
The Roles of Obesity and ASB4 in Preeclampsia Pathogenesis

 ,
,
Abstract
:1. Introduction
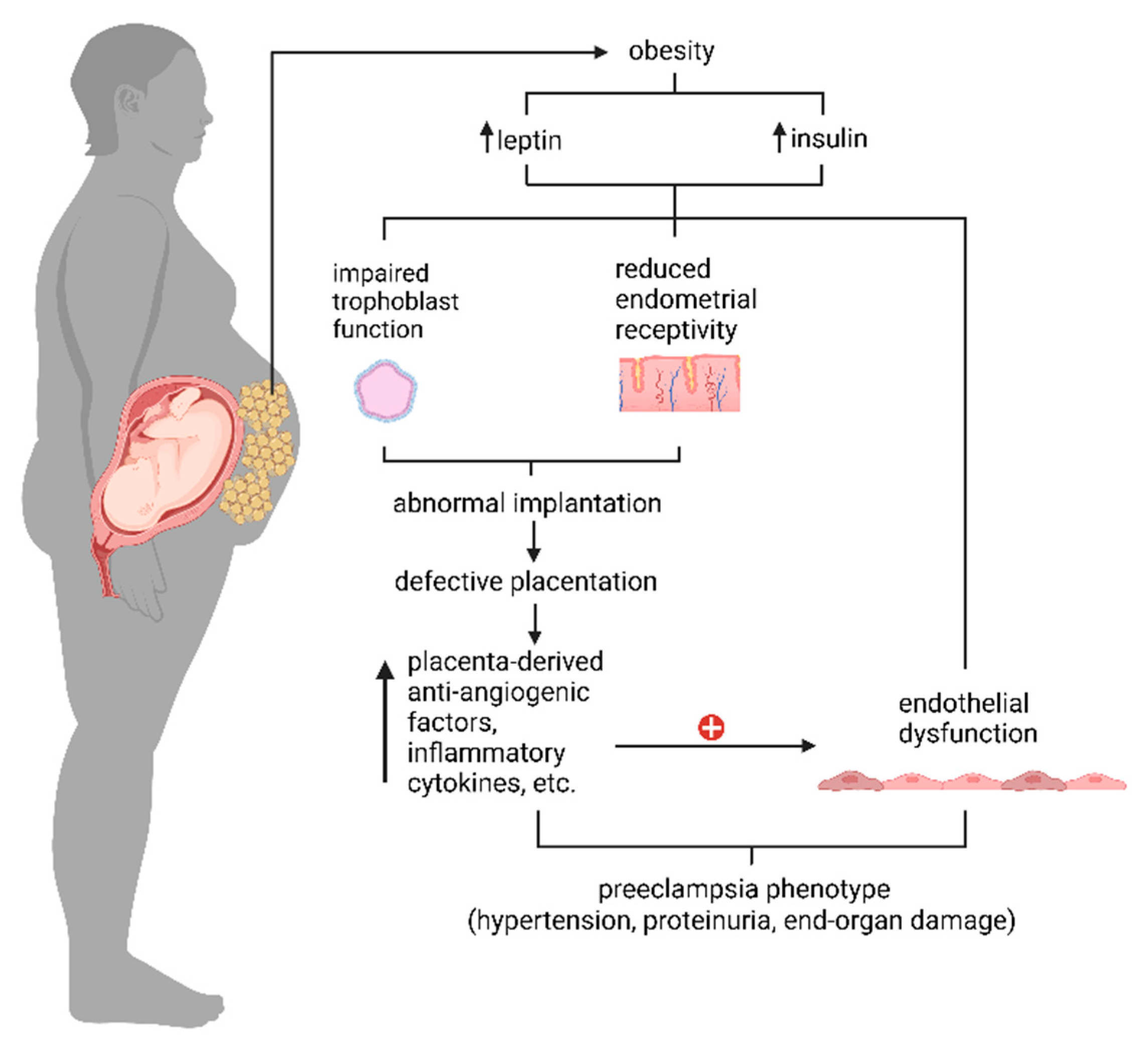
2. Preeclampsia
3. Obesity and Preeclampsia
3.1. Insulin
3.1.1. Insulin and Implantation
3.1.2. Excess Insulin in Maternal Endometrial Receptivity
3.1.3. Excess Insulin in Trophoblast Cells
3.1.4. Insulin in Maternal Endothelium
3.2. Leptin
3.2.1. Leptin and Implantation
3.2.2. Excess Leptin in Maternal Endometrial Receptivity
3.2.3. Excess Leptin in Trophoblast Cells
3.2.4. Leptin in Maternal Endothelium
4. ASB4
4.1. ASB4 and Obesity
4.2. ASB4 and Vascular Development
4.3. ASB4 and Preeclampsia
4.3.1. ASB4, Implantation, and Preeclampsia
4.3.2. ASB4, Obesity, and Preeclampsia
5. Conclusions
6. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saftlas, A.F.; Olson, D.R.; Franks, A.L.; Atrash, H.K.; Pokras, R. Epidemiology of preeclampsia and eclampsia in the United States, 1979–1986. Am. J. Obstet. Gynecol. 1990, 163, 460–465. [Google Scholar] [CrossRef]
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ 2013, 347, f6564. [Google Scholar] [CrossRef] [PubMed]
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am. J. Hypertens. 2008, 21, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2022, 226, S907–S927. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Bodnar, L.M.; Patrick, T.E.; Powers, R.W. The Role of Obesity in Preeclampsia. Pregnancy Hypertens. 2011, 1, 6–16. [Google Scholar] [CrossRef]
- Ferguson, J.E.; Wu, Y.; Smith, K.; Charles, P.; Powers, K.; Wang, H.; Patterson, C. ASB4 Is a Hydroxylation Substrate of FIH and Promotes Vascular Differentiation via an Oxygen-Dependent Mechanism. Mol. Cell. Biol. 2023, 27, 6407–6419. [Google Scholar] [CrossRef]
- Townley-Tilson, W.H.D.; Wu, Y.; Ferguson, J.E.; Patterson, C. The Ubiquitin Ligase ASB4 Promotes Trophoblast Differentiation through the Degradation of ID2. PLoS ONE 2014, 9, e89451. [Google Scholar] [CrossRef]
- Li, F.; Fushima, T.; Oyanagi, G.; Townley-Tilson, H.W.; Sato, E.; Nakada, H.; Oe, Y.; Hagaman, J.R.; Wilder, J.; Li, M.; et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc. Natl. Acad. Sci. USA 2016, 113, 13450–13455. [Google Scholar] [CrossRef]
- Kayashima, Y.; Townley-Tilson, W.H.D.; Vora, N.L.; Boggess, K.; Homeister, J.W.; Maeda-Smithies, N.; Li, F. Insulin Elevates ID2 Expression in Trophoblasts and Aggravates Preeclampsia in Obese ASB4-Null Mice. Int. J. Mol. Sci. 2023, 24, 2149. [Google Scholar] [CrossRef]
- Raymond, D.; Peterson, E. A critical review of early-onset and late-onset preeclampsia. Obs. Gynecol. Surv. 2011, 66, 497–506. [Google Scholar] [CrossRef]
- Benedetti, T.J.; Kates, R.; Williams, V. Hemodynamic observations in severe preeclampsia complicated by pulmonary edema. Am. J. Obstet. Gynecol. 1985, 152, 330–334. [Google Scholar] [CrossRef]
- Drislane, F.W.; Wang, A.M. Multifocal cerebral hemorrhage in eclampsia and severe pre-eclampsia. J. Neurol. 1997, 244, 194–198. [Google Scholar] [CrossRef]
- Morriss, M.C.; Twickler, D.M.; Hatab, M.R.; Clarke, G.D.; Peshock, R.M.; Cunningham, F.G. Cerebral blood flow and cranial magnetic resonance imaging in eclampsia and severe preeclampsia. Obstet. Gynecol. 1997, 89, 561–568. [Google Scholar] [PubMed]
