
STAT3 Increases CVB3 Replication and Acute Pancreatitis and Myocarditis Pathology via Impeding Nuclear Translocation of STAT1 and Interferon-Stimulated Gene Expression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
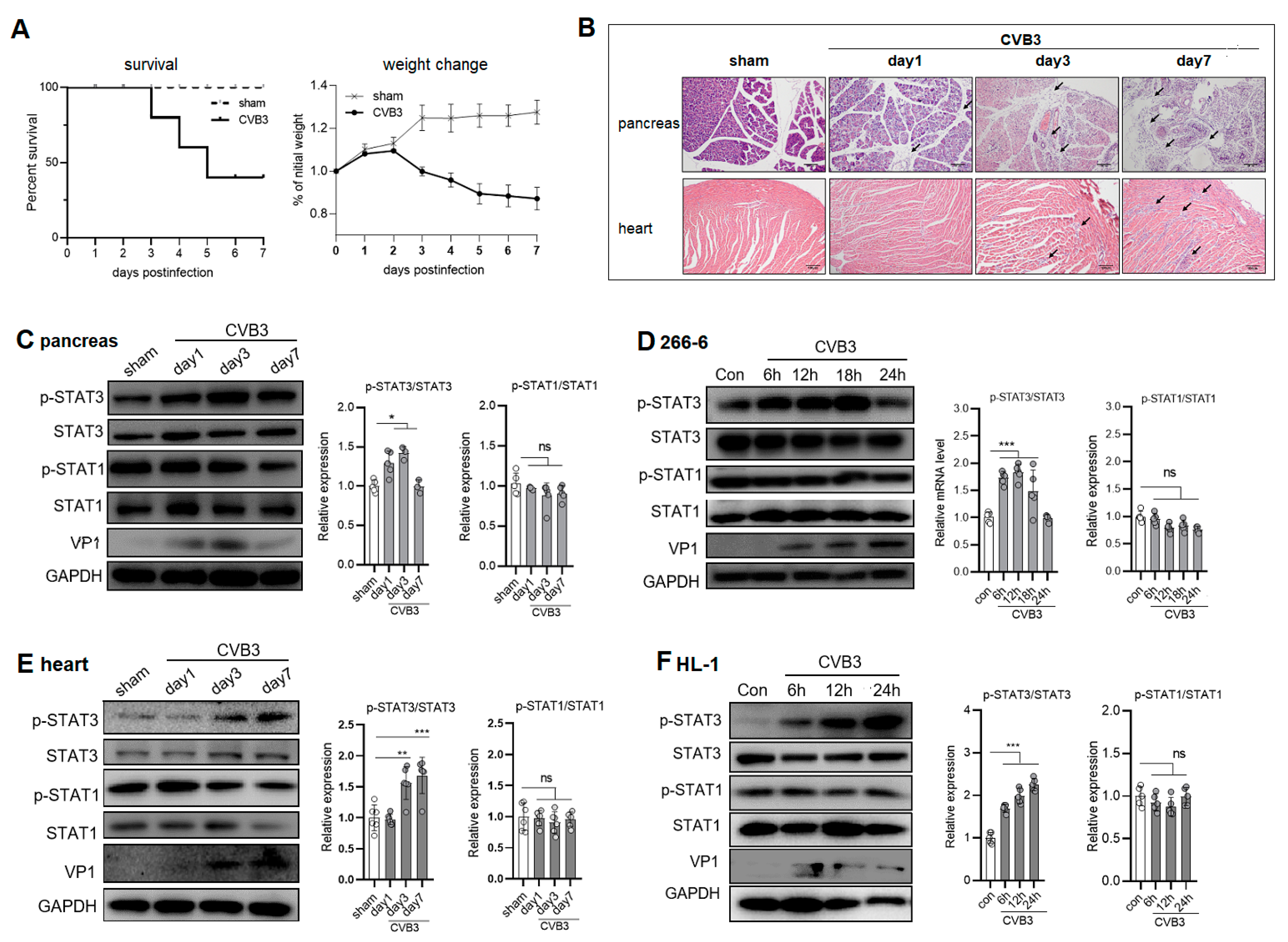
2.1. CVB3 Upregulates p-STAT3 but Not p-STAT1 Expression in the Pancreases and Hearts of Mice
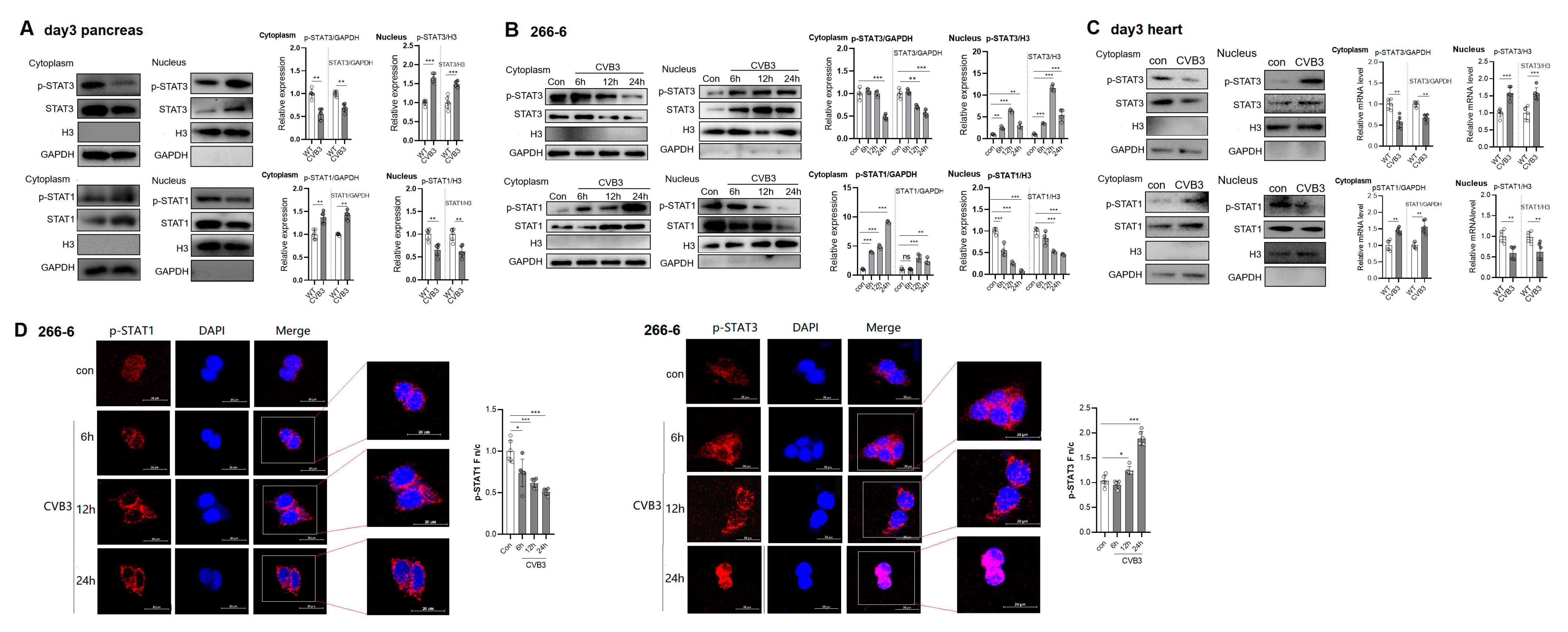
2.2. CVB3 Infection Increases Nuclear Import of p-STAT3 but Blunts Nuclear Translocation of p-STAT1
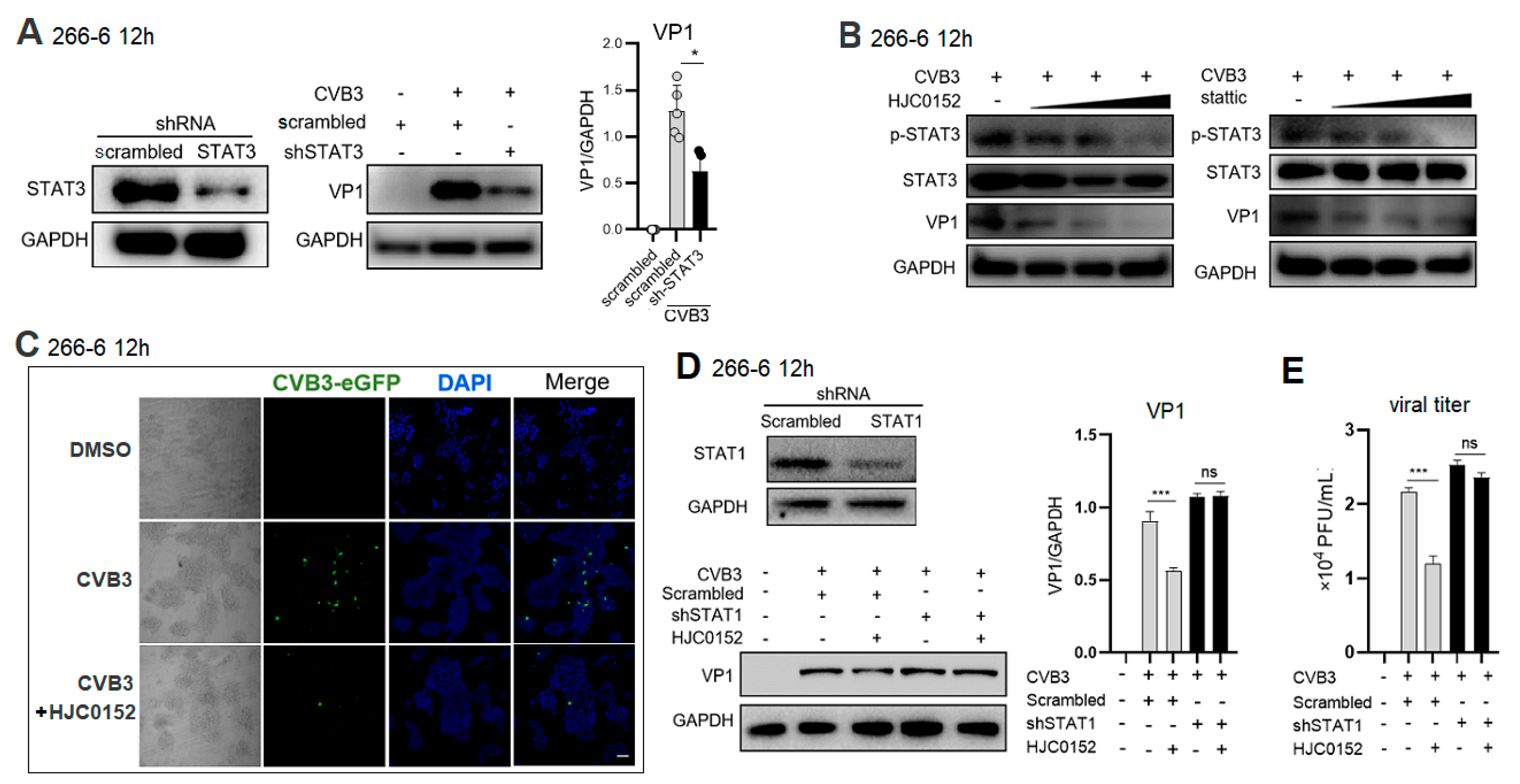
2.3. STAT3 Inhibition Rescues Nuclear Translocation of p-STAT1 and Enhances ISG Expression in CVB3-Infected Cell
2.4. STAT3 Inhibitor Restricts CVB3 Replication via STAT1-Dependent Way
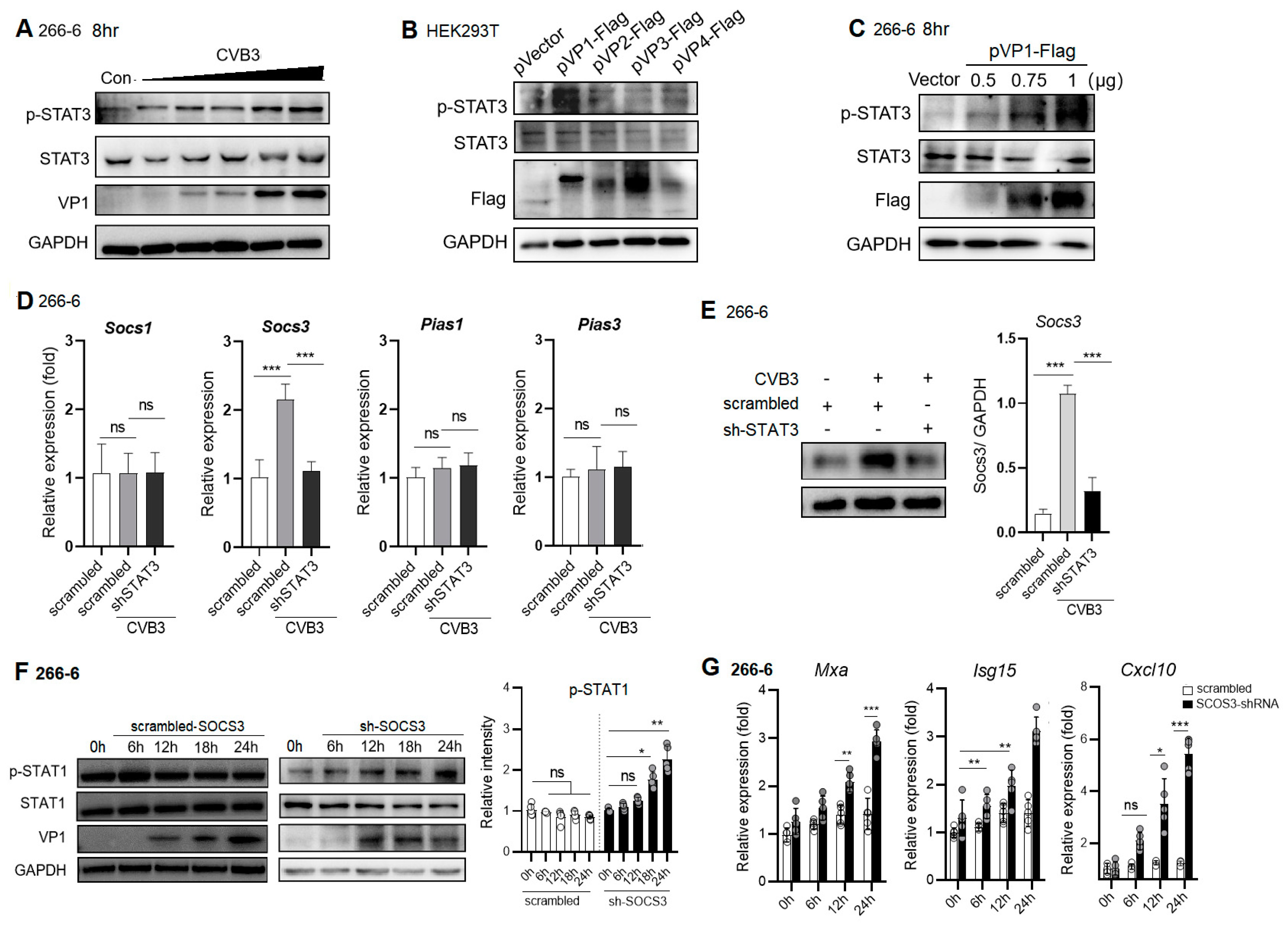
2.5. CVB3 VP1 Induces STAT3 Activation, Which Induces Expression of Negative Regulator, SOCS3, to Block pSTAT1/ISG Expression
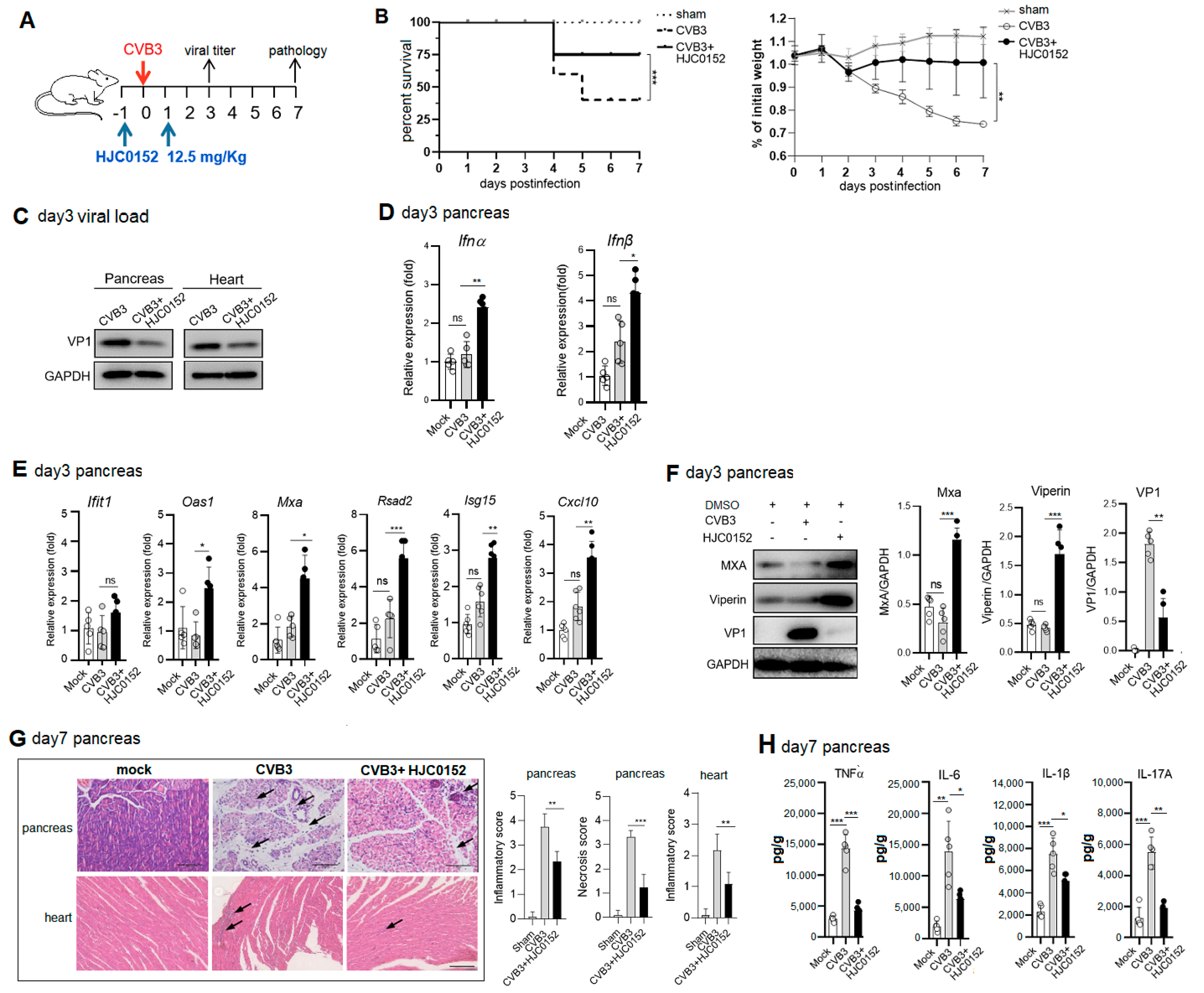
2.6. Therapeutic STAT3 Inhibitor Treatment Mitigates AP and VMC Pathology via Rescuing ISG Induction
3. Discussion
4. Materials and Methods
4.1. Animals, Virus and Cells
4.2. CVB3-Induced Acute Pancreatitis and Histopathology Evaluation
4.3. Cell Treatment and Virus Infection
4.4. Plaque Assay
4.5. Western Blot
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Nucleic and Cytoplasmic Fraction Purification
4.8. Luciferase Reporter Assay
4.9. shRNA Knockdown
4.10. Quantitative Real-Time PCR (qPCR)
4.11. STAT3 Inhibitor Treatment Experiment
4.12. Immunofluorescent Staining
4.13. Confocal Laser Scanning Microscopy
