PyP5+, Impairs the Migration and Invasion of Non-Small Cell Lung Cancer Cells, Either Alone or Combined with Cisplatin. Cancers 2023, 15, 3814. [Google Scholar] [CrossRef] [PubMed]
- Aston, K.; Rath, N.; Naik, A.; Slomczynska, U.; Schall, O.F.; Riley, D.P. Computer-aided design (CAD) of Mn(II) complexes: Superoxide dismutase mimetics with catalytic activity exceeding the native enzyme. Inorg. Chem. 2001, 40, 1779–1789. [Google Scholar] [CrossRef]
- Glenn, J.K.; Akileswaran, L.; Gold, M.H. Mn(II) oxidation is the principal function of the extracellular Mn-peroxidase from Phanerochaete chrysosporium. Arch. Biochem. Biophys. 1986, 251, 688–696. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Berlett, B.S.; Chock, P.B. Manganese-dependent disproportionation of hydrogen peroxide in bicarbonate buffer. Proc. Natl. Acad. Sci. USA 1990, 87, 384–388. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Vitturi, D.; Batinic-Haberle, I.; Fridovich, I.; Goldstein, S.; Czapski, G.; Radi, R. Reactions of manganese porphyrins with peroxynitrite and carbonate radical anion. J. Biol. Chem. 2003, 278, 27432–27438. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Ollosson, R.; Stewart, V.C.; Clark, J.B. Oxidation of nitric oxide by oxomanganese-salen complexes: A new mechanism for cellular protection by superoxide dismutase/catalase mimetics. Biochem. J. 2002, 366 Pt 1, 97–107. [Google Scholar] [CrossRef]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Tocco, G.; Malfroy, E.; Adinolfi, C.A.; Kruk, H.; Baker, K.; Lazarowych, N.; Mascarenhas, J.; et al. Salen-manganese complexes as catalytic scavengers of hydrogen peroxide and cytoprotective agents: Structure-activity relationship studies. J. Med. Chem. 2002, 45, 4549–4558. [Google Scholar] [CrossRef]
- Murphy, C.K.; Fey, E.G.; Watkins, B.A.; Wong, V.; Rothstein, D.; Sonis, S.T. Efficacy of superoxide dismutase mimetic M40403 in attenuating radiation-induced oral mucositis in hamsters. Clin. Cancer Res. 2008, 14, 4292–4297. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.S.; Chu, Y.; Glass, J.; Tapp, A.A.; Brown, S.A. The manganese superoxide dismutase mimetic, M40403, protects adult mice from lethal total body irradiation. Free Radic. Res. 2010, 44, 529–540. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Flippo, K.H.; Schoenfeld, J.D.; Riley, D.P.; Strack, S.; Hejleh, T.A.; Furqan, M.; Monga, V.; Domann, F.E.; Buatti, J.M.; et al. Mitochondrial Superoxide Increases Age-Associated Susceptibility of Human Dermal Fibroblasts to Radiation and Chemotherapy. Cancer Res. 2017, 77, 5054–5067. [Google Scholar] [CrossRef]
- Anderson, C.M.; Lee, C.M.; Kelley, J.R.; Walker, G.V.; Dunlap, N.E.; Bar-Ad, V.C.; Miller, D.A.; King, V.J.; Peddada, A.V.; Ciuba, D.F.; et al. ROMAN: Phase 3 trial of avasopasem manganese (GC4419) for severe oral mucositis (SOM) in patients receiving chemoradiotherapy (CRT) for locally advanced, nonmetastatic head and neck cancer (LAHNC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 6005. [Google Scholar] [CrossRef]
- Anderson, C.M.; Lee, C.M.; Saunders, D.P.; Curtis, A.; Dunlap, N.; Nangia, C.; Lee, A.S.; Gordon, S.M.; Kovoor, P.; Arevalo-Araujo, R.; et al. Phase IIb, Randomized, Double-Blind Trial of GC4419 Versus Placebo to Reduce Severe Oral Mucositis Due to Concurrent Radiotherapy and Cisplatin For Head and Neck Cancer. J. Clin. Oncol. 