
Exploring Mitochondrial Heterogeneity and Evolutionary Dynamics in Thelephora ganbajun through Population Genomics
 and
and
Abstract
:1. Introduction
2. Results
2.1. General Features of 40 Mitogenomes of T. ganbajun
2.2. Structure of Standard Protein-Coding Genes
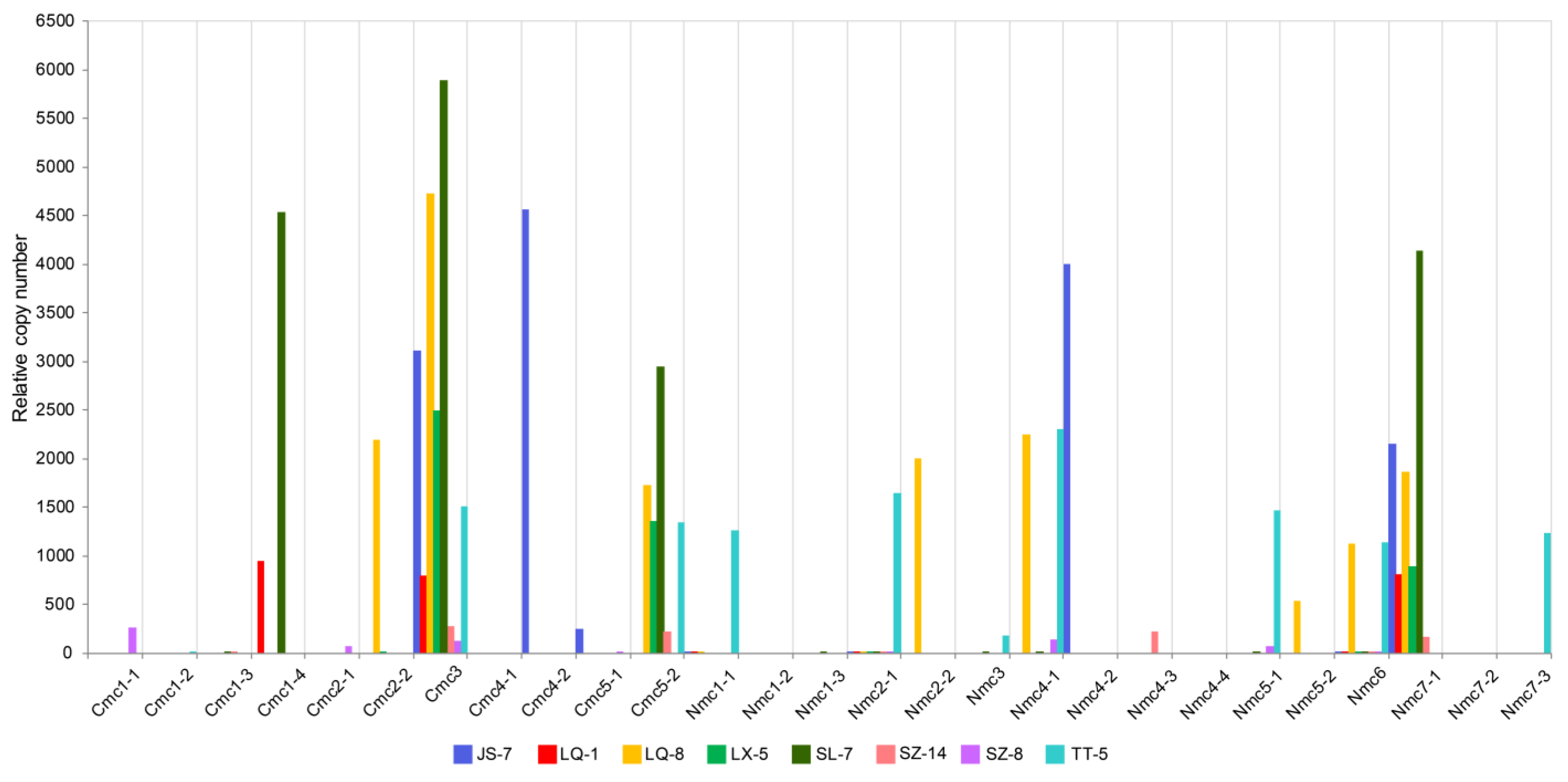
2.3. Multicopy HTs Verification in cox1 and nad5
2.4. Intron and HEs Dynamics of Mitogenomes
2.4.1. Intron Distribution
2.4.2. Intronic Polymorphism
2.5. Correlation Analysis of the Main Characteristics of the Mitochondrial Genomes
2.6. Horizontal Gene Transfer of HEs
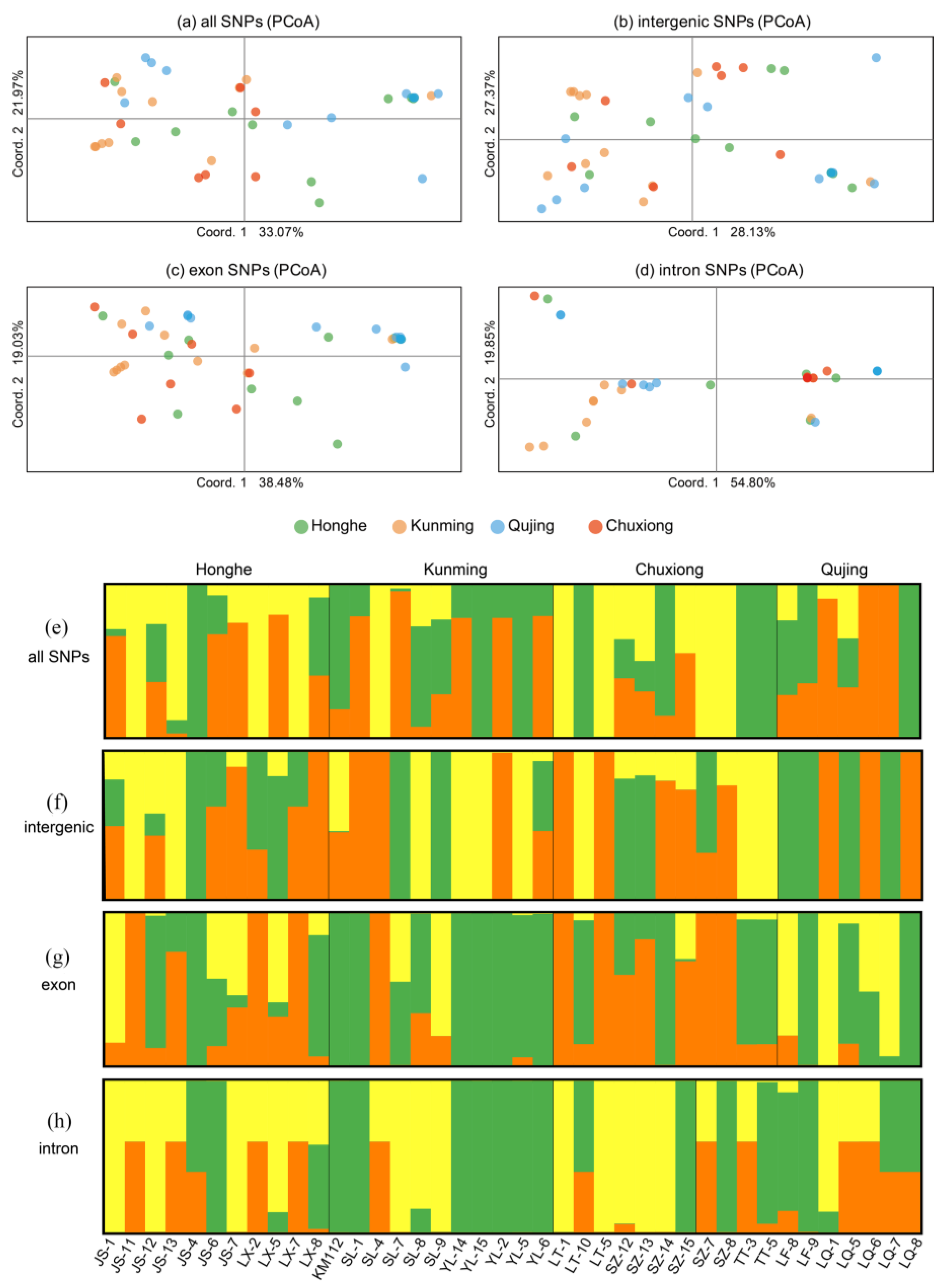
2.7. Population Genetics
3. Discussion
3.1. Variations of HTs
3.2. The Effect of Heterogeneity
3.3. Intron Dynamics
3.4. Population Structure
4. Materials and Methods
4.1. Sampling, DNA Isolation, and Genome Sequencing
4.2. De Novo Assembly, Annotation, and Comparative Analysis of Mitogenomes
4.3. Existence of Multicopy Heterogeneity in the nad5 and cox1 Genes Verified by qPCR
4.4. Population Genetics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atlante, A.; Valenti, D. Mitochondria Have Made a Long Evolutionary Path from Ancient Bacteria Immigrants within Eukaryotic Cells to Essential Cellular Hosts and Key Players in Human Health and Disease. Curr. Issues Mol. Biol. 2023, 45, 4451–4479. [Google Scholar] [CrossRef] [PubMed]
- Araújo, D.S.; De-Paula, R.B.; Tomé, L.M.R.; Quintanilha-Peixoto, G.; Salvador-Montoya, C.A.; Del-Bem, L.-E.; Badotti, F.; Azevedo, V.A.C.; Brenig, B.; Aguiar, E.R.G.R.; et al. Comparative mitogenomics of Agaricomycetes: Diversity, abundance, impact and coding potential of putative open-reading frames. Mitochondrion 2021, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sandor, S.; Zhang, Y.; Xu, J. Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 2018, 102, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Pramateftaki, P.V.; Kouvelis, V.N.; Lanaridis, P.; Typas, M.A. The mitochondrial genome of the wine yeast Hanseniaspora uvarum: A unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 2006, 6, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cai, Y.; Zhang, Q.; Chen, L.; Shu, F.; Ma, X.; Bian, Y. The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 2020, 143, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Rogozin, I.B.; Koonin, E.V. MitoCOGs: Clusters of orthologous genes from mitochondria and implications for the evolution of eukaryotes. BMC Evol. Biol. 2014, 14, 237. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, G.; Ozkilinc, H. Pan-Mitogenomics Approach Discovers Diversity and Dynamism in the Prominent Brown Rot Fungal Pathogens. Front. Microbiol. 2021, 12, 647989. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Feng, H.; Tu, W.; Xiong, C.; Jin, X.; Li, P.; Wang, X.; Li, Q. Comparative Mitogenomic Analysis Reveals Dynamics of Intron Within and Between Tricholoma Species and Phylogeny of Basidiomycota. Front. Genet. 2021, 12, 534871. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, P.; Li, L.; Feng, H.; Tu, W.; Bao, Z.; Xiong, C.; Gui, M.; Huang, W. The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 2021, 172, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Longchamps, R.J.; Castellani, C.A.; Yang, S.Y.; Newcomb, C.E.; Sumpter, J.A.; Lane, J.; Grove, M.L.; Guallar, E.; Pankratz, N.; et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS ONE 2020, 15, e0228166. [Google Scholar] [CrossRef]
- Xu, J.; Wang, P. Mitochondrial inheritance in basidiomycete fungi. Fungal Biol. Rev. 2015, 29, 209–219. [Google Scholar] [CrossRef]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Manfredi, G.; Hirano, M.; Schon, E.A.; Fox, T.D. Maintenance of Human Rearranged Mitochondrial DNAs in Long-Term Cultured Transmitochondrial Cell Lines. Mol. Biol. Cell 2000, 11, 2349–2358. [Google Scholar] [CrossRef]
- Whiston, E.; Taylor, J.W. Genomics in Coccidioides: Insights into evolution, ecology, and pathogenesis. Med. Mycol. 2013, 52, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Borkovich, K.A.; Kanzi, A.M.; Wingfield, B.D.; Steenkamp, E.T.; Naidoo, S.; van der Merwe, N.A. Intron Derived Size Polymorphism in the Mitochondrial Genomes of Closely Related Chrysoporthe Species. PLoS ONE 2016, 11, e0156104. [Google Scholar] [CrossRef]
- Megarioti, A.H.; Kouvelis, V.N.; Embley, M. The Coevolution of Fungal Mitochondrial Introns and Their Homing Endonucleases (GIY-YIG and LAGLIDADG). Genome Biol. Evol. 2020, 12, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.M.; Sondhi, S.; Ziesel, A.; Paliwal, S.; Fiumera, H.L. Mitochondrial-nuclear coadaptation revealed through mtDNA replacements in Saccharomyces cerevisiae. BMC Evol. Biol. 