
The GPR30 Receptor Is Involved in IL-6-Induced Metastatic Properties of MCF-7 Luminal Breast Cancer Cells

 ,
,  and
and
Abstract
:1. Introduction
2. Results
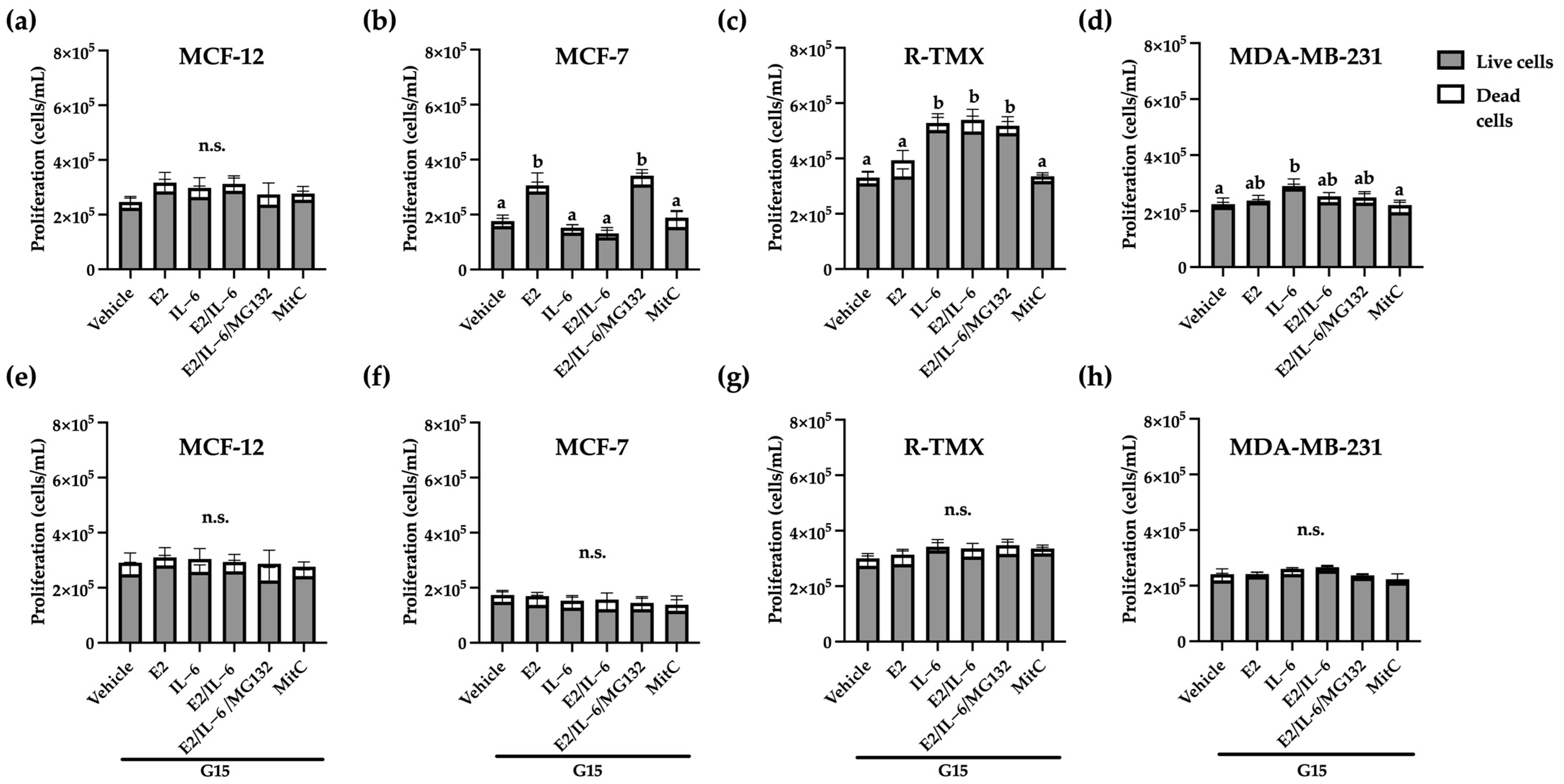
2.1. GPR30 Regulates the Proliferative Effects of E2 and IL-6 in Breast Cancer Cells
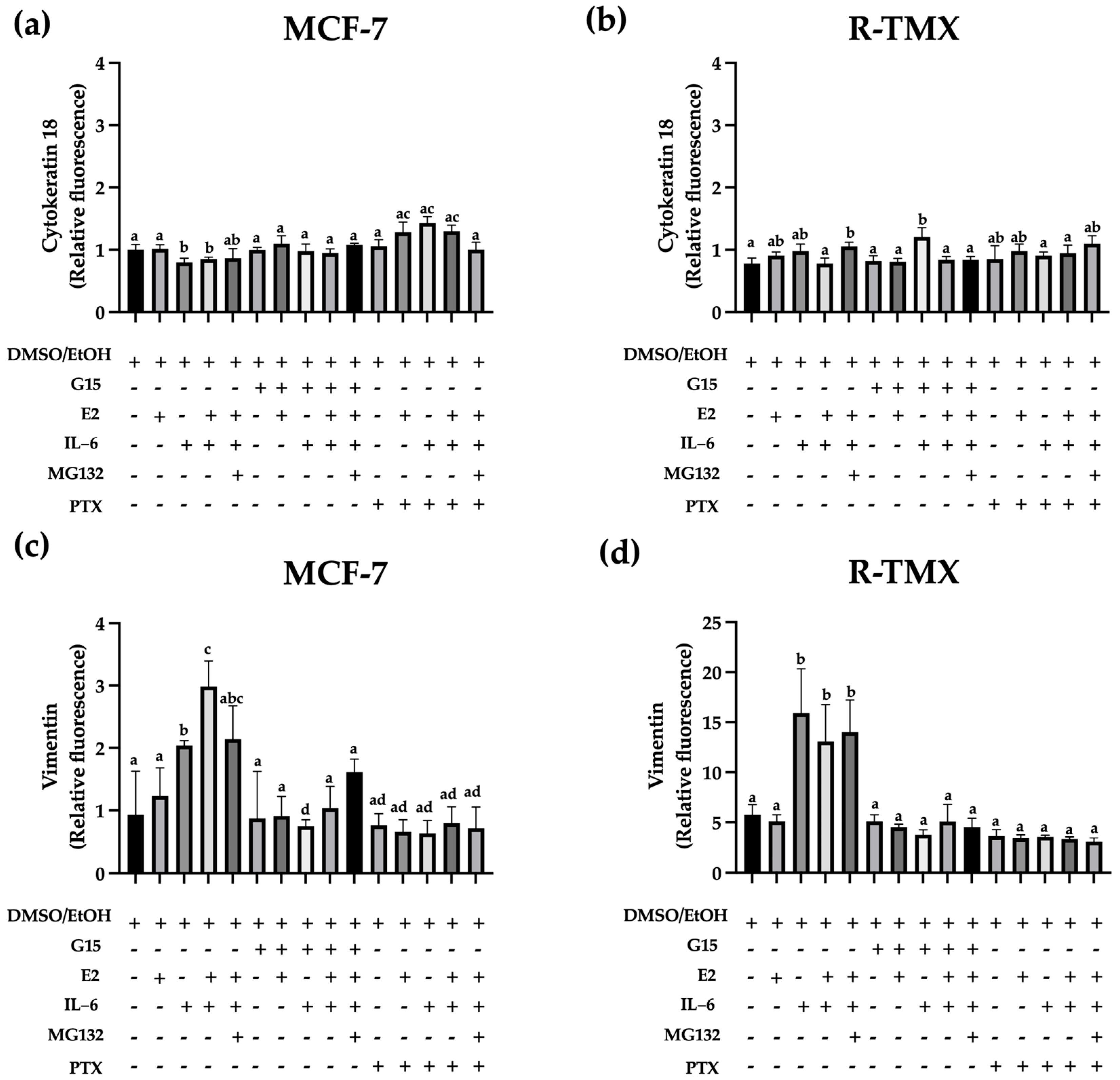
2.2. IL-6 Promotes the Expression of Vimentin through GPR30
2.3. GPR30 Regulates Basal and IL-6-Induced Migration in Breast Cancer Cells
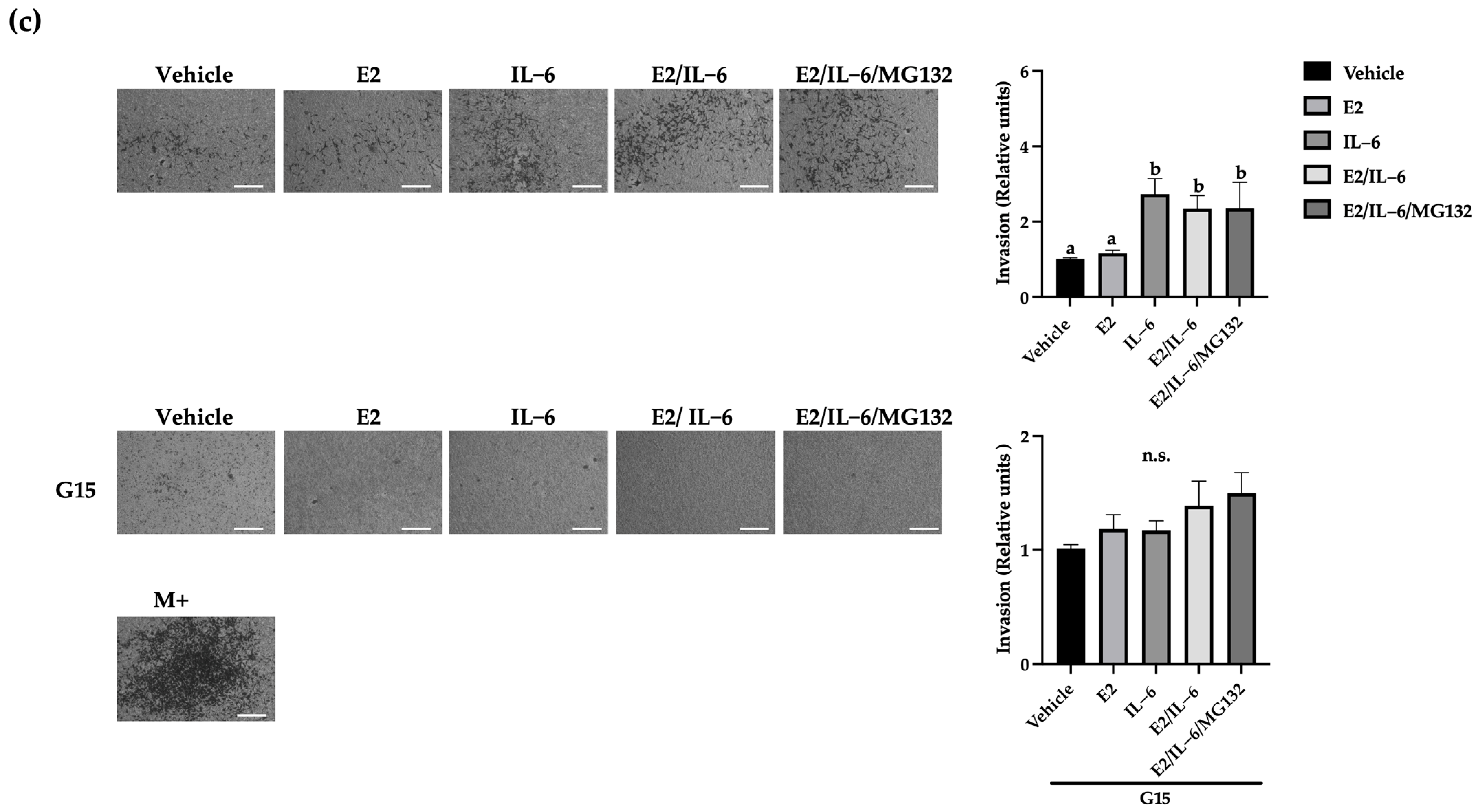
2.4. IL-6 Promotes Breast Cancer Cell Invasion through the GPR30 Receptor
2.5. GPR30 Is Also Involved in the IL-6-Induced TMX Resistance in Luminal Breast Cancer
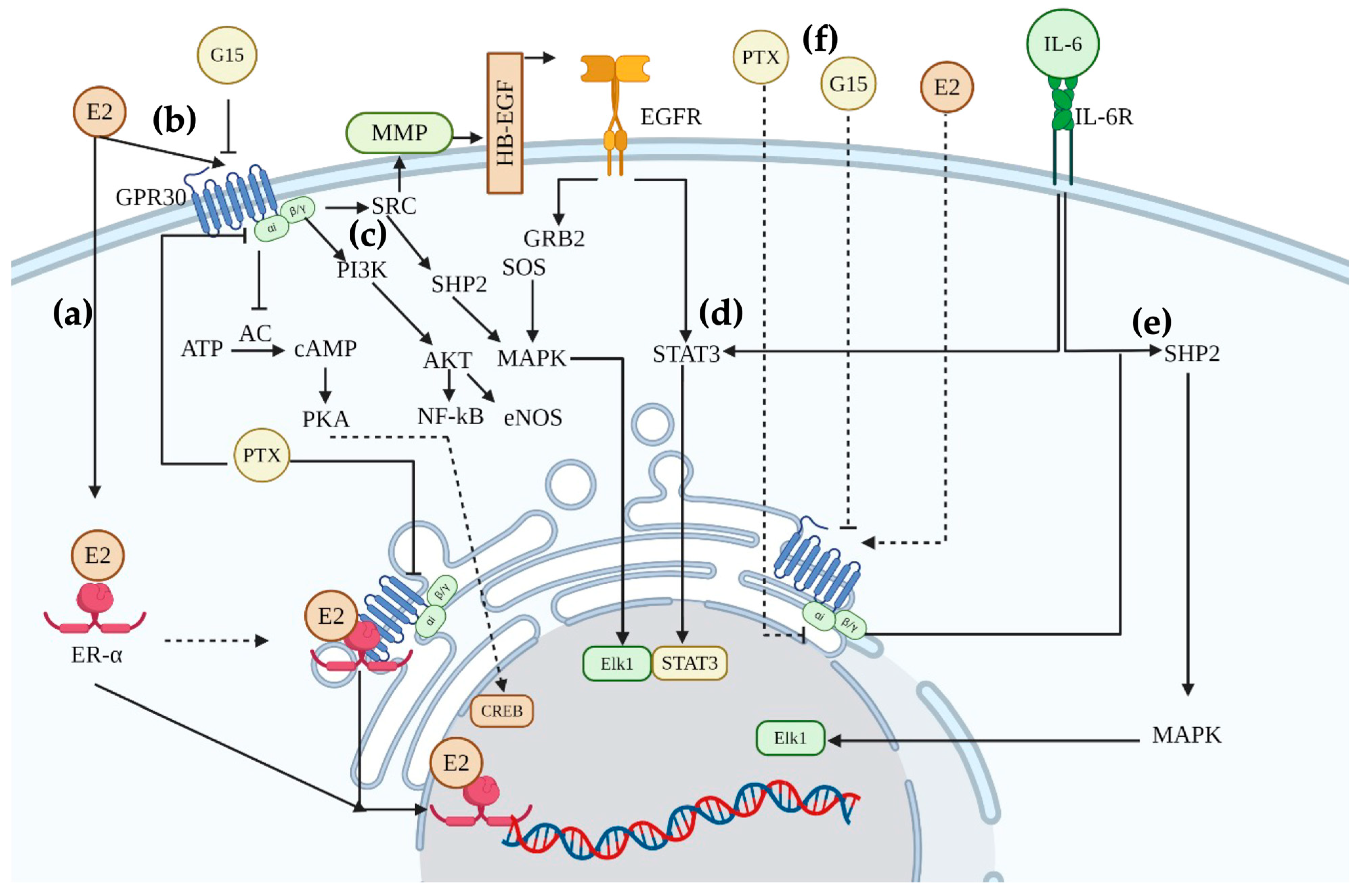
3. Discussion
4. Materials and Methods
4.1. MCF-12, MCF-7, and MDA-MB-231 Cell Culture
4.2. Treatments
4.3. Tamoxifen Resistant Line (R-TMX)
4.4. Proliferation and TMX Cytotoxicity
4.5. Flow Cytometry
4.6. Wound Healing Assay
4.7. Evaluation of Cell Invasion by Transwell Chamber
4.8. Statistic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morra, A.; Jung, A.Y.; Behrens, S.; Keeman, R.; Ahearn, T.U.; Anton-Culver, H.; Arndt, V.; Augustinsson, A.; Auvinen, P.K.; Beane, L.E.; et al. Breast cancer risk factors and survival by tumor subtype: Pooled analyses from the breast cancer association consortium. Cancer Epidemiol. Biomark. Prev. 2021, 30, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Kuske, A.; Joosse, A.A.; Yigit, G.; Sflomos, G.; Thaler, S.; Smit, D.J.; Werner, S.; Borgmann, K.; Gärtner, S.; et al. Characterization of circulating breast cancer cells with tumorigenic and metastatic capacity. EMBO Mol. Med. 2020, 12, e11908. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Zhang, Y.; Merkher, L.; Chen, N.; Liu, S.; Leonov, Y.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Szostakowska, M.; Trębińska-Stryjewska, E.A.; Grzybowska, A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, M.; Gun, X.Y.; Hong, X. Induced tamoxifen resistance is mediated by increased methylation of E-cadherin in estrogen receptor-expressing breast cancer cells. Sci. Rep. 2019, 9, 14140. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Sheikholeslami, S.A.; Jalili, A.; Bereimipour, S.; Sharbati, V.; Kaveh, S.; Salari, S. Investigating the molecular mechanisms of tamoxifen on the EMT pathway among patients with breast cancer. J. Med. Life 2022, 15, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, J.; Lv, W.; Zhao, Y.; Xia, Y.; Wu, Q.; Zhang, Q. The pleiotropic roles of adipocyte secretome in remodeling breast cancer. J. Exp. Clin. Cancer Res. 2022, 41, 203. [Google Scholar] [CrossRef] [PubMed]
- Abaurrea, A.M.; Araujo, M.M.; Caffarel, M. The role of the IL-6 cytokine family in epithelial-mesenchymal plasticity in cancer progression. Int. J. Mol. Sci. 2021, 22, 8334. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Man, E.P.S.; Chau, K.M.; Khoo, U.S. Targeting the IL-6/STAT3 signalling cascade to reverse tamoxifen resistance in estrogen receptor positive breast cancer. Cancers 2021, 13, 1511. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Nakayama, Y.; Yamaguchi, Y. Down-regulation of Forkhead box protein A1 (FOXA1) leads to cancer stem cell-like properties in tamoxifen-resistant breast cancer cells through induction of interleukin-6. J. Biol. Chem. 2017, 292, 8136–8148. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Qi, L.; Kong, Z.; Wang, Y.; Fang, J.; Wang, J. Identification of the significant genes regulated by estrogen receptor in estrogen receptor-positive breast cancer and their expression pattern changes when tamoxifen or fulvestrant resistance occurs. Front. Genet. 2020, 11, 538734. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A. GPER1 and microRNA: Two players in breast cancer progression. Int. J. Mol. Sci. 2020, 22, 98. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, G.E.; Forcados, A.P.; Yusuf, M.B.; Abubakar, I.Z.; Sadiq, I.; Elhussin, M.A.T.; Siddique, S.; Aminu, R.B.; Suleiman, Y.S.; Abubakar, B.S.; et al. Comparative G-protein-coupled estrogen receptor (GPER) systems in diabetic and cancer conditions: A Review. Molecules 2022, 27, 8943. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Liu, M.; Yang, H.; Luo, H.; Li, G.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Xiong, W.; Wang, M.; Yang, J.; Shi, H.; Chen, H.; Gang, N. Nuclear G Protein-Coupled oestrogen receptor (GPER) predicts poor survival in patients with ovarian cancer. J. Int. Med. Res. 2018, 46, 723–731. [Google Scholar] [CrossRef]
- Xu, S.; Yu, D.; Dong, L.; Lee, T. G protein-coupled estrogen receptor: A potential therapeutic target in cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, M.; Jaymand, S.; Kolahian, T.; Javaheri, P.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Srivastava, A.; Pateriya, M.S.; Tomar, A.K.; Mishra, A.; Shrivastava, A. Metabolic reprogramming confers tamoxifen resistance in breast cancer. Chem.