
Genome Resequencing for Autotetraploid Rice and Its Closest Relatives Reveals Abundant Variation and High Potential in Rice Breeding

 ,
,
Abstract
1. Introduction
2. Results
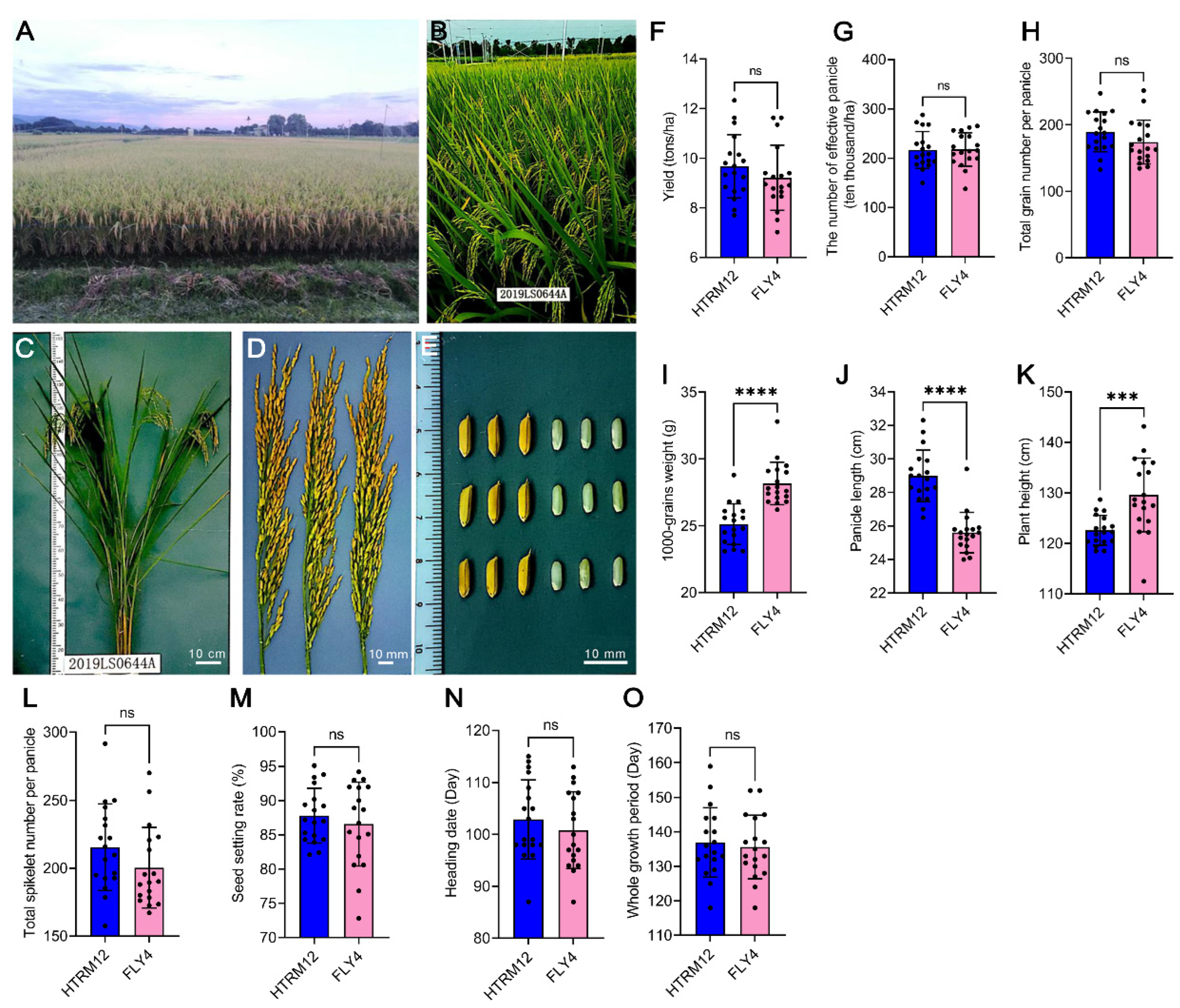
2.1. Breeding and Field Performance of Superior Hybrid Rice HTRM12
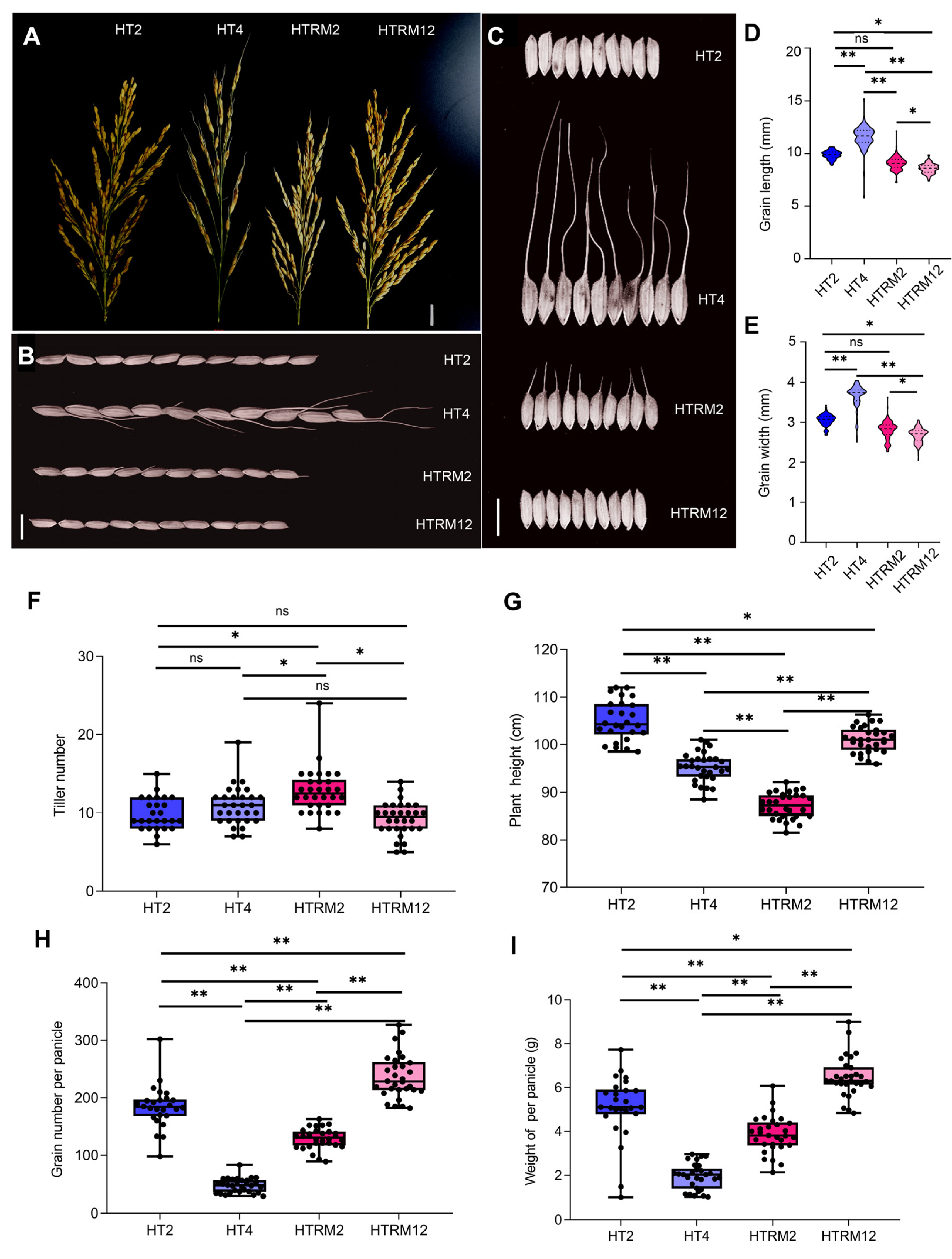
2.2. Comparison of Agronomic Traits of HT2, HT4, HTRM2, and HTRM12
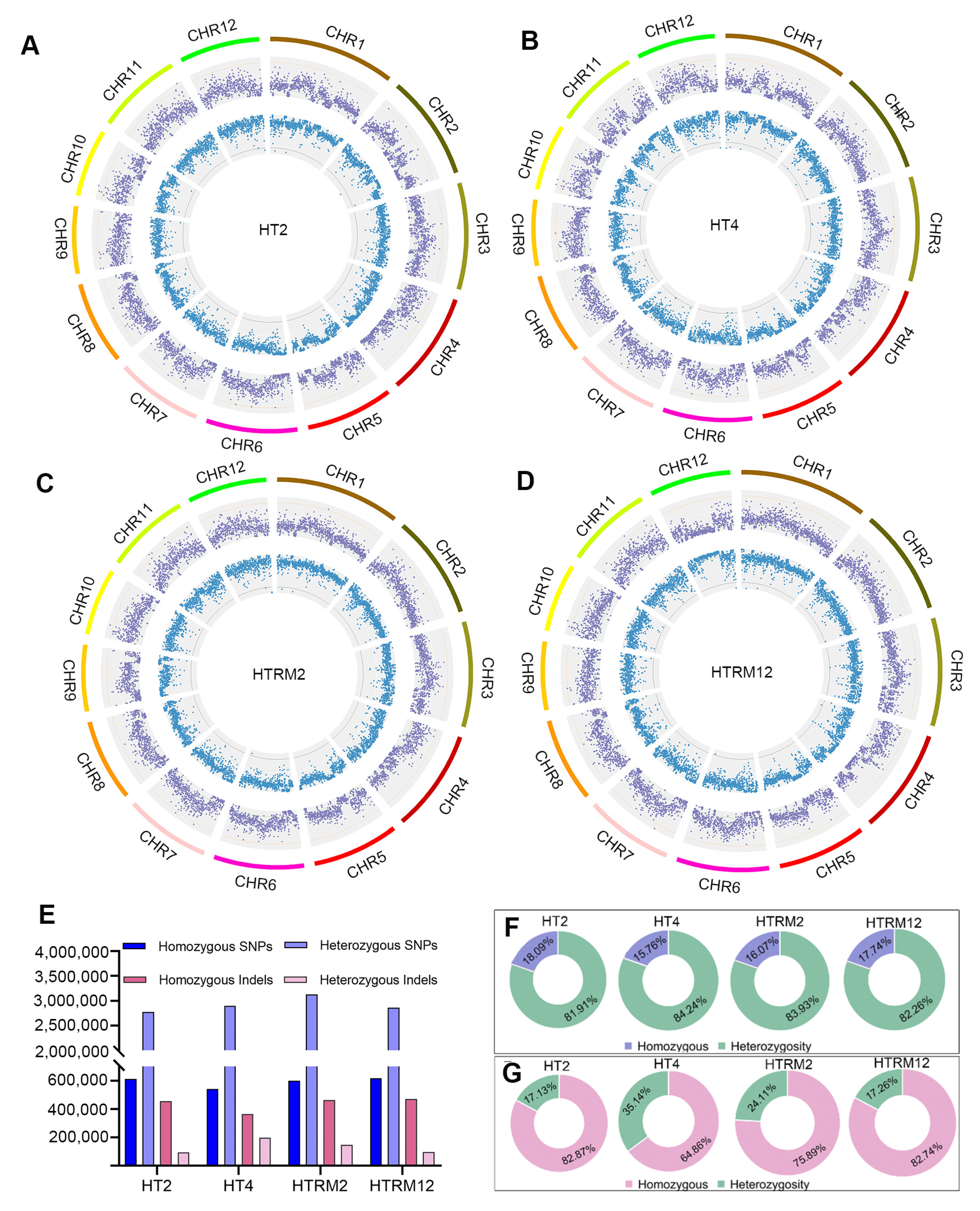
2.3. Genome-Wide Polymorphism Detection in HT2, HT4, HTRM2, and HTRM12
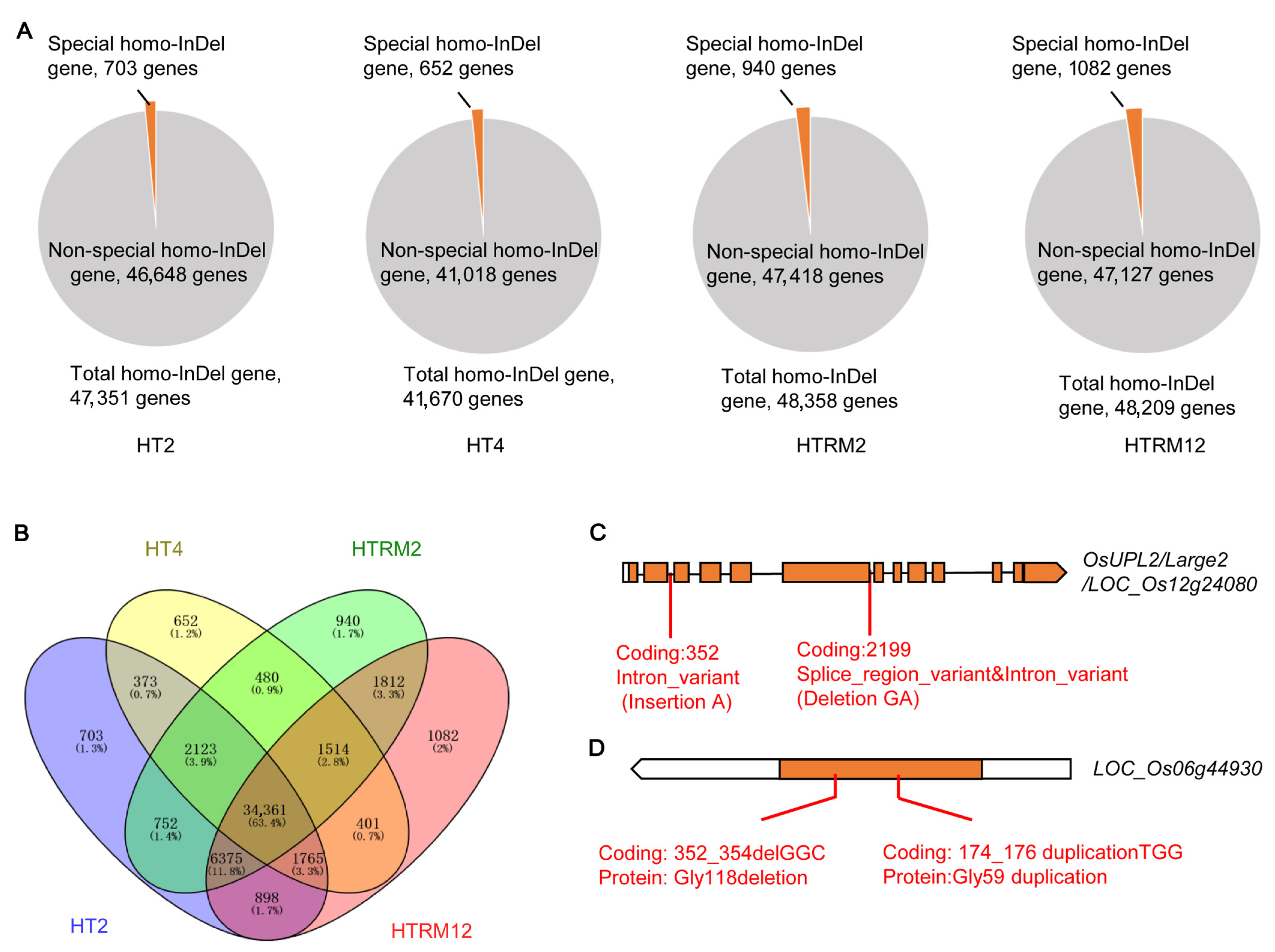
2.4. SNP and InDel Site Annotation and Mutation Type Analysis of HT2, HT4, HTRM2, and HTRM12
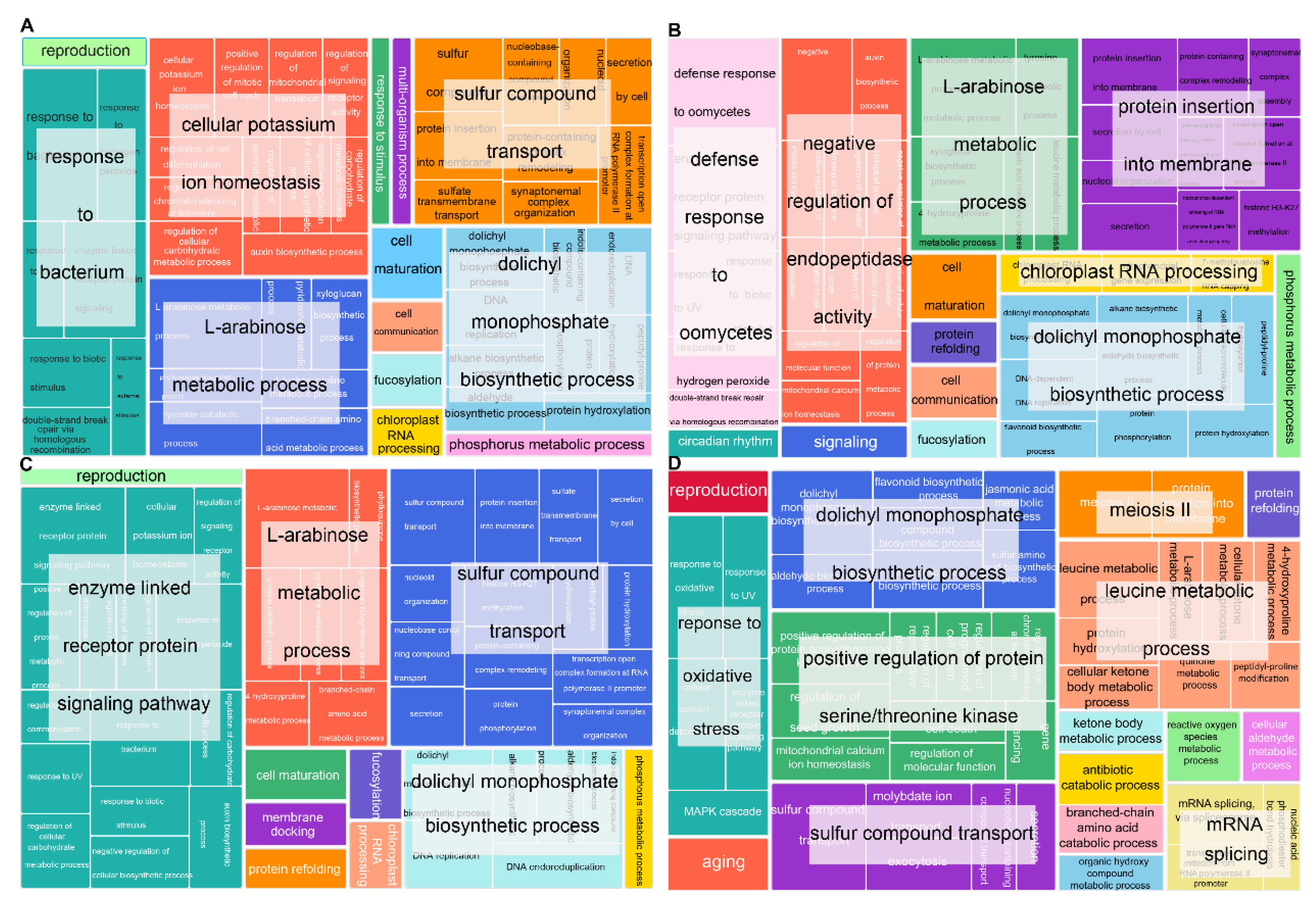
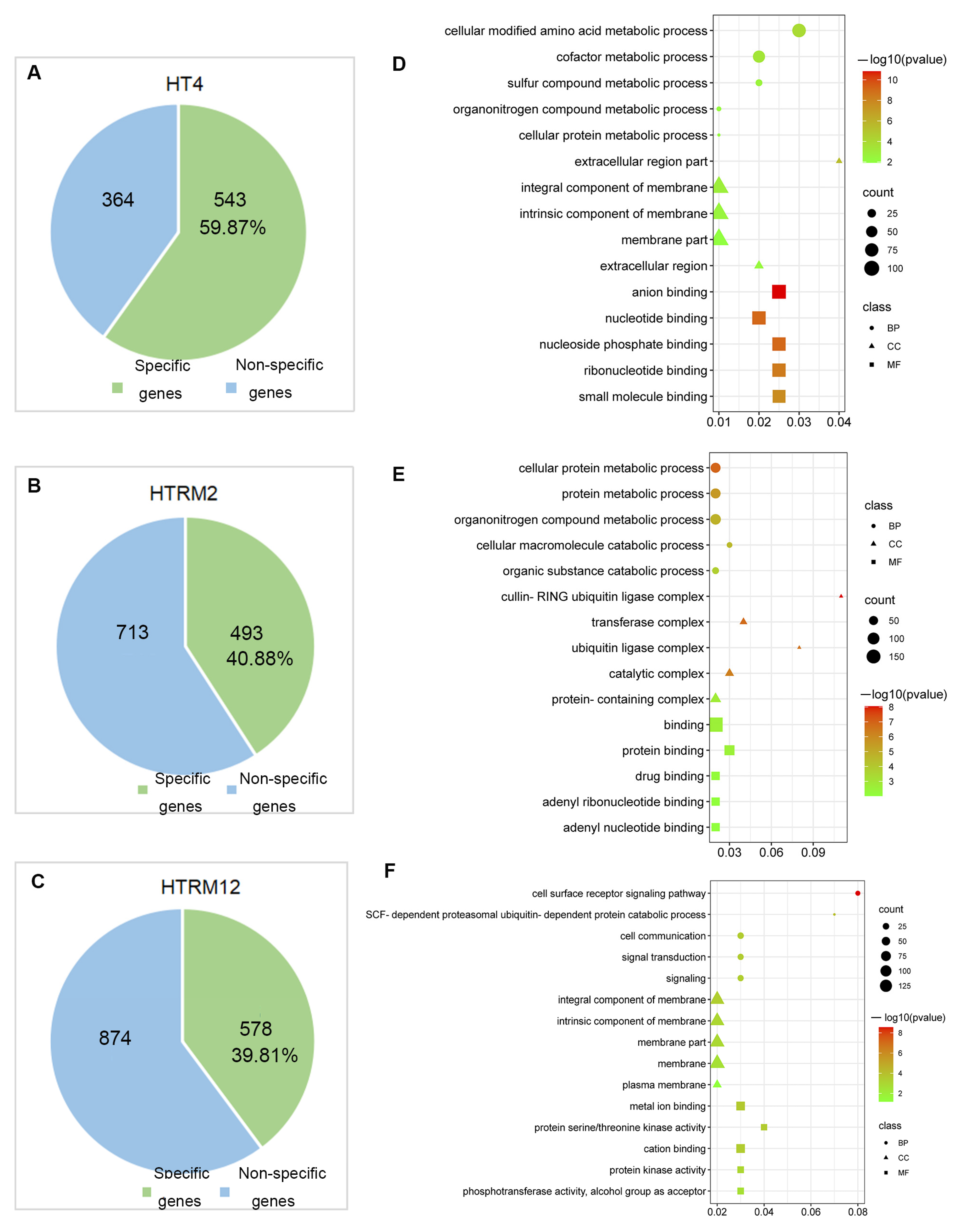
2.5. GO Terms and KEGG Enrichment Analysis of SNP Mutant Genes of HT2, HT4, HTRM2, and HTRM12
2.6. GO Terms and KEGG Enrichment Analysis of InDel Mutant Genes of HT2, HT4, HTRM2, and HTRM12
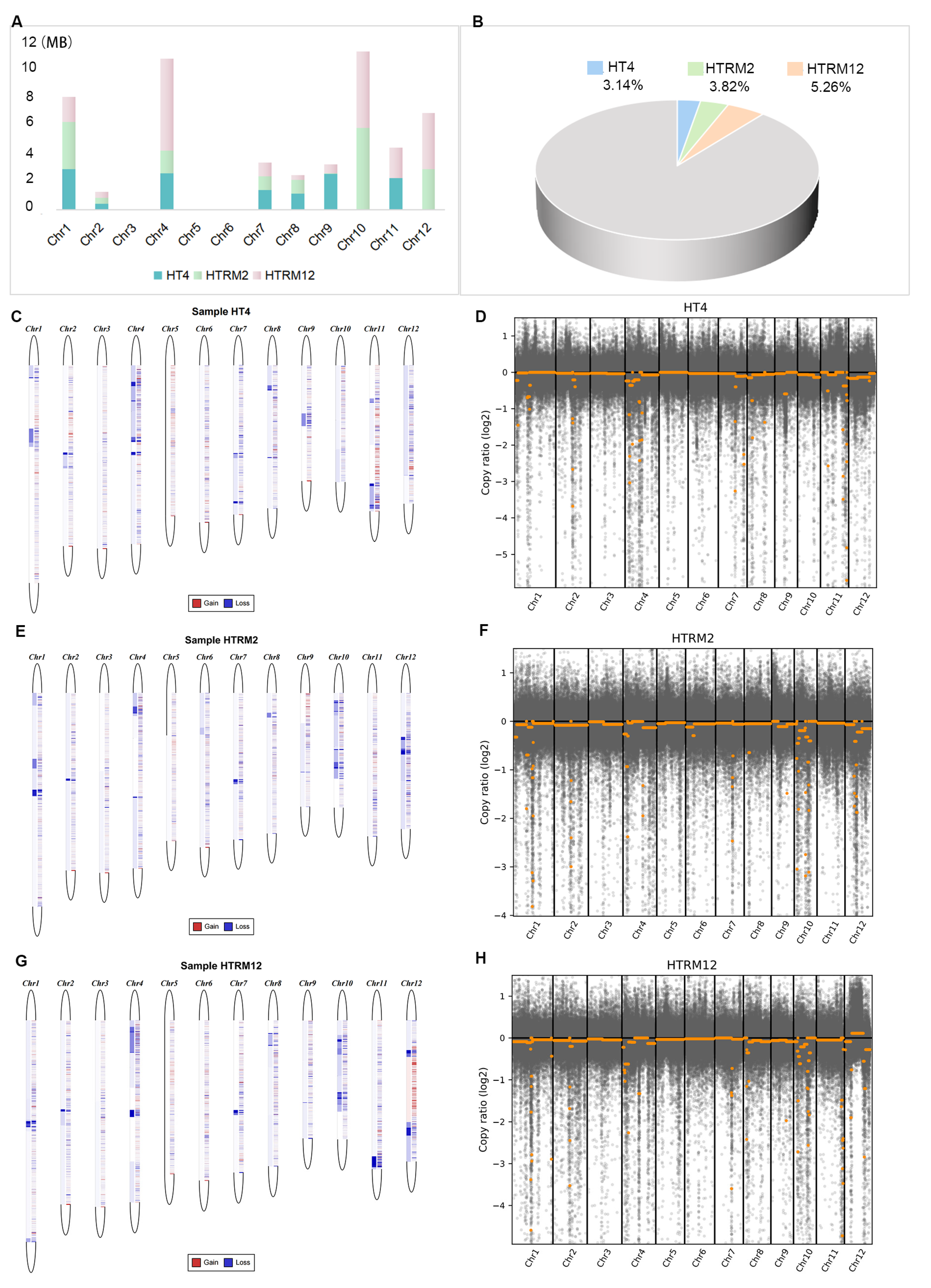
2.7. Identification and Comparison of Copy Number Variation (CNV) for HT4, HTRM2, and HTRM12
3. Discussion
4. Materials and Methods
4.1. Rice Material and Phenotype Examination
4.2. The Disease and Pest Resistance Test
4.3. The High Temperature Resistance Test
4.4. Genome Sequencing and Data Quality Assessment
4.5. Analysis of Variation Information
4.6. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analyses
4.7. GWAS Phenotype Enrichment Analysis
4.8. Identification and Comparative Analysis of Copy Number Variation Intervals
4.9. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mishra, P.; Devi, M.; Fatih, C.; Devi, M.; Williams, A.J. Forecasting of Rice Production using the Meteorological Factor in Major States in India and its Role in Food Security. Int. J. Agric. Environ. Biotechnol. 2021, 14, 51–62. [Google Scholar]
- Khush, G.S. Green revolution: The way forward. Nat. Rev. Genet. 2001, 2, 815–822. [Google Scholar] [CrossRef]