2019, 37, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Sonis, S.T.; Lee, C.M.; Adkins, D.; Allen, B.G.; Sun, W.; Agarwala, S.S.; Venigalla, M.L.; Chen, Y.; Zhen, W.; et al. Phase 1b/2a Trial of the Superoxide Dismutase Mimetic GC4419 to Reduce Chemoradiotherapy-Induced Oral Mucositis in Patients With Oral Cavity or Oropharyngeal Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Vasquez Martinez, G.; Pulliam, C.F.; Petronek, M.S.; Steinbach, E.J.; Monga, V.; Furqan, M.; Jetton, J.G.; Saunders, D.P.; Pearce, A.; et al. Avasopasem manganese (GC4419) protects against cisplatin-induced chronic kidney disease: An exploratory analysis of renal metrics from a randomized phase 2b clinical trial in head and neck cancer patients. Redox Biol. 2023, 60, 102599. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Pulliam, C.F.; Tomanek-Chalkley, A.; Rastogi, P.; Wen, H.; Dayal, S.; Griffin, B.R.; Zepeda-Orozco, D.; Sindler, A.L.; Anderson, C.M.; et al. The antioxidant and anti-inflammatory activities of avasopasem manganese in age-associated, cisplatin-induced renal injury. Redox Biol. 2024, 70, 103022. [Google Scholar] [CrossRef] [PubMed]
- El-Mahdy, M.A.; Alzarie, Y.A.; Hemann, C.; Badary, O.A.; Nofal, S.; Zweier, J.L. The novel SOD mimetic GC4419 increases cancer cell killing with sensitization to ionizing radiation while protecting normal cells. Free Radic. Biol. Med. 2020, 160, 630–642. [Google Scholar] [CrossRef]
- Sishc, B.J.; Ding, L.; Nam, T.K.; Heer, C.D.; Rodman, S.N.; Schoenfeld, J.D.; Fath, M.A.; Saha, D.; Pulliam, C.F.; Langen, B.; et al. Avasopasem manganese synergizes with hypofractionated radiation to ablate tumors through the generation of hydrogen peroxide. Sci. Transl. Med. 2021, 13, eabb3768. [Google Scholar] [CrossRef]
- Zaher, A.; Mapuskar, K.A.; Petronek, M.S.; Tanas, M.R.; Isaacson, A.L.; Dodd, R.D.; Milhem, M.; Furqan, M.; Spitz, D.R.; Miller, B.J.; et al. Superoxide Dismutase Mimetic Avasopasem Manganese Enhances Radiation Therapy Effectiveness in Soft Tissue Sarcomas and Accelerates Wound Healing. Antioxidants 2024, 13, 587. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Liu, G.S.; Thompson, E.W.; Dusting, G.J.; Peshavariya, H.M. Differential effects of superoxide dismutase and superoxide dismutase/catalase mimetics on human breast cancer cells. Breast Cancer Res. Treat. 2015, 150, 523–534. [Google Scholar] [CrossRef]
- Heer, C.D.; Davis, A.B.; Riffe, D.B.; Wagner, B.A.; Falls, K.C.; Allen, B.G.; Buettner, G.R.; Beardsley, R.A.; Riley, D.P.; Spitz, D.R. Superoxide Dismutase Mimetic GC4419 Enhances the Oxidation of Pharmacological Ascorbate and Its Anticancer Effects in an H(2)O(2)-Dependent Manner. Antioxidants 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
| Damage Mechanism | Outcome | Reference |
|---|---|---|
| Mitochondrial Dysfunction | Decreased ATP synthesis, impaired mitochondrial function. | [9] |
| Fenton Chemistry | DNA, lipid, and protein damage. | [10] |
| Protein Modification | Irreversible protein oxidation and fragmentation, mitochondrial dysfunction, hyperactivation or inactivation of essential proteins (e.g., kinases). | [11,12,13] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaher, A.; Petronek, M.S.; Allen, B.G.; Mapuskar, K.A. Balanced Duality: H2O2-Based Therapy in Cancer and Its Protective Effects on Non-Malignant Tissues. Int. J. Mol. Sci. 2024, 25, 8885. https://doi.org/10.3390/ijms25168885
Zaher A, Petronek MS, Allen BG, Mapuskar KA. Balanced Duality: H2O2-Based Therapy in Cancer and Its Protective Effects on Non-Malignant Tissues. International Journal of Molecular Sciences. 2024; 25(16):8885. https://doi.org/10.3390/ijms25168885
Chicago/Turabian StyleZaher, Amira, Michael S. Petronek, Bryan G. Allen, and Kranti A. Mapuskar. 2024. "Balanced Duality: H2O2-Based Therapy in Cancer and Its Protective Effects on Non-Malignant Tissues" International Journal of Molecular Sciences 25, no. 16: 8885. https://doi.org/10.3390/ijms25168885