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boxhoorn, L.; Voermans, R.P.; Bouwense, S.A.; Bruno, M.J.; Verdonk, R.C.; Boermeester, M.A.; Van Santvoort, H.C.; Besselink, M.G. Acute pancreatitis. Lancet 2020, 396, 726–734. [Google Scholar] [CrossRef]
- Habtezion, A. Inflammation in acute and chronic pancreatitis. Curr. Opin. Gastroenterol. 2015, 31, 395–399. [Google Scholar] [CrossRef]
- Huber, S.; Ramsingh, A.I. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004, 17, 358–369. [Google Scholar] [CrossRef]
- Szatmary, P.; Grammatikopoulos, T.; Cai, W.; Huang, W.; Mukherjee, R.; Halloran, C.; Beyer, G.; Sutton, R. Acute Pancreatitis: Diagnosis and Treatment. Drugs 2022, 82, 1251–1276. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Schoggins, J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Research 2018, 7, 309. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Shiratori, T.; Imaizumi, T.; Hirono, K.; Kawaguchi, S.; Matsumiya, T.; Seya, K.; Tasaka, S. ISG56 is involved in CXCL10 expression induced by TLR3 signaling in BEAS-2B bronchial epithelial cells. Exp. Lung. Res. 2020, 46, 195–202. [Google Scholar] [CrossRef]
- Dzimianski, J.V.; Scholte, F.E.M.; Bergeron, É.; Pegan, S.D. ISG15: It’s Complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef]
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Överby, A.K. Viperin Restricts Zika Virus and Tick-Borne Encephalitis Virus Replication by Targeting NS3 for Proteasomal Degradation. J. Virol. 2018, 92, e02054-17. [Google Scholar] [CrossRef]