- Masterson, J. Stomatal size in fossil plants: Evidence for polyploidy in majority of angiosperms. Science 1994, 264, 421–424. [Google Scholar] [CrossRef]
- Dubcovsky, J.; Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 2007, 316, 1862–1866. [Google Scholar] [CrossRef]
- Doebley, J.; Bacigalupo, A.; Stec, A. Inheritance of Kernel Weight in Two Maize-Teosinte Hybrid Populations: Implications for Crop Evolution. J. Hered. 1994, 85, 191–195. [Google Scholar]
- Begna, T. Effects of crop evolution under domestication and narrowing genetic bases of crop species. Open J. Plant Sci. 2021, 6, 49–54. [Google Scholar]
- Yoo, M.J.; Liu, X.X.; Pires, J.C.; Soltis, P.S.; Soltis, D.E. Nonadditive Gene Expression in Polyploids. Annu. Rev. Genet. 2014, 48, 485–517. [Google Scholar] [CrossRef]
- Fridman, E. Consequences of hybridization and heterozygosity on plant vigor and phenotypic stability. Plant Sci. 2015, 232, 35–40. [Google Scholar] [CrossRef]
- Cai, D.T.; Yuan, L.P.; Lu, X.G. A new strategy of rice breeding in the 21st century: II. searching a new pathway of rice breeding by utilization of double heterosis of wide cross and polyploidization. Acta Agron. Sin. 2001, 27, 110–116. [Google Scholar]
- Hattersley, P.W.; Watson, L. Diversification of photosynthesis. In Chapman GP (ed) Grass Evolution and Domestication; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Nieto Feliner, G.; Casacuberta, J.; Wendel, J.F. Genomics of evolutionary novelty in hybrids and polyploids. Front. Genet. 2020, 11, 792. [Google Scholar] [CrossRef]
- Koide, Y.; Kuniyoshi, D.; Kishima, Y. Fertile Tetraploids: New Resources for Future Rice Breeding? Front. Plant Sci. 2020, 11, 1231. [Google Scholar] [CrossRef]
- Chen, R.R.; Feng, Z.Y.; Zhang, X.H.; Song, Z.J.; Cai, D.T. A New Way of Rice Breeding: Polyploid Rice Breeding. Plants 2021, 10, 422. [Google Scholar] [CrossRef]