2020, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Lian, K.; Furulund, B.M.N.; Tveita, A.A.; Haugen, P.; Johansen, S.D. Mobile group I introns at nuclear rDNA position L2066 harbor sense and antisense homing endonuclease genes intervened by spliceosomal introns. Mob. DNA 2022, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, J.Y.; Münch, K.; Schotanus, K.; Stukenbrock, E.H.; Kahmann, R. The insertion of a mitochondrial selfish element into the nuclear genome and its consequences. Ecol. Evol. 2020, 10, 11117–11132. [Google Scholar] [CrossRef] [PubMed]
- Parenteau, J.; Maignon, L.; Berthoumieux, M.; Catala, M.; Gagnon, V.; Abou Elela, S. Introns are mediators of cell response to starvation. Nature 2019, 565, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, B.L. Homing endonucleases from mobile group I introns: Discovery to genome engineering. Mob. DNA 2014, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Morange, M. What history tells us XLIV: The construction of the zinc finger nucleases. J. Biosci. 2017, 42, 527–530. [Google Scholar] [CrossRef]
- Schröer, K.; Arakrak, F.; Bremke, A.; Ehrhardt, A.; Zhang, W. HEHR: Homing Endonuclease-Mediated Homologous Recombination for Efficient Adenovirus Genome Engineering. Genes 2022, 13, 2129. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Thompson, S.R.L.; Lee, D.K.; Lachance, M.-A.; Smith, D.R. Mutational Effects of Mobile Introns on the Mitochondrial Genomes of Metschnikowia Yeasts. Front. Genet. 2021, 12, 785218. [Google Scholar] [CrossRef]
- Repar, J.; Warnecke, T. Mobile Introns Shape the Genetic Diversity of Their Host Genes. Genetics 2017, 205, 1641–1648. [Google Scholar] [CrossRef]
- Fonseca, P.L.C.; Badotti, F.; De-Paula, R.B.; Araújo, D.S.; Bortolini, D.E.; Del-Bem, L.-E.; Azevedo, V.A.; Brenig, B.; Aguiar, E.R.G.R.; Góes-Neto, A. Exploring the Relationship Among Divergence Time and Coding and Non-coding Elements in the Shaping of Fungal Mitochondrial Genomes. Front. Microbiol. 2020, 11, 765. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, J.; Wang, S.; Wang, P.; Rao, W.; Hou, B.; Zhang, Y. Genetic Differentiation and Widespread Mitochondrial Heteroplasmy among Geographic Populations of the Gourmet Mushroom T. ganbajun from Yunnan, China. Genes 2022, 13, 854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, S.; Li, H.; Liu, C.; Mi, F.; Wang, R.; Mo, M.; Xu, J. Evidence for Persistent Heteroplasmy and Ancient Recombination in the Mitochondrial Genomes of the Edible Yellow Chanterelles From Southwestern China and Europe. Front. Microbiol. 2021, 12, 699598. [Google Scholar] [CrossRef]
- Wang, P.; Sha, T.; Zhang, Y.; Cao, Y.; Mi, F.; Liu, C.; Yang, D.; Tang, X.; He, X.; Dong, J.; et al. Frequent heteroplasmy and recombination in the mitochondrial genomes of the basidiomycete mushroom T. ganbajun. Sci. Rep. 2017, 7, 1626. [Google Scholar] [CrossRef]
- Cheng, J.; Luo, Q.; Ren, Y.; Luo, Z.; Liao, W.; Wang, X.; Li, Q. Panorama of intron dynamics and gene rearrangements in the phylum Basidiomycota as revealed by the complete mitochondrial genome of Turbinellus floccosus. Appl. Microbiol. Biotechnol. 2021, 105, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Mi, F.; Dong, J.; Zhang, Y.; Wu, J. Changes of Genome Size of Fungal Mitochondria. J. Anhui Agric. 2016, 44, 36–39. [Google Scholar] [CrossRef]
- Yildiz, G.; Ozkilinc, H. First characterization of the complete mitochondrial genome of fungal plant-pathogen Monilinia laxa which represents the mobile intron rich structure. Sci. Rep. 2020, 10, 13644. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Tikochinski, Y.; Carreras, C.; Tikochinski, G.; Vilaça, S.T. Population-specific signatures of intra-individual mitochondrial DNA heteroplasmy and their potential evolutionary advantages. Sci. Rep. 2020, 10, 211. [Google Scholar] [CrossRef]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Models Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef] [PubMed]
- Ladoukakis, E.D.; Zouros, E. Evolution and inheritance of animal mitochondrial DNA: Rules and exceptions. J. Biol. Res. 2017, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. The inheritance of organelle genes and genomes: Patterns and mechanisms. Genome 2005, 48, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, C.S.; Keepers, K.G.; Nadiadi, A.Y.; Bailey, D.W.; Lendemer, J.C.; Tripp, E.A.; Kane, N.C. Genome streamlining via complete loss of introns has occurred multiple times in lichenized fungal mitochondria. Ecol. Evol. 2019, 9, 4245–4263. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Hu, L.; Shen, P.; Li, R.; Liu, D. SMRT Sequencing Revealed Mitogenome Characteristics and Mitogenome-Wide DNA Modification Pattern in Ophiocordyceps sinensis. Front. Microbiol. 2017, 8, 1422. [Google Scholar] [CrossRef] [PubMed]