- Cunningham, F.G.; Fernandez, C.O.; Hernandez, C. Blindness associated with preeclampsia and eclampsia. Am. J. Obstet. Gynecol. 1995, 172, 1291–1298. [Google Scholar] [CrossRef]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: Risk factors and outcomes associated with early- versus late-onset disease. Am. J. Obstet. Gynecol. 2013, 209, 544.e541–544.e512. [Google Scholar] [CrossRef]
- Odegard, R.A.; Vatten, L.J.; Nilsen, S.T.; Salvesen, K.A.; Austgulen, R. Preeclampsia and fetal growth. Obstet. Gynecol. 2000, 96, 950–955. [Google Scholar] [PubMed]
- Xiao, R.; Sorensen, T.K.; Williams, M.A.; Luthy, D.A. Influence of pre-eclampsia on fetal growth. J. Matern. Fetal Neonatal Med. 2003, 13, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.M.; Mercer, B.M.; Schiff, E.; Friedman, S.A. Aggressive versus expectant management of severe preeclampsia at 28 to 32 weeks’ gestation: A randomized controlled trial. Am. J. Obstet. Gynecol. 1994, 171, 818–822. [Google Scholar] [CrossRef]
- Roberts, J.M.; Gammill, H. Pre-eclampsia and cardiovascular disease in later life. Lancet 2005, 366, 961–962. [Google Scholar] [CrossRef]
- Fields, J.A.; Garovic, V.D.; Mielke, M.M.; Kantarci, K.; Jayachandran, M.; White, W.M.; Butts, A.M.; Graff-Radford, J.; Lahr, B.D.; Bailey, K.R.; et al. Preeclampsia and cognitive impairment later in life. Am. J. Obstet. Gynecol. 2017, 217, 74.e71–74.e11. [Google Scholar] [CrossRef]
- Wojczakowski, W.; Kimber-Trojnar, Ż.; Dziwisz, F.; Słodzińska, M.; Słodziński, H.; Leszczyńska-Gorzelak, B. Preeclampsia and Cardiovascular Risk for Offspring. J. Clin. Med. 2021, 10, 3154. [Google Scholar] [CrossRef]
- Dimitriadis, E.; Rolnik, D.L.; Zhou, W.; Estrada-Gutierrez, G.; Koga, K.; Francisco, R.P.V.; Whitehead, C.; Hyett, J.; da Silva Costa, F.; Nicolaides, K.; et al. Pre-eclampsia. Nat. Rev. Dis. Primers 2023, 9, 8. [Google Scholar] [CrossRef]
- Chang, K.-J.; Seow, K.-M.; Chen, K.-H. Preeclampsia: Recent Advances in Predicting, Preventing, and Managing the Maternal and Fetal Life-Threatening Condition. Int. J. Environ. Res. Public. Health 2023, 20, 2994. [Google Scholar] [CrossRef]
- Odigboegwu, O.; Pan, L.J.; Chatterjee, P. Use of Antihypertensive Drugs During Preeclampsia. Front. Cardiovasc. Med. 2018, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Catov, J.M.; Ness, R.B.; Kip, K.E.; Olsen, J. Risk of early or severe pre-eclampsia related to pre-existing conditions. Int. J. Epidemiol. 2007, 36, 412–419. [Google Scholar] [CrossRef]
- Abdelaal, M.; le Roux, C.W.; Docherty, N.G. Morbidity and mortality associated with obesity. Ann. Transl. Med. 2017, 5, 161. [Google Scholar] [CrossRef]
- Hill, B.; Skouteris, H.; Teede, H.J.; Bailey, C.; Baxter, J.-A.B.; Bergmeier, H.J.; Borges, A.L.V.; Harrison, C.L.; Jack, B.; Jorgensen, L.; et al. Health in Preconception, Pregnancy and Postpartum Global Alliance: International Network Preconception Research Priorities for the Prevention of Maternal Obesity and Related Pregnancy and Long-Term Complications. J. Clin. Med. 2019, 8, 2119. [Google Scholar] [CrossRef] [PubMed]
- Ogunwole, S.M.; Zera, C.A.; Stanford, F.C. Obesity Management in Women of Reproductive Age. JAMA 2021, 325, 433–434. [Google Scholar] [CrossRef]
- Meldrum, D.R.; Morris, M.A.; Gambone, J.C. Obesity pandemic: Causes, consequences, and solutions-but do we have the will? Fertil. Steril. 2017, 107, 833–839. [Google Scholar] [CrossRef]
- Rahman, M.M.; Abe, S.K.; Kanda, M.; Narita, S.; Rahman, M.S.; Bilano, V.; Ota, E.; Gilmour, S.; Shibuya, K. Maternal body mass index and risk of birth and maternal health outcomes in low- and middle-income countries: A systematic review and meta-analysis. Obes. Rev. 2015, 16, 758–770. [Google Scholar] [CrossRef]
- Mbah, A.K.; Kornosky, J.L.; Kristensen, S.; August, E.M.; Alio, A.P.; Marty, P.J.; Belogolovkin, V.; Bruder, K.; Salihu, H.M. Super-obesity and risk for early and late pre-eclampsia. BJOG 2010, 117, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Ness, R.B.; Markovic, N.; Roberts, J.M. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann. Epidemiol. 2005, 15, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Corvera, S.; Solivan-Rivera, J.; Yang Loureiro, Z. Angiogenesis in adipose tissue and obesity. Angiogenesis 2022, 25, 439–453. [Google Scholar] [CrossRef]
- Savini, I.; Catani, M.; Evangelista, D.; Gasperi, V.; Avigliano, L. Obesity-Associated Oxidative Stress: Strategies Finalized to Improve Redox State. Int. J. Mol. Sci. 2013, 14, 10497–10538. [Google Scholar] [CrossRef]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Portincasa, P.; Gómez-Ambrosi, J. Normalization of adiponectin concentrations by leptin replacement in ob/ob mice is accompanied by reductions in systemic oxidative stress and inflammation. Sci. Rep. 2017, 7, 2752. [Google Scholar] [CrossRef] [PubMed]
- Fryk, E.; Olausson, J.; Mossberg, K.; Strindberg, L.; Schmelz, M.; Brogren, H.; Gan, L.-M.; Piazza, S.; Provenzani, A.; Becattini, B.; et al. Hyperinsulinemia and insulin resistance in the obese may develop as part of a homeostatic response to elevated free fatty acids: A mechanistic case-control and a population-based cohort study. EBioMedicine 2021, 65, 103264. [Google Scholar] [CrossRef]
- Magkos, F.; Wang, X.; Mittendorfer, B. Metabolic actions of insulin in men and women. Nutrition 2010, 26, 686–693. [Google Scholar] [CrossRef]
- Sekulovski, N.; Whorton, A.E.; Shi, M.; Hayashi, K.; MacLean, J.A. Insulin signaling is an essential regulator of endometrial proliferation and implantation in mice. FASEB J. 2021, 35, e21440. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Chaudhry, P.; Asselin, E. Bridging endometrial receptivity and implantation: Network of hormones, cytokines, and growth factors. J. Endocrinol. 2011, 210, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Rabaglino, M.B.; Post Uiterweer, E.D.; Jeyabalan, A.; Hogge, W.A.; Conrad, K.P. Bioinformatics approach reveals evidence for impaired endometrial maturation before and during early pregnancy in women who developed preeclampsia. Hypertension 2015, 65, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef]
- Kasumov, T.; Solomon, T.P.; Hwang, C.; Huang, H.; Haus, J.M.; Zhang, R.; Kirwan, J.P. Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity 2015, 23, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.M.; Han, J.E.; Seok, H.H.; Lee, D.R.; Yoon, T.K.; Lee, W.S. Insulin resistance does not affect early embryo development but lowers implantation rate in in vitro maturation—In vitro fertilization-embryo transfer cycle. Clin. Endocrinol. 2013, 79, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Piltonen, T.T. Polycystic ovary syndrome: Endometrial markers. Best. Pract. Res. Clin. Obs. Gynaecol. 2016, 37, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, J.; He, J.; Wang, Y.; Liu, X.; Chen, X.; Tong, C.; Ding, Y.; Su, Y.; Chen, W.; et al. Mice endometrium receptivity in early pregnancy is impaired by maternal hyperinsulinemia. Mol. Med. Rep. 2017, 15, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, Y.; Caron, K.M. Haploinsufficiency for Adrenomedullin Reduces Pinopodes and Diminishes Uterine Receptivity in Mice1. Biol. Reprod. 2008, 79, 1169–1175. [Google Scholar] [CrossRef]
- Ujvari, D.; Jakson, I.; Oldmark, C.; Attarha, S.; Alkasalias, T.; Salamon, D.; Gidlöf, S.; Hirschberg, A.L. Prokineticin 1 is up-regulated by insulin in decidualizing human endometrial stromal cells. J. Cell. Mol. Med. 2017, 22, 163–172. [Google Scholar] [CrossRef]
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertil. Steril. 2019, 111, 489–496.e485. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; Nunes, C.; Correia-Branco, A.; Araújo, J.R.; Martel, F. Insulin Exhibits an Antiproliferative and Hypertrophic Effect in First Trimester Human Extravillous Trophoblasts. Reprod. Sci. 2017, 24, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, L.; Shao, Y.; Du, R.; Li, L.; Shi, X.; Bai, Y. Calcium/calmodulin dependent protein kinase IV in trophoblast cells under insulin resistance: Functional and metabolomic analyses. Mol. Med. 2023, 29, 82. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-a.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase– and MAP kinase–mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Bakris, G.L. Insulin and Endothelin: An Interplay Contributing to Hypertension Development? J. Clin. Endocrinol. Metab. 2007, 92, 379–385. [Google Scholar] [CrossRef]
- Cervero, A.; Horcajadas, J.A.; Dominguez, F.; Pellicer, A.; Simon, C. Leptin system in embryo development and implantation: A protein in search of a function. Reprod. Biomed. Online 2005, 10, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.M.; Carter, N.D.; Murray, J.F.; Scaramuzzi, R.J.; Wilson, C.A.; Stock, M.J. Leptin Requirement for Conception, Implantation, and Gestation in the Mouse. Endocrinology 2001, 142, 5198–5202. [Google Scholar] [CrossRef]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Lollmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Mendieta Zeron, H.; Garcia Solorio, V.J.; Nava Diaz, P.M.; Garduno Alanis, A.; Santillan Benitez, J.G.; Dominguez Garcia, V.; Escobar Briones, C.; Denova Gutierrez, E. Hyperleptinemia as a prognostic factor for preeclampsia: A cohort study. Acta Medica (Hradec Kral. ) 2012, 55, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, F.A.; Ngala, R.A.; Bawah, A.T.; Asare-Anane, H.; Alidu, H.; Hamid, A.M.; Wumbee, J.D.K. Adiposity and hyperleptinemia during the first trimester among pregnant women with preeclampsia. Int. J. Womens Health 2017, 9, 449–454. [Google Scholar] [CrossRef]