- Wang, N.N.; Fan, X.H.; Lin, Y.J.; Li, Z.; Wang, Y.K.; Zhou, Y.M.; Meng, W.L.; Peng, Z.W.; Zhang, C.Y.; Ma, J. Alkaline Stress Induces Different Physiological, Hormonal and Gene Expression Responses in Diploid and Autotetraploid Rice. Int. J. Mol. Sci. 2022, 23, 5561. [Google Scholar] [CrossRef]
- Wu, J.W.; Hu, C.Y.; Shahid, M.Q.; Guo, H.B.; Zeng, Y.X.; Liu, X.D.; Lu, Y.G. Analysis on genetic diversification and heterosis in autotetraploid rice. Springerplus 2013, 2, 439. [Google Scholar] [CrossRef]
- Tu, S.B.; Kong, F.L.; Xu, Q.F.; He, T. Study on new system of heterosis utilization in autotetraploid rice. Bull. Chin. Acad. Sci. 2003, 6, 426–428. (In Chinese) [Google Scholar]
- Shahid, M.Q.; Sun, J.F.; Wei, C.M.; Zhang, P.; Liu, X.D. Studies on the Abnormality of Embryo Sac and Pollen Fertility in Autotetraploid Rice during Different Growing Seasons. Pak. J. Bot. 2010, 42, 7–19. [Google Scholar]
- He, Y.C.; Wei, Q.O.; Ge, J.; Jiang, A.M.; Gan, L.; Song, Z.J.; Cai, D.T. Genome duplication effects on pollen development and the interrelated physiological substances in tetraploid rice with polyploid meiosis stability. Planta 2010, 232, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Shahid, M.Q.; Chen, L.; Chen, Z.X.; Wang, L.; Liu, X.D.; Lu, Y.G. Polyploidy Enhances F1 Pollen Sterility Loci Interactions That Increase Meiosis Abnormalities and Pollen Sterility in Autotetraploid Rice. Plant Physiol. 2015, 169, 2700–2717. [Google Scholar] [CrossRef]
- Li, X.; Yu, H.; Jiao, Y.M.; Shahid, M.Q.; Wu, J.W.; Liu, X.D. Genome-wide analysis of DNA polymorphisms, the methylome and transcriptome revealed that multiple factors are associated with low pollen fertility in autotetraploid rice. PLoS ONE 2018, 13, e0201854. [Google Scholar] [CrossRef]
- Ghouri, F.; Zhu, J.N.; Yu, H.; Wu, J.W.; Baloch, F.S.; Liu, X.D.; Shahid, M.Q. Deciphering global DNA variations and embryo sac fertility in autotetraploid rice line. Turk. J. Agric. For. 2019, 43, 554–568. [Google Scholar] [CrossRef]