- Férandon, C.; Xu, J.; Barroso, G. The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 2013, 55, 85–91. [Google Scholar] [CrossRef]
- Badger, J.H.; Férandon, C.; Moukha, S.; Callac, P.; Benedetto, J.-P.; Castroviejo, M.; Barroso, G. The Agaricus bisporus cox1 Gene: The Longest Mitochondrial Gene and the Largest Reservoir of Mitochondrial Group I Introns. PLoS ONE 2010, 5, e14048. [Google Scholar] [CrossRef]
- Ma, M.-Y.; Che, X.-R.; Porceddu, A.; Niu, D.-K. Evaluation of the mechanisms of intron loss and gain in the social amoebae Dictyostelium. BMC Evol. Biol. 2015, 15, 286. [Google Scholar] [CrossRef] [PubMed]
- Himmelstrand, K.; Brandström Durling, M.; Karlsson, M.; Stenlid, J.; Olson, Å. Multiple rearrangements and low inter- and intra-species mitogenome sequence variation in the Heterobasidion annosum s.l. species complex. Front. Microbiol. 2023, 14, 1159811. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Mardanov, A.V.; Beletsky, A.V.; Kadnikov, V.V.; Ignatov, A.N.; Ravin, N.V. The 203 kbp Mitochondrial Genome of the Phytopathogenic Fungus Sclerotinia borealis Reveals Multiple Invasions of Introns and Genomic Duplications. PLoS ONE 2014, 9, e107536. [Google Scholar] [CrossRef]
- Husnik, F.; McCutcheon, J.P. Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 2017, 16, 67–79. [Google Scholar] [CrossRef]
- Mayers, C.G.; Harrington, T.C.; Wai, A.; Hausner, G. Recent and Ongoing Horizontal Transfer of Mitochondrial Introns Between Two Fungal Tree Pathogens. Front. Microbiol. 2021, 12, 656609. [Google Scholar] [CrossRef] [PubMed]
- Shinozuka, H.; Hettiarachchige, I.K.; Shinozuka, M.; Cogan, N.O.I.; Spangenberg, G.C.; Cocks, B.G.; Forster, J.W.; Sawbridge, T.I. Horizontal transfer of a ß-1,6-glucanase gene from an ancestral species of fungal endophyte to a cool-season grass host. Sci. Rep. 2017, 7, 9024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bai, X.; Ren, L.Y.; Sun, H.H.; Tang, H.P.; Vaario, L.M.; Xu, J.; Zhang, Y.J. Dynamic evolution of eukaryotic mitochondrial and nuclear genomes: A case study in the gourmet pine mushroom Tricholoma matsutake. Environ. Microbiol. 2021, 23, 7214–7230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hao, A.-J.; Zhao, Y.-X.; Zhang, X.-Y.; Zhang, Y.-J. Comparative mitochondrial genomics toward exploring molecular markers in the medicinal fungus Cordyceps militaris. Sci. Rep. 2017, 7, 40219. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Bondarenko, V.S.; Gelfand, M.S. Evolution of the Exon-Intron Structure in Ciliate Genomes. PLoS ONE 2016, 11, e0161476. [Google Scholar] [CrossRef]
- Muzafar, S.; Sharma, R.D.; Chauhan, N.; Prasad, R. Intron distribution and emerging role of alternative splicing in fungi. FEMS Microbiol. Lett. 2021, 368, fnab135. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, G.; Fang, M.; Deng, C.; Zhang, K.-Q.; Yu, Z.; Xu, J. Comparative Analyses of Mitochondrial Genomes Provide Evolutionary Insights Into Nematode-Trapping Fungi. Front. Microbiol. 2020, 11, 617. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2004, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Michael, F. Corrgrams: Exploratory displays for correlation matrices. The American Statistician 2002, 56, 316–324. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Computing 2013, 1, 12–21. [Google Scholar]
- Nicklas, J.A.; Brooks, E.M.; Hunter, T.C.; Single, R.; Branda, R.F. Development of a quantitative PCR (TaqMan) assay for relative mitochondrial DNA copy number and the common mitochondrial DNA deletion in the rat. Environ. Mol. Mutagen. 2004, 44, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sha, T.A.O.; Li, Y.C.; Zhao, Z.W.; Yang, Z.L. Recombination and genetic differentiation among natural populations of the ectomycorrhizal mushroom Tricholoma matsutake from southwestern China. Mol. Ecol. 2008, 17, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Genome Size (bp) | GC Content (%) | Standard Protein-Encoding Genes (PCGs) Size (bp) | Tot. Multicopy Genes Size (bp) | HEGs Size (bp) | Normalintrons Size (bp) (No.) | No. HE | Tot. Introns | GenBank Accession |
|---|---|---|---|---|---|---|---|---|---|
| JS-1 | 65,668 | 26.04 | 22,096 | 11,172 | 4022 | 1567 (5) | 11 | 16 | PQ126529 |
| JS-11 | 65,543 | 25.90 | 24,854 | 12,013 | 4022 | 1397 (6) | 11 | 17 | PQ149496 |
| JS-12 | 64,773 | 25.89 | 22,177 | 11,135 | 4121 | 1850 (8) | 11 | 19 | PQ149497 |
| JS-13 | 66,588 | 26.04 | 24,816 | 11,975 | 4723 | 2078 (8) | 13 | 21 | PQ149498 |
| JS-4 | 64,130 | 25.85 | 22,505 | 8428 | 3456 | 2101 (6) | 9 | 15 | PQ149493 |
| JS-6 | 65,686 | 25.86 | 23,799 | 11,968 | 3150 | 1500 (7) | 9 | 16 | PQ149494 |
| JS-7 | 68,725 | 25.94 | 24,078 | 10,039 | 4116 | 2396 (8) | 12 | 20 | PQ149495 |
| KM112 | 64,682 | 25.86 | 21,813 | 9868 | 4121 | 1940 (6) | 11 | 18 | PQ149499 |
| LF-8 | 65,622 | 26.00 | 23,710 | 10,746 | 4022 | 1129 (4) | 11 | 15 | PQ149500 |
| LF-9 | 64,911 | 25.88 | 23,941 | 10,813 | 3162 | 2497 (9) | 9 | 18 | PQ149501 |
| LQ-1 | 67,472 | 25.93 | 25,158 | 12,040 | 4425 | 1980 (6) | 12 | 18 | PQ149502 |
| LQ-5 | 65,082 | 25.87 | 20,697 | 9655 | 4431 | 1157 (6) | 12 | 18 | PQ149503 |
| LQ-6 | 64,913 | 25.93 | 23,545 | 9925 | 4121 | 2286 (7) | 11 | 18 | PQ149504 |
| LQ-7 | 68,859 | 25.95 | 25,190 | 11,679 | 4022 | 2396 (8) | 11 | 19 | PQ149505 |
| LQ-8 | 67,071 | 25.76 | 23,873 | 10,000 | 3107 | 2085 (7) | 8 | 15 | PQ149506 |
| LT-1 | 65,543 | 25.89 | 24,854 | 12,013 | 4022 | 1397 (6) | 11 | 17 | PQ149507 |
| LT-10 | 65,796 | 26.03 | 23,723 | 9874 | 4121 | 2343 (8) | 11 | 19 | PQ149509 |
| LT-5 | 66,733 | 26.03 | 24,854 | 12,013 | 4376 | 1397 (6) | 11 | 17 | PQ149508 |
| LX-2 | 65,573 | 26.03 | 25,012 | 12,171 | 4022 | 1408 (6) | 11 | 17 | PQ149510 |
| LX-5 | 65,635 | 25.98 | 24,381 | 11,127 | 4061 | 2430 (9) | 11 | 20 | PQ149511 |
| LX-7 | 65,709 | 26.05 | 24,952 | 12,111 | 4022 | 1300 (5) | 11 | 16 | PQ149512 |
| LX-8 | 64,567 | 26.23 | 19,515 | 9205 | 3580 | 1164 (6) | 11 | 17 | PQ149513 |
| SL-1 | 64,806 | 25.86 | 23,091 | 9868 | 4121 | 2383 (8) | 11 | 19 | PQ149514 |
| SL-4 | 64,475 | 26.11 | 22,798 | 10,746 | 4332 | 1206 (5) | 12 | 17 | PQ149515 |
| SL-7 | 67,542 | 25.92 | 22,654 | 10,585 | 4949 | 2059 (8) | 14 | 22 | PQ149516 |
| SL-8 | 65,207 | 25.83 | 21,530 | 8998 | 3249 | 1806 (6) | 9 | 15 | PQ149517 |
| SL-9 | 65,631 | 26.00 | 23,710 | 10,746 | 4022 | 1129 (4) | 11 | 15 | PQ149518 |
| SZ-12 | 65,060 | 25.93 | 25,335 | 11,461 | 4407 | 1941 (6) | 12 | 18 | PQ149521 |
| SZ-13 | 66,778 | 26.07 | 24,842 | 10,746 | 4236 | 1760 (7) | 12 | 19 | PQ149522 |
| SZ-14 | 67,314 | 25.99 | 20,216 | 9322 | 4071 | 2688 (8) | 12 | 20 | PQ149523 |
| SZ-15 | 64,879 | 25.95 | 22,862 | 9356 | 3800 | 2134 (7) | 11 | 18 | PQ149524 |
| SZ-7 | 65,570 | 26.03 | 24,854 | 12,013 | 4332 | 1397 (6) | 12 | 18 | PQ149519 |
| SZ-8 | 65,539 | 26.04 | 24,854 | 12,013 | 4022 | 1397 (6) | 11 | 17 | PQ149520 |
| TT-3 | 65,809 | 25.90 | 23,779 | 9874 | 4121 | 2351 (7) | 11 | 18 | PQ149525 |
| TT-5 | 65,824 | 25.90 | 25,053 | 11,148 | 4121 | 2733 (9) | 11 | 20 | PQ149526 |