-Biol. Interact. 2021, 347, 109602. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Kyuno, D.; Takasawa, A.; Kikuchi, I.; Takemas, M.; Osanai, T.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Menz, A.; Weitbrecht, T.; Gorbokon, N.; Büscheck, F.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Hinsch, D.; Höflmayer, S.; Weidemann, C.; et al. Diagnostic and prognostic impact of cytokeratin 18 expression in human tumors: A tissue microarray study on 11,952 Molecular Medicine. Mol. Med. 2021, 27, 16. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, F1000 Faculty Rev-1796. [Google Scholar] [CrossRef] [PubMed]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer invasion: Patterns and mechanisms. Acta Naturae 2015, 7, 17–28. [Google Scholar] [CrossRef]
- Xie, G.; Ji, A.; Yuan, Q.; Jin, Z.; Yuan, Y.; Ren, C.; Guo, Q.; Yao, K.; Yang, X.; Lin, L.; et al. Tumour-initiating capacity is independent of epithelial-mesenchymal transition status in breast cancer cell lines. Br. J. Cancer 2014, 110, 2514–2523. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Weng, X.; Sharma, S.; Awate, P. Pleiotropic mechanisms drive endocrine resistance in the three-dimensional bone microenvironment. Cancer Res. 2020, 81, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the understanding of the mechanism of tamoxifen resistance in breast cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wei, Y.; Yang, Q.; Huang, Y.; Chen, K.; Zeng, J.; Chen, J. IL-6: The link between inflammation, immunity and breast cancer. Front. Oncol. 2022, 12, 903800. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Liao, Q.; Wang, Q. Activation of GPER by E2 promotes proliferation, invasion and migration of breast cancer cells by regulating the miR-124/CD151 pathway. Oncol. Lett. 2021, 21, 432. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.N.; Gorelick, D.A. Crosstalk between nuclear and G protein-coupled estrogen receptors. Gen. Comp. Endocrinol. 2018, 261, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Wang, C.; Fu, N.; Liu, L.; Zhu, D.; Wei, Z.; Zhang, H.; Xing, J.; Wang, Y. Downregulation of cytokeratin 18 enhances BCRP-mediated multidrug resistance through induction of epithelial-mesenchymal transition and predicts poor prognosis in breast cancer. Oncol. Rep. 2019, 41, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ma, D.; Chen, S.; Tang, R.; Yang, J.; Meng, C.; Feng, Y.; Liu, L.; Wang, J.; Luo, H.; et al. High GPER expression in triple-negative breast cancer is linked to pro-metastatic pathways and predicts poor patient outcomes. NPJ Breast Cancer 2022, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Tutzauer, J.; Sjöström, M.; Bendahl, P.-O.; Rydén, L.; Fernö, M.; Leeb-Lundberg, L.M.F.; Alkner, S. Plasma membrane expression of G protein-coupled estrogen receptor (GPER)/G protein-coupled receptor 30 (GPR30) is associated with worse outcome in metachronous contralateral breast cancer. PLoS ONE 2020, 15, e0231786. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER function in breast cancer: An overview. Frontiers 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Zhou, X.; Chen, X.; Santos, G.S.; Peuget, S.; Cheng, Q.; Rihani, A.; Arnér, E.S.J.; Hartman, J.; Selivanova1, G. Identification and targeting of selective vulnerability rendered by tamoxifen resistance. Breast Cancer Res. 2020, 22, 80. [Google Scholar] [CrossRef] [PubMed]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous exposure of breast cancer cells to tamoxifen upregulates GPER-1 and increases cell proliferation. Front. Endocrinol. 2020, 11, 563165. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Conditions | IC50 TMX (μM) |
|---|---|
| MCF-7 | 3.8 |
| MCF-7 + G15 | 4.26 |
| MCF-7 + IL-6 (24 h) | 10.98 * |
| MCF-7 + IL-6 (48 h) | 12.69 * |
| MCF-7 + IL-6 (24 h) + G15 | 3.87 |
| MCF-7 + IL-6 (48 h) + G15 | 5.08 * |
| R-TMX | >20 * |
| R-TMX + G15 | 9.05 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirado-Garibay, A.C.; Ruiz-Barcenas, B.; Rescala-Ponce de León, J.I.; Ochoa-Zarzosa, A.; López-Meza, J.E. The GPR30 Receptor Is Involved in IL-6-Induced Metastatic Properties of MCF-7 Luminal Breast Cancer Cells. Int. J. Mol. Sci. 2024, 25, 8988. https://doi.org/10.3390/ijms25168988
Tirado-Garibay AC, Ruiz-Barcenas B, Rescala-Ponce de León JI, Ochoa-Zarzosa A, López-Meza JE. The GPR30 Receptor Is Involved in IL-6-Induced Metastatic Properties of MCF-7 Luminal Breast Cancer Cells. International Journal of Molecular Sciences. 2024; 25(16):8988. https://doi.org/10.3390/ijms25168988
Chicago/Turabian StyleTirado-Garibay, Ana Carolina, Betzabe Ruiz-Barcenas, Julia Isabel Rescala-Ponce de León, Alejandra Ochoa-Zarzosa, and Joel E. López-Meza. 2024. "The GPR30 Receptor Is Involved in IL-6-Induced Metastatic Properties of MCF-7 Luminal Breast Cancer Cells" International Journal of Molecular Sciences 25, no. 16: 8988. https://doi.org/10.3390/ijms25168988