- Grabowski, F.; Kochańczyk, M.; Korwek, Z.; Czerkies, M.; Prus, W.; Lipniacki, T. Antagonism between viral infection and innate immunity at the single-cell level. PLoS Pathog. 2023, 19, e1011597. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Kalinke, U. Interferon-beta expression and type I interferon receptor signaling of hepatocytes prevent hepatic necrosis and virus dissemination in Coxsackievirus B3-infected mice. PLoS Pathog. 2018, 14, e1007235. [Google Scholar]
- Liu, H.; Li, M.; Song, Y.; Xu, W. TRIM21 Restricts Coxsackievirus B3 Replication, Cardiac and Pancreatic Injury via Interacting With MAVS and Positively Regulating IRF3-Mediated Type-I Interferon Production. Front. Immunol. 2018, 9, 2479. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Rauch, U. Protease-activated receptor-2 regulates the innate immune response to viral infection in a coxsackievirus B3-induced myocarditis. J. Am. Coll. Cardiol. 2013, 62, 1737–1745. [Google Scholar]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Mdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; De Groot, R.J.; Van Kuppeveld, F.J.M. Essential Role of Enterovirus 2A Protease in Counteracting Stress Granule Formation and the Induction of Type I Interferon. J. Virol. 2019, 93, e00222-19. [Google Scholar] [CrossRef]
- Ruppert, V.; Meyer, T.; Pankuweit, S.; Jonsdottir, T.; Maisch, B. Activation of STAT1 transcription factor precedes up-regulation of coxsackievirus-adenovirus receptor during viral myocarditis. Cardiovasc. Pathol. 2008, 17, 81–92. [Google Scholar] [CrossRef]
- Antoniak, S.; Owens, A.P.; Baunacke, M.; Williams, J.C.; Lee, R.D.; Weithäuser, A.; Sheridan, P.A.; Malz, R.; Luyendyk, J.P.; Esserman, D.A.; et al. PAR-1 contributes to the innate immune response during viral infection. J. Clin. Investig. 2013, 123, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Carcereri De Prati, A.; Mariotto, S. Redox Regulation of STAT1 and STAT3 Signaling. Int. J. Mol. Sci. 2020, 21, 7034. [Google Scholar] [CrossRef] [PubMed]
- Regis, G.; Pensa, S.; Boselli, D.; Novelli, F.; Poli, V. Ups and downs: The STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin. Cell. Dev. Biol. 2008, 19, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.N.; Tassiulas, I.; Hu, Y.; Mecklenbräuker, I.; Tarakhovsky, A.; Ivashkiv, L.B. IFN-alpha priming results in a gain of proinflammatory function by IL-10: Implications for systemic lupus erythematosus pathogenesis. J. Immunol. 2004, 172, 6476–6481. [Google Scholar] [CrossRef] [PubMed]
- Herrero, C.; Hu, X.; Li, W.P.; Samuels, S.; Sharif, M.N.; Kotenko, S.; Ivashkiv, L.B. Reprogramming of IL-10 activity and signaling by IFN-gamma. J. Immunol. 2003, 171, 5034–5041. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, Q.; Ye, Q.; Wang, J.; Dong, Q.; Chen, Y.; Wang, M.; Fu, Y.; Wu, R.; Wu, T. STAT3 Suppresses Cardiomyocytes Apoptosis in CVB3-Induced Myocarditis via Survivin. Front. Pharmacol. 2020, 11, 613883. [Google Scholar] [CrossRef]
- Ho, H.H.; Ivashkiv, L.B. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef]
- Tsai, M.H.; Pai, L.M.; Lee, C.K. Fine-Tuning of Type I Interferon Response by STAT3. Front. Immunol. 2019, 10, 448. [Google Scholar] [CrossRef]
- Wang, W.B.; Levy, D.E.; Lee, C.K. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, M.; Wang, S.; Zhang, L.; Zhang, R.; Zou, X.; Wang, X.; Chen, D.; Wu, Z. STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1. Int. J. Mol. Sci. 2019, 20, 4870. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, H.; Wang, P.; Wang, J.; Zou, L. The roles of SOCS3 and STAT3 in bacterial infection and inflammatory diseases. Scand. J. Immunol. 2018, 88, e12727. [Google Scholar] [CrossRef]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef]
- Weber, M.; Sediri, H.; Felgenhauer, U.; Binzen, I.; Bänfer, S.; Jacob, R.; Brunotte, L.; García-Sastre, A.; Schmid-Burgk, J.L.; Schmidt, T.; et al. Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 2015, 17, 309–319. [Google Scholar] [CrossRef]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H.; et al. Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q.; Jia, X.; Cheng, C.Q.; Wang, Y.; Li, Y.Y.; Kong, L.D.; Li, Q.Q.; Xie, F.; Yu, Y.L.; He, Y.T.; et al. Capsaicin functions as a selective degrader of STAT3 to enhance host resistance to viral infection. Acta Pharmacol. Sin. 2023, 44, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2017, 7, e1001311. [Google Scholar]
- Andrews, D.D.T.; Vlok, M.; Akbari Bani, D.; Hay, B.N.; Mohamud, Y.; Foster, L.J.; Luo, H.; Overall, C.M.; Jan, E. Cleavage of 14-3-3ε by the enteroviral 3C protease dampens RIG-I-mediated antiviral signaling. J. Virol. 2023, 97, e0060423. [Google Scholar] [CrossRef]
- Tolomeo, M.; Cavalli, A.; Cascio, A. STAT1 and Its Crucial Role in the Control of Viral Infections. Int. J. Mol. Sci. 2022, 23, 4095. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Zhuang, Z.; Cai, S.; Zhao, Z.; Zhou, L.; Zhang, J.; Wang, P.H.; Zhao, J.; Cui, J. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct. Target. Ther. 2020, 5, 221. [Google Scholar] [CrossRef]
- Lan, K.H.; Lan, K.L.; Lee, W.P.; Sheu, M.L.; Chen, M.Y.; Lee, Y.L.; Yen, S.H.; Chang, F.Y.; Lee, S.D. HCV NS5A inhibits interferon-alpha signaling through suppression of STAT1 phosphorylation in hepatocyte-derived cell lines. J. Hepatol. 2007, 46, 59–67. [Google Scholar] [CrossRef]
- Sun, M.; Lin, Q.; Wang, C.; Xing, J.; Yan, K.; Liu, Z.; Jin, Y.; Cardona, C.J.; Xing, Z. Enterovirus A71 2B Inhibits Interferon-Activated JAK/STAT Signaling by Inducing Caspase-3-Dependent Karyopherin-α1 Degradation. Front. Microbiol. 2021, 12, 762869. [Google Scholar] [CrossRef]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Kuchipudi, S.V. The Complex Role of STAT3 in Viral Infections. J. Immunol. Res. 2015, 2015, 272359. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Nemati, M.; Jafarzadeh, S. Contribution of STAT3 to the pathogenesis of COVID-19. Microb. Pathog. 2021, 154, 104836. [Google Scholar] [CrossRef] [PubMed]
- Hsia, H.C.; Stopford, C.M.; Zhang, Z.; Damania, B.; Baldwin, A.S. Signal transducer and activator of transcription 3 (Stat3) regulates host defense and protects mice against herpes simplex virus-1 (HSV-1) infection. J. Leukoc. Biol. 2017, 101, 1053–1064. [Google Scholar] [CrossRef]
- Chang, Z.; Wang, Y.; Bian, L.; Liu, Q.; Long, J.E. Enterovirus 71 antagonizes the antiviral activity of host STAT3 and IL-6R with partial dependence on virus-induced miR-124. J. Gen. Virol. 2017, 98, 3008–3025. [Google Scholar] [CrossRef] [PubMed]