| YL-14 | 64,831 | 25.86 | 23,091 | 9868 | 3586 | 2428 (9) | 10 | 19 | PQ149530 |
| YL-15 | 64,414 | 25.69 | 20,413 | 8468 | 3456 | 1263 (5) | 9 | 14 | PQ149531 |
| YL-2 | 64,840 | 25.85 | 22,642 | 11,069 | 3237 | 1729 (8) | 9 | 17 | PQ149527 |
| YL-5 | 66,855 | 25.80 | 24,501 | 10,707 | 3772 | 2217 (7) | 10 | 17 | PQ149528 |
| YL-6 | 64,799 | 25.87 | 21,441 | 9868 | 4121 | 1612 (7) | 11 | 18 | PQ149529 |
| Geographic Population | Sampling Site | Long. (E) | Lat. (N) | Region Sample Size | Local Geographic Population Sample Size |
|---|---|---|---|---|---|
| Honghe (HH) | Luxi | 103.76 | 24.52 | 11 | 4 |
| Jianshui | 102.79 | 23.64 | 7 | ||
| Kunming (KM) | Kunming | 102.83 | 24.88 | 11 | 1 |
| Yiliang | 103.08 | 24.55 | 5 | ||
| Shilin | 103.2 | 24.45 | 5 | ||
| Qujing (QJ) | Longtan | 104.01 | 26.41 | 11 | 3 |
| Tangtang | 104.21 | 26.48 | 2 | ||
| Shizong | 103.59 | 24.49 | 6 | ||
| Chuxiong (CX) | Lufeng | 102.04 | 25.09 | 7 | 2 |
| Luquan | 102.28 | 25.33 | 5 |
| SNPs Type | All Sites | Parsimony Information Sites | Singleton Variable Sites | Nucleotide Diversity (Pi) | Pi of per Population | |||
|---|---|---|---|---|---|---|---|---|
| Honghe | Kunming | Qujing | Chuxiong | |||||
| All SNPs | 2463 | 1911 | 552 | 0.230 | 0.240 | 0.206 | 0.222 | 0.246 |
| Intergenic SNPs | 1467 | 1160 | 310 | 0.233 | 0.215 | 0.199 | 0.235 | 0.240 |
| Exon SNPs | 921 | 692 | 229 | 0.263 | 0.282 | 0.215 | 0.199 | 0.259 |
| Intron SNPs | 66 | 53 | 13 | 0.252 | 0.227 | 0.254 | 0.258 | 0.199 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Liang, T.; Liu, Y.; Wang, P.; Wang, S.; Zhao, M.; Zhang, Y. Exploring Mitochondrial Heterogeneity and Evolutionary Dynamics in Thelephora ganbajun through Population Genomics. Int. J. Mol. Sci. 2024, 25, 9013. https://doi.org/10.3390/ijms25169013
Li H, Liang T, Liu Y, Wang P, Wang S, Zhao M, Zhang Y. Exploring Mitochondrial Heterogeneity and Evolutionary Dynamics in Thelephora ganbajun through Population Genomics. International Journal of Molecular Sciences. 2024; 25(16):9013. https://doi.org/10.3390/ijms25169013
Chicago/Turabian StyleLi, Haixia, Tong Liang, Yongju Liu, Pengfei Wang, Shaojuan Wang, Min Zhao, and Ying Zhang. 2024. "Exploring Mitochondrial Heterogeneity and Evolutionary Dynamics in Thelephora ganbajun through Population Genomics" International Journal of Molecular Sciences 25, no. 16: 9013. https://doi.org/10.3390/ijms25169013