- Laird, S.M.; Quinton, N.D.; Anstie, B.; Li, T.C.; Blakemore, A.I.F. Leptin and leptin-binding activity in women with recurrent miscarriage: Correlation with pregnancy outcome. Hum. Reprod. 2001, 16, 2008–2013. [Google Scholar] [CrossRef]
- Lin, X.-H.; Liu, M.-E.; Xu, H.-Y.; Chen, X.-J.; Wang, H.; Tian, S.; Sheng, J.-Z.; Huang, H.-F. Leptin down-regulates γ-ENaC expression: A novel mechanism involved in low endometrial receptivity. Fertil. Steril. 2015, 103, 228–235.e223. [Google Scholar] [CrossRef]
- Tanaka, T.; Utsunomiya, T.; Bai, T.; Nakajima, S.; Umesaki, N. Leptin inhibits decidualization and enhances cell viability of normal human endometrial stromal cells. Int. J. Mol. Med. 2003, 12, 95–98. [Google Scholar] [CrossRef]
- Zeng, S.; Liu, Y.; Fan, P.; Yang, L.; Liu, X. Role of leptin in the pathophysiology of preeclampsia. Placenta 2023, 142, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.C.; Choy, M.Y.; Deng, W.; Wong, H.L.; Lau, W.M.; Cheung, A.N.; Ngan, H.Y.; Tsao, S.W. Establishment and characterization of a human first-trimester extravillous trophoblast cell line (TEV-1). J. Soc. Gynecol. Investig. 2005, 12, e21–e32. [Google Scholar] [CrossRef]
- Liu, H.; Wu, Y.; Qiao, F.; Gong, X. Effect of leptin on cytotrophoblast proliferation and invasion. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 631–636. [Google Scholar] [CrossRef]
- Al-Sultan, A.I.; Al-Elq, A.H. Leptin levels in normal weight and obese saudi adults. J. Fam. Community Med. 2006, 13, 97–102. [Google Scholar]
- Fan, M.; Dong, L.; Meng, Y.; Wang, Y.; Zhen, J.; Qiu, J.; Huang, M.-H. Leptin Promotes HTR-8/SVneo Cell Invasion via the Crosstalk between MTA1/WNT and PI3K/AKT Pathways. Dis. Markers 2022, 2022, 7052176. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, H.; Shao, Q.; Dong, Z.; Xie, Q.; Zhao, L.; Wang, Q.; Kong, B.; Qu, X. Leptin-Promoted Human Extravillous Trophoblast Invasion Is MMP14 Dependent and Requires the Cross Talk Between Notch1 and PI3K/Akt Signaling1. Biol. Reprod. 2014, 90, 78. [Google Scholar] [CrossRef]
- Korda, M.; Kubant, R.; Patton, S.; Malinski, T. Leptin-induced endothelial dysfunction in obesity. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1514–H1521. [Google Scholar] [CrossRef]
- Manuel-Apolinar, L.; Lopez-Romero, R.; Zarate, A.; Damasio, L.; Ruiz, M.; Castillo-Hernandez, C.; Guevara, G.; Mera-Jimenez, E. Leptin mediated ObRb receptor increases expression of adhesion intercellular molecules and cyclooxygenase 2 on murine aorta tissue inducing endothelial dysfunction. Int. J. Clin. Exp. Med. 2013, 6, 192–196. [Google Scholar]
- Mizuno, Y.; Sotomaru, Y.; Katsuzawa, Y.; Kono, T.; Meguro, M.; Oshimura, M.; Kawai, J.; Tomaru, Y.; Kiyosawa, H.; Nikaido, I.; et al. Asb4, Ata3, and Dcn Are Novel Imprinted Genes Identified by High-Throughput Screening Using RIKEN cDNA Microarray. Biochem. Biophys. Res. Commun. 2002, 290, 1499–1505. [Google Scholar] [CrossRef]
- Kohroki, J.; Nishiyama, T.; Nakamura, T.; Masuho, Y. ASB proteins interact with Cullin5 and Rbx2 to form E3 ubiquitin ligase complexes. FEBS Lett. 2005, 579, 6796–6802. [Google Scholar] [CrossRef]
- Linossi, E.M.; Nicholson, S.E. The SOCS box—Adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 2012, 64, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, A.; Katsanakis, K.D.; Bheda, F.; Pillay, T.S. Asb6, an Adipocyte-specific Ankyrin and SOCS Box Protein, Interacts with APS to Enable Recruitment of Elongins B and C to the Insulin Receptor Signaling Complex. J. Biol. Chem. 2004, 279, 38881–38888. [Google Scholar] [CrossRef]
- Kile, B.T.; Viney, E.M.; Willson, T.A.; Brodnicki, T.C.; Cancilla, M.R.; Herlihy, A.S.; Croker, B.A.; Baca, M.; Nicola, N.A.; Hilton, D.J.; et al. Cloning and characterization of the genes encoding the ankyrin repeat and SOCS box-containing proteins Asb-1, Asb-2, Asb-3 and Asb-4. Gene 2000, 258, 31–41. [Google Scholar] [CrossRef]
- Li, J.-Y.; Chai, B.; Zhang, W.; Wu, X.; Zhang, C.; Fritze, D.; Xia, Z.; Patterson, C.; Mulholland, M.W. Ankyrin repeat and SOCS box containing protein 4 (Asb-4) colocalizes with insulin receptor substrate 4 (IRS4) in the hypothalamic neurons and mediates IRS4 degradation. BMC Neurosci. 2011, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Mulholland, M.W.; Wang, H.; Zhang, W.; Chai, B.-X.; Li, J.-Y. Expression of Ankyrin Repeat and Suppressor of Cytokine Signaling Box Protein 4 (Asb-4) in Proopiomelanocortin Neurons of the Arcuate Nucleus of Mice Produces a Hyperphagic, Lean Phenotype. Endocrinology 2010, 151, 134–142. [Google Scholar] [CrossRef]
- Vagena, E.; Crneta, J.; Engström, P.; He, L.; Yulyaningsih, E.; Korpel, N.L.; Cheang, R.T.; Bachor, T.P.; Huang, A.; Michel, G.; et al. ASB4 modulates central melanocortinergic neurons and calcitonin signaling to control satiety and glucose homeostasis. Sci. Signal. 2022, 15, eabj8204. [Google Scholar] [CrossRef]
- Li, J.Y.; Kuick, R.; Thompson, R.C.; Misek, D.E.; Lai, Y.M.; Liu, Y.Q.; Chai, B.X.; Hanash, S.M.; Gantz, I. Arcuate Nucleus Transcriptome Profiling Identifies Ankyrin Repeat and Suppressor of Cytokine Signalling Box-Containing Protein 4 as a Gene Regulated by Fasting in Central Nervous System Feeding Circuits. J. Neuroendocrinol. 2005, 17, 394–404. [Google Scholar] [CrossRef]
- Li, J.-Y.; Chai, B.-X.; Zhang, W.; Liu, Y.-Q.; Ammori, J.B.; Mulholland, M.W. Akyrin repeat and SOCS box containing protein 4 (Asb-4) interacts with GPS1 (CSN1) and inhibits c-Jun NH2-terminal kinase activity. Cell. Signal. 2007, 19, 1185–1192. [Google Scholar] [CrossRef]
- Au, V.; H Tsang, F.; Man, K.; Tat Fan, S.; Tp Poon, R.; P Lee, N. Expression of ankyrin repeat and SOCS box containing 4 (ASB4) confers migration and invasion properties of hepatocellular carcinoma cells. BioScience Trends 2014, 8, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Siani, A. Role of microRNAs in obesity and obesity-related diseases. Genes. Nutr. 2017, 12, 23. [Google Scholar] [CrossRef]
