
Antioxidant Role of Probiotics in Inflammation-Induced Colorectal Cancer


Abstract
1. Introduction
2. Reactive Oxygen Species, Oxidative Stress, and Colorectal Cancer (CRC)
2.1. Antioxidants, Nrf2-Keap1, and Carcinogenesis
2.2. Inflammatory Signaling, Oxidative Stress, and Carcinogenesis
3. Gut Microbiota and Colorectal Cancer (CRC)
3.1. Microbiota, Reactive Oxygen Species, and Colorectal Cancer
Antibiotics, Redox Balance, and Colorectal Cancer
3.2. Microbiota Metabolites, Reactive Oxygen Species, and Colorectal Cancer
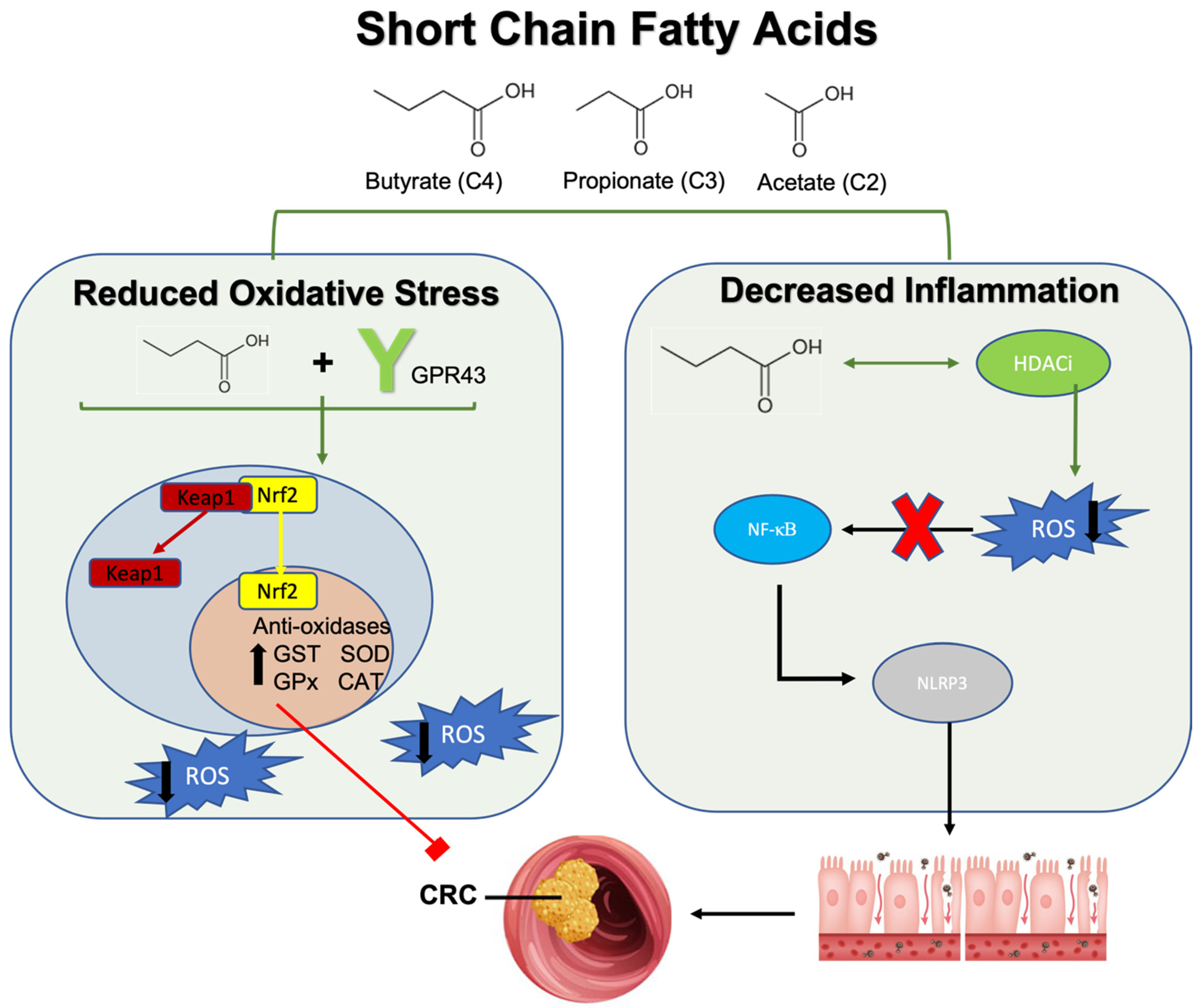
3.2.1. Short Chain Fatty Acids (SCFAs), Reactive Oxygen Species, and Colorectal Cancer
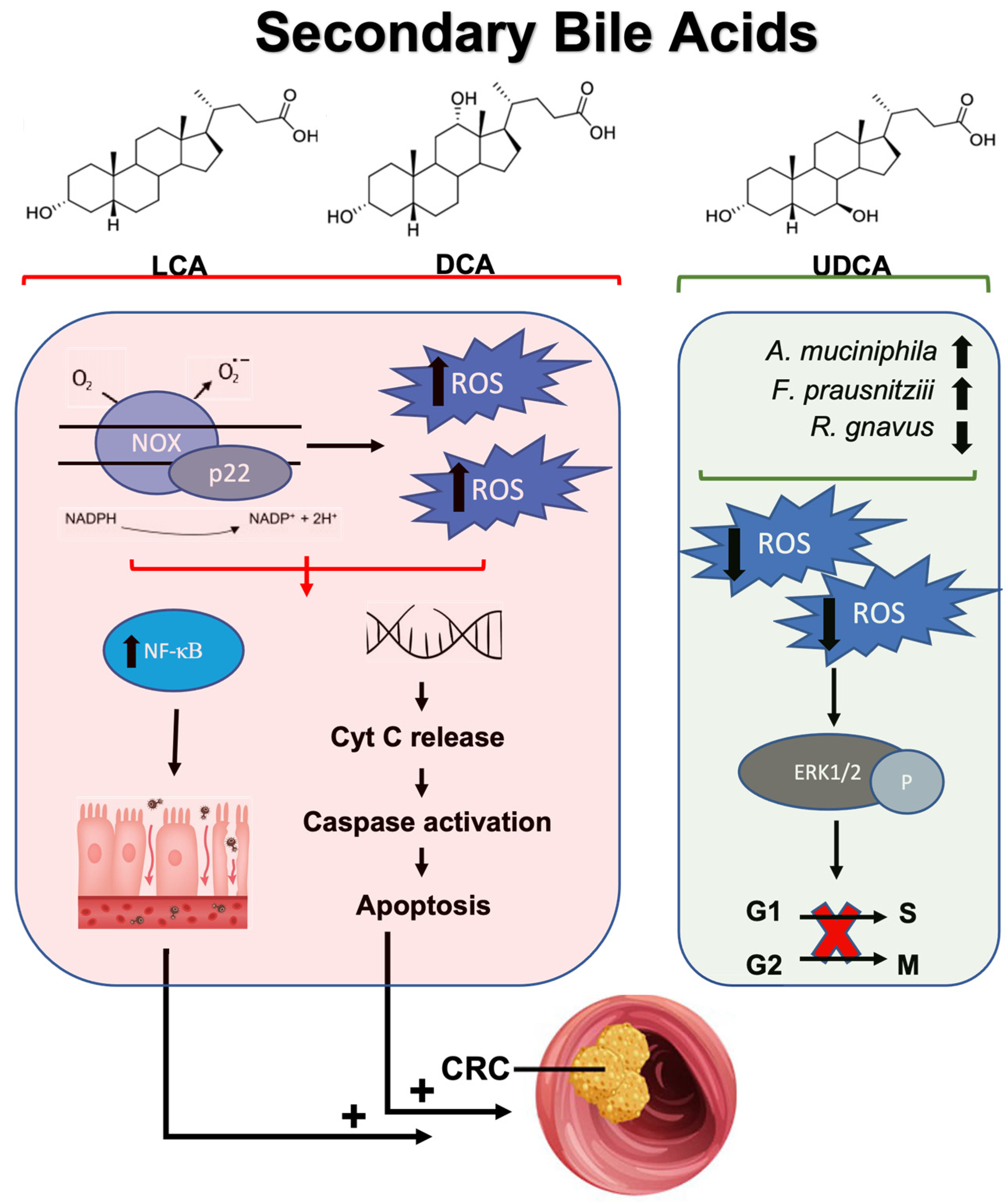
3.2.2. Secondary Bile Acids, Reactive Oxygen Species, and Colorectal Cancer
3.3. Trimethylamine-N-Oxide, Oxidative Stress, and Colorectal Cancer
4. Probiotics, Antioxidant Properties, and Colorectal Cancer
4.1. Probiotics, Antioxidant Enzymes, and Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probiotic | Results/Implications | Reference | |
|---|---|---|---|
| Antioxidant Enzymes | Lactococcus lactis | Increased catalase activity resulting in significantly decreased ROS-mediated inflammatory damage and tumor appearance | [255] |
| Lactobacillus plantarum | Administration into DMH-induced colon cancer cells, increased antioxidant enzymes (SOD, CAT, and GST) to reduce mean tumor volume and size | [27] | |
| Lactobacillus acidophilus | Produced metabolic derivative, EPS, which increased antioxidant enzymes (SOD, CAT, and GPx) to mitigate DMH-induced colon cancer | [256] | |
| Lactobacillus delbruecki | EPS reduced lipid peroxidation with concurrent increases in antioxidant enzyme activity (SOD, CAT, GPx, and GSH) Ameliorated Inflammatory damage to colonic epithelium | [258] | |
| Nrf2-Keap1 | Lactobacillus casei | Reduces oxidative and inflammatory stress in enterocytes by activating the Nrf2-Keap1 and NF-KB pathways Nrf2 activation reduced ROS accumulation through the upregulation of GPx | [248] |