- Mahony, R.; Gargan, S.; Roberts, K.L. A novel anti-viral role for STAT3 in IFN-α signalling responses. Cell Mol. Life Sci. 2017, 74, 1755–1764. [Google Scholar] [CrossRef]
- Gan, C.J.; Li, W.F.; Li, C.N.; Li, L.L.; Zhou, W.Y.; Peng, X.M. EGF receptor inhibitors comprehensively suppress hepatitis B virus by downregulation of STAT3 phosphorylation. Biochem. Biophys. Rep. 2020, 22, 100763. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Wang, Y.; Liu, P.; Liu, K.; Sun, J.; Zhang, P.; Wang, X.; Liu, X.; Xu, X. Influenza A virus infection activates STAT3 to enhance SREBP2 expression, cholesterol biosynthesis, and virus replication. iScience 2024, 27, 110424. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Che, X.; Rajamani, J.; Zerboni, L.; Sung, P.; Ptacek, J.; Arvin, A.M. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 600–6055. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, J.M.; Sato, H.; Nevels, M.; Terhune, S.S.; Paulus, C. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J. Virol. 2013, 87, 10763–10776. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, Y.; Luo, Y.; Chen, X.; Gong, T.; Wu, D.; Feng, Y.; Zheng, X.; Wang, H.; Zhang, G.; et al. African Swine Fever Virus Envelope Glycoprotein CD2v Interacts with Host CSF2RA to Regulate the JAK2-STAT3 Pathway and Inhibit Apoptosis to Facilitate Virus Replication. J. Virol. 2023, 97, e0188922. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Lindner, D.; Hilbrandt, M.; Marggraf, K.; Becher, P.M.; Hilfiker-Kleiner, D.; Klingel, K.; Pauschinger, M.; Schultheiss, H.P.; Tschöpe; Westermann, D. Protective Function of STAT3 in CVB3-Induced Myocarditis. Cardiol. Res. Pract. 