- Tu, S.B.; Luan, L.; Liu, Y.H.; Long, W.B.; Kong, F.L.; He, T.; Xu, Q.F.; Yan, W.N.; Yu, M. Production and heterosis analysis of rice autotetraploid hybrids. Crop Sci. 2007, 47, 2356–2363. [Google Scholar] [CrossRef]
- Cai, D.T.; Chen, J.G.; Chen, D.L.; Dai, B.C.; Zhang, W.; Song, Z.J.; Yang, Z.F.; Du, C.Q.; Tang, Z.Q.; He, Y.C.; et al. The breeding of two polyploid rice lines with the characteristic of polyploid meiosis stability. Sci. China Ser. C Life Sci. 2007, 50, 356–366. [Google Scholar] [CrossRef]
- Guo, H.B.; Shahid, M.Q.; Zhao, J.; Li, Y.J.; Wang, L.; Liu, X.D. Agronomic Traits and Cytogenetic Evaluation of Newly Developed Autotetraploid Rice Line. Pak. J. Agric. Sci. 2016, 53, 291–301. [Google Scholar] [CrossRef]
- Guo, H.B.; Mendrikahy, J.N.; Xie, L.; Deng, J.F.; Lu, Z.J.; Wu, J.W.; Li, X.; Shahid, M.Q.; Liu, X.D. Transcriptome analysis of neo-tetraploid rice reveals specific differential gene expressions associated with fertility and heterosis. Sci. Rep. 2017, 7, 40139. [Google Scholar] [CrossRef]
- Bei, X.; Shahid, M.Q.; Wu, J.; Chen, Z.; Liu, X. Re-sequencing and transcriptome analysis reveal rich DNA variations and differential expressions of fertility-related genes in neo-tetraploid rice. PLoS ONE 2019, 14, e0214953. [Google Scholar]
- Yu, H.; Shahid, M.Q.; Li, Q.H.; Li, Y.D.; Li, C.; Lu, Z.J.; Wu, J.W.; Zhang, Z.M.; Liu, X.D. Production Assessment and Genome Comparison Revealed High Yield Potential and Novel Specific Alleles Associated with Fertility and Yield in Neo-Tetraploid Rice. Rice 2020, 13, 32. [Google Scholar] [CrossRef]
- Ghaleb, M.A.A.; Li, C.; Shahid, M.Q.; Yu, H.; Liang, J.H.; Chen, R.X.; Wu, J.W.; Liu, X.D. Heterosis analysis and underlying molecular regulatory mechanism in a wide-compatible neo-tetraploid rice line with long panicles. BMC Plant Biol. 2020, 20, 83. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zuo, B.; Song, Z.J.; Wang, W.; He, Y.C.; Liu, Y.H.; Cai, D.T. Breeding and study of two new photoperiod- and thermo-sensitive genic male sterile lines of polyploid rice (Oryza sativa L.). Sci Rep. 2017, 7, 14744. [Google Scholar] [CrossRef]
- Chen, Y.; Shahid, M.Q.; Wu, J.W.; Deng, R.L.; Chen, Z.X.; Wang, L.; Liu, G.Q.; Zhou, H.; Liu, X.D. Thermo-Sensitive Genic Male Sterile Lines of Neo-Tetraploid Rice Developed through Gene Editing Technology Revealed High Levels of Hybrid Vigor. Plants 2022, 11, 1390. [Google Scholar] [CrossRef]
- Bassil, N.V.; McCallum, S.; Graham, J.; Olmstead, J.W.; Itle, R.A.; Brown, A.; Buck, E.J.; Wiedow, C.; Finn, C.E.; Hancock, J.F.; et al. Progress toward a Universal Linkage Map in Tetraploid Highbush Blueberry. In Proceedings of the 2011 ASHS Annual Conference, Kohala, HI, USA, 25–28 September 2011. [Google Scholar]
- Xiong, X.Y.; Xiao, X.C.; Lei, X.F. Breeding Utilization of the Diploid Rice in Autopolyploid Self Returning Progeny. J. Anhui Agric. Sci. 2016, 44, 28+51. (In Chinese) [Google Scholar]
- Kamara, N.; Jiao, Y.M.; Lu, Z.J.; Aloryi, K.D.; Wu, J.W.; Liu, X.D.; Shahid, M.Q. Cytological Observations and Bulked-Segregant Analysis Coupled Global Genome Sequencing Reveal Two Genes Associated with Pollen Fertility in Tetraploid Rice. Int. J. Mol. Sci. 2021, 22, 841. [Google Scholar] [CrossRef]
- Yu, H.; Li, Q.H.; Li, Y.D.; Yang, H.J.; Lu, Z.J.; Wu, J.W.; Zhang, Z.M.; Shahid, M.Q.; Liu, X.D. Genomics Analyses Reveal Unique Classification, Population Structure and Novel Allele of Neo-Tetraploid Rice. Rice 2021, 14, 16. [Google Scholar] [CrossRef]
- Song, Z.J.; Du, C.Q.; Zhang, X.H.; Chen, D.L.; He, Y.C.; Cai, D.T. Studies on awns in polyploid rice (Oryza sativa L.) and preliminary cross experiments of a special awnless tetraploid rice. Genet. Resour. Crop Evol. 2014, 61, 797–807. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Wang, M.; Xie, L.J.; Wu, Z.X.; Yu, J.M.; Wang, Y.C.; Zhang, Z.Q.; Jia, Y.F.; Liu, Q.P. The miR528-D3 Module Regulates Plant Height in Rice by Modulating the Gibberellin and Abscisic Acid Metabolisms. Rice 2022, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.Y.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Xu, L.L. Characters of Cysteine Endopeptidases in Wheat Endosperm during Seed Germination and Subsequent Seedling Growth. J. Integr. Plant Biol. 2009, 51, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Höwing, T.; Huesmann, C.; Hoefle, C.; Nagel, M.K.; Isono, E.; Hückelhoven, R.; Gietl, C. Endoplasmic reticulum KDEL-tailed cysteine endopeptidase 1 of Arabidopsis (AtCEP1) is involved in pathogen defense. Front. Plant Sci. 2014, 5, 58. [Google Scholar] [CrossRef]
- Yap, K.; Makeyev, E.V. Regulation of gene expression in mammalian nervous system through alternative pre-mRNA splicing coupled with RNA quality control mechanisms. Mol. Cell. Neurosci. 2013, 56, 420–428. [Google Scholar] [CrossRef]
- Hunter, N.; Verma, V. Cell division| Meiosis. In Encyclopedia of Biological Chemistry, 3rd ed.; Elsevier: Amsterdam, the Netherlands, 2021; pp. 49–56. [Google Scholar]
- Deamer, D.W. Bioenergetics and Primitive Metabolic Pathways. In Assembling Life; Oxford Academic: Oxford, UK, 2019. [Google Scholar]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Kao, Y.T.; Gonzalez, K.L.; Bartel, B. Peroxisome Function, Biogenesis, and Dynamics in Plants. Plant Physiol. 2018, 176, 162–177. [Google Scholar] [CrossRef]
- Wang, J.; Qi, M.; Liu, J.; Zhang, Y. CARMO: A Comprehensive Annotation Platform for Functional Exploration of Rice Multi-Omics Data. Plant J. 2015, 83, 359–374. [Google Scholar]
- Huang, L.; Hua, K.; Xu, R.; Zeng, D.; Wang, R.; Dong, G.; Zhang, G.; Lu, X.; Fang, N.; Wang, D.; et al. The LARGE2-APO1/APO2 regulatory module controls panicle size and grain number in rice. Plant Cell 2021, 33, 1212–1228. [Google Scholar]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar]