- Bode, M.; Wu, Y.; Pi, X.; Lockyer, P.; Dechyapirom, W.; Portbury, A.L.; Patterson, C. Regulation of ankyrin repeat and suppressor of cytokine signalling box protein 4 expression in the immortalized murine endothelial cell lines MS1 and SVR: A role for tumour necrosis factor alpha and oxygen. Cell Biochem. Funct. 2011, 29, 334–341. [Google Scholar] [CrossRef]
- Khanna, D.; Khanna, S.; Khanna, P.; Kahar, P.; Patel, B.M. Obesity: A Chronic Low-Grade Inflammation and Its Markers. Cureus 2022, 14, e22711. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, Inflammation, and Metabolic Disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef]
- Ling, F.; Kang, B.; Sun, X.H. Id proteins: Small molecules, mighty regulators. Curr. Top. Dev. Biol. 2014, 110, 189–216. [Google Scholar] [CrossRef]
- Liu, Y.P.; Burleigh, D.; Durning, M.; Hudson, L.; Chiu, I.M.; Golos, T.G. Id2 is a primary partner for the E2-2 basic helix-loop-helix transcription factor in the human placenta. Mol. Cell Endocrinol. 2004, 222, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Tsunedomi, R.; Iizuka, N.; Tamesa, T.; Sakamoto, K.; Hamaguchi, T.; Somura, H.; Yamada, M.; Oka, M. Decreased ID2 promotes metastatic potentials of hepatocellular carcinoma by altering secretion of vascular endothelial growth factor. Clin. Cancer Res. 2008, 14, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Rothschild, G.; Yokota, Y.; Russell, R.G.; Iavarone, A. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol. Cell Biol. 2005, 25, 3563–3574. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Visceral Adipose Tissue (g) | P-Cholesterol (mg/dL) | P-Triglyceride (mg/dL) | P-Glucose (mg/dL) | P-Insulin (mg/dL) | P-Leptin (mg/dL) | Fetal Number (#) | Fetal Weight (g) | Placental Weight (g) | |
|---|---|---|---|---|---|---|---|---|---|
| NC-WT | - | - | - | - | - | - | - | - | - |
| HFD-WT | - | - | - | - | - | ↑ [74] | - | - | - |
| NC-Asb4−/− | - | - | - | - | - | - | ↓ | - | ↑ |
| HFD-Asb4−/− | ↑ | ↑ | - | - | ↑ | ↑↑ | ↓↓ | - | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Ssengonzi, R.; Townley-Tilson, W.H.D.; Kayashima, Y.; Maeda-Smithies, N.; Li, F. The Roles of Obesity and ASB4 in Preeclampsia Pathogenesis. Int. J. Mol. Sci. 2024, 25, 9017. https://doi.org/10.3390/ijms25169017
Wang Y, Ssengonzi R, Townley-Tilson WHD, Kayashima Y, Maeda-Smithies N, Li F. The Roles of Obesity and ASB4 in Preeclampsia Pathogenesis. International Journal of Molecular Sciences. 2024; 25(16):9017. https://doi.org/10.3390/ijms25169017
Chicago/Turabian StyleWang, Yuye, Rebecca Ssengonzi, W. H. Davin Townley-Tilson, Yukako Kayashima, Nobuyo Maeda-Smithies, and Feng Li. 2024. "The Roles of Obesity and ASB4 in Preeclampsia Pathogenesis" International Journal of Molecular Sciences 25, no. 16: 9017. https://doi.org/10.3390/ijms25169017
APA StyleWang, Y., Ssengonzi, R., Townley-Tilson, W. H. D., Kayashima, Y., Maeda-Smithies, N., & Li, F. (2024). The Roles of Obesity and ASB4 in Preeclampsia Pathogenesis. International Journal of Molecular Sciences, 25(16), 9017. https://doi.org/10.3390/ijms25169017