2012, 2012, 437623. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Lee, C.K. STAT3 Cooperates With Phospholipid Scramblase 2 to Suppress Type I Interferon Response. Front. Immunol. 2018, 9, 1886. [Google Scholar] [CrossRef]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, T.; Zhang, Z.; Bai, Z.; Xu, L.; Xu, W. STAT3 Increases CVB3 Replication and Acute Pancreatitis and Myocarditis Pathology via Impeding Nuclear Translocation of STAT1 and Interferon-Stimulated Gene Expression. Int. J. Mol. Sci. 2024, 25, 9007. https://doi.org/10.3390/ijms25169007
Liang T, Zhang Z, Bai Z, Xu L, Xu W. STAT3 Increases CVB3 Replication and Acute Pancreatitis and Myocarditis Pathology via Impeding Nuclear Translocation of STAT1 and Interferon-Stimulated Gene Expression. International Journal of Molecular Sciences. 2024; 25(16):9007. https://doi.org/10.3390/ijms25169007
Chicago/Turabian StyleLiang, Tianming, Zhipeng Zhang, Zhenxin Bai, Le Xu, and Wei Xu. 2024. "STAT3 Increases CVB3 Replication and Acute Pancreatitis and Myocarditis Pathology via Impeding Nuclear Translocation of STAT1 and Interferon-Stimulated Gene Expression" International Journal of Molecular Sciences 25, no. 16: 9007. https://doi.org/10.3390/ijms25169007
APA StyleLiang, T., Zhang, Z., Bai, Z., Xu, L., & Xu, W. (2024). STAT3 Increases CVB3 Replication and Acute Pancreatitis and Myocarditis Pathology via Impeding Nuclear Translocation of STAT1 and Interferon-Stimulated Gene Expression. International Journal of Molecular Sciences, 25(16), 9007. https://doi.org/10.3390/ijms25169007