- Palacios, R.; Jáuregui, C.G.; Flores, M.; Palacios-Flores, K. Copy Number Variation. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Cacabelos, R.; Tellado, I.; Cacabelos, P. The Epigenetic Machinery in the Life Cycle and Pharmacoepigenetics. In Pharmacoepigenetics; Cacabelos, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 10, pp. 1–100. [Google Scholar]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Xing, S.; Zhou, K. Genetic study on the self-bred progenies of special autopolyploid rice. J. Sichuan Agric. Univ. 2000, 18, 308–310+314. (In Chinese) [Google Scholar]
- Li, L.; Zhou, X.; Wang, J.; Mei, X.; Zhang, X.; Song, Z.; Cai, D. Analysis of the Grain Quality of Elite Diploid Rice Lines Reverted from Polyploid Rice. Hans J. Agric. Sci. 2021, 11, 184–190. (In Chinese) [Google Scholar]
- Wendel, J.F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 2015, 102, 1753–1756. [Google Scholar] [CrossRef]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef]
- Zmienko, A.; Samelak, A.; Kozlowski, P.; Figlerowicz, M. Copy number polymorphism in plant genomes. Theor. Appl. Genet. 2014, 127, 1–18. [Google Scholar] [CrossRef]
- Lye, Z.N.; Purugganan, M.D. Copy Number Variation in Domestication. Trends Plant Sci 2019, 24, 352–365. [Google Scholar] [CrossRef]
- Luan, L.; Long, W.-B.; Wang, X.; Chen, Y.; Qiu, L.; Tu, S.-B.; Kong, F.-L.; Xiao, X.-Y. Research on creating auto-tetraploid rice using colchicum to induce the panicle callus of diploid rice. J. Sichuan Univ. (Nat. Sci. Ed.) 2009, 46, 829–837. (In Chinese) [Google Scholar]
- NY/T 1300 2007; Technical Procedures for Rice Variety Trials. The Ministry of Agriculture of the People’s Republic of China: Beijing, China, 2007.
- NY/T2646-2014; Technical Specification for the Identification and Evaluation of Blast Resistance in Rice Variety Testing. The Ministry of Agriculture of the People’s Republic of China: Beijing, China, 2014.
- HNZ070-2015; Technical Regulations of Bacterial Blight Resistance of Rice Varieties. Department of Agriculture and Rural Affairs of Hunan: Changsha, China, 2015.
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Supek, F.; Bonjak, M.; Kunca, N.; Muc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variety | Susceptibility Index of Rice Blast | Susceptibility Index of Bacterial Leaf Blight | Susceptibility Index of Brown Planthopper |
|---|---|---|---|
| HTRM12 | 5.2 | 5 | 9 |
| FLY4 (CK) | 6.5 | 5 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Du, A.; Tong, L.; Yan, G.; Lu, L.; Yin, Y.; Fu, X.; Yang, H.; Li, H.; Huang, W.; et al. Genome Resequencing for Autotetraploid Rice and Its Closest Relatives Reveals Abundant Variation and High Potential in Rice Breeding. Int. J. Mol. Sci. 2024, 25, 9012. https://doi.org/10.3390/ijms25169012
Zhang Y, Du A, Tong L, Yan G, Lu L, Yin Y, Fu X, Yang H, Li H, Huang W, et al. Genome Resequencing for Autotetraploid Rice and Its Closest Relatives Reveals Abundant Variation and High Potential in Rice Breeding. International Journal of Molecular Sciences. 2024; 25(16):9012. https://doi.org/10.3390/ijms25169012
Chicago/Turabian StyleZhang, Yachun, Anping Du, Liqi Tong, Gui Yan, Longxiang Lu, Yanni Yin, Xingyue Fu, Huixin Yang, Hui Li, Weizao Huang, and et al. 2024. "Genome Resequencing for Autotetraploid Rice and Its Closest Relatives Reveals Abundant Variation and High Potential in Rice Breeding" International Journal of Molecular Sciences 25, no. 16: 9012. https://doi.org/10.3390/ijms25169012
APA StyleZhang, Y., Du, A., Tong, L., Yan, G., Lu, L., Yin, Y., Fu, X., Yang, H., Li, H., Huang, W., Cai, D., Song, Z., Zhang, X., He, Y., & Tu, S. (2024). Genome Resequencing for Autotetraploid Rice and Its Closest Relatives Reveals Abundant Variation and High Potential in Rice Breeding. International Journal of Molecular Sciences, 25(16), 9012. https://doi.org/10.3390/ijms25169012