| Bifidobacterium bifidum, Lactobacillus gasseri | Activating effects on Nrf2 in combination with vitamin D3 to increase GST and inhibit the onset of colorectal carcinogenesis | [261] | |
| NF-κB | Faecalibacterium prausnatzii | Inhibited NF-κB activation to attenuate the proliferation of CRC cell lines Reduced lipid peroxidation and oxidative stress | [114] |
| Lactobacillus fermentum | Attenuated NF-κB signaling in DSS-induced colorectal cancer Decreased pro-oxidant cyclo-oxygenase 2 | [262] | |
| Lactiplantibacillus plantarum-12 | EPS production alleviated inflammation through inhibition of NF-κB signaling pathway Related reduction in pro-inflammatory cytokines to inhibit inflammation-induced CRC development | [263] | |
| Clostridium butyricum | Decreases the phosphorylation of NF-κB to decrease cytokine activation, ultimately reducing tumor incidence and size in a colitis-induced CRC model Improves microbial composition in the same CRC model | [264] | |
| Bifidobacterium longum | Diminished NF-κB induction in CRC cells to attenuate development of aberrant crypt foci | [265] | |
| VSL#3 | Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus casei, Lactobacillus bulgaricus, Bifidobacterium breve, Bifidobacterium longum, Bifidobacterium infantis, Streptococcus thermophilus | Reduced the expression of pro-oxidant enzymes such as cyclo-oxygenase 2 in DSS-induced mice Attenuate IL-6, IL-1β and increased concentrations of regulatory IL-10 | [266] |
| Attenuated ROS concentrations to reduce pro-inflammatory chemokines and colitis symptoms Reduced barrier permeability | [267] | ||
| Promotes butyrate production, which is found to increase the expression of antioxidants in colitis-associated CRC models | [268] | ||
| Immuno-therapy Augmentation | Lactobacillus rhamnosus GG | Incorporated as part of a probiotic-CRISPR cancer immunotherapy system that can penetrate hypoxic tumor environments Allowed CRISPR system to reach tumor microenvironment to enhance ROS release and promote cell death | [269] |
| Lactobacillus reuteri | Metabolite, reuterin, alters tumor microenvironment oxidative status Selectively enhances ROS expression within colorectal cancer cells to suppress tumor growth | [244] | |
| Enterococcus faecalis | Reduced NLRP3 inflammasome activity through attenuation of phagocytosis, which is necessary for activation Prevented the onset of CRC development | [270] | |
| Enterococcus durans | Nanoparticle treatment of probiotic cultures increased folate concentrations ROS-producing capabilities of the treated probiotic decreased CRC viability by 19% | [271] | |
| Roseburia intestinalis | Induction of CD8+ T-cell mediated cytotoxicity through metabolite, butyrate Augmented PD-1 therapy against CRC | [251] | |
| Radiotherapy Augmentation | Roseburia intestinalis | Sensitized inflammation-induced CRC cells to radiotherapy treatment through butyrate production Butyrate facilitated autophagy in irradiated cells to induce cell death | [272] |
4.2. Probiotics, Antioxidant Signaling Pathways, and Colorectal Cancer
4.3. Probiotics, ROS-Mediated Inflammatory Signaling Pathways, and Colorectal Cancer
4.4. Probiotics, Antioxidants and Colorectal Cancer Immunotherapy, and Treatment
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Patel, S.G.; Karlitz, J.J.; Yen, T.; Lieu, C.H.; Boland, C.R. The rising tide of early-onset colorectal cancer: A comprehensive review of epidemiology, clinical features, biology, risk factors, prevention, and early detection. Lancet Gastroenterol. Hepatol. 2022, 7, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- Murphy, C.C.; Zaki, T.A. Changing epidemiology of colorectal cancer—Birth cohort effects and emerging risk factors. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 25–34. [Google Scholar] [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Gausman, V.; Dornblaser, D.; Anand, S.; Hayes, R.B.; O’Connell, K.; Du, M.; Liang, P.S. Risk Factors Associated With Early-Onset Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 2752–2759.e2752. [Google Scholar] [CrossRef]
- Feizi, H.; Rezaee, M.A.; Ghotaslou, R.; Sadrkabir, M.; Jadidi-Niaragh, F.; Gholizadeh, P.; Taghizadeh, S.; Ghanbarov, K.; Yousefi, M.; Kafil, H.S. Gut Microbiota and Colorectal Cancer Risk Factors. Curr. Pharm. Biotechnol. 2023, 24, 1018–1034. [Google Scholar] [CrossRef]
- Bai, X.; Wei, H.; Liu, W.; Coker, O.O.; Gou, H.; Liu, C.; Zhao, L.; Li, C.; Zhou, Y.; Wang, G.; et al. Cigarette smoke promotes colorectal cancer through modulation of gut microbiota and related metabolites. Gut 2022, 71, 2439–2450. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e132. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Wang, M.; Ajibade, A.A.; Tan, P.; Fu, C.; Chen, L.; Zhu, M.; Hao, Z.Z.; Chu, J.; Yu, X.; et al. Microbiota regulate innate immune signaling and protective immunity against cancer. Cell Host Microbe 2021, 29, 959–974.e957. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Oh, J.; Xue, M.; Huh, W.J.; Wang, J.; Gonzalez-Hernandez, J.A.; Rice, T.A.; Martin, A.L.; Song, D.; Crawford, J.M.; et al. Commensal microbiota from patients with inflammatory bowel disease produce genotoxic metabolites. Science 2022, 378, eabm3233. [Google Scholar] [CrossRef]
- Singh, V.; Shirbhate, E.; Kore, R.; Vishwakarma, S.; Parveen, S.; Veerasamy, R.; Tiwari, A.K.; Rajak, H. Microbial Metabolites-Induced Epigenetic Modifications for Inhibition of Colorectal Cancer: Current Status and Future Perspectives. Mini Rev. Med. Chem. 2024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.S.; Xie, Y.L.; Xiao, X.Y.; Kang, Z.R.; Lin, X.L.; Zhang, L.; Li, C.S.; Qian, Y.; Xu, P.P.; Leng, X.X.; et al. Fusobacterium nucleatum-derived succinic acid induces tumor resistance to immunotherapy in colorectal cancer. Cell Host Microbe 2023, 31, 781–797.e789. [Google Scholar] [CrossRef] [PubMed]
- Lamaudière, M.T.F.; Arasaradnam, R.; Weedall, G.D.; Morozov, I.Y. The Colorectal Cancer Microbiota Alter Their Transcriptome To Adapt to the Acidity, Reactive Oxygen Species, and Metabolite Availability of Gut Microenvironments. mSphere 2023, 8, e0062722. [Google Scholar] [CrossRef]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Treviño, S.; Rosas-Murrieta, N.H.; Millán-Perez-Peña, L.; Maycotte, P. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.I.; Jo, Y.; Yuk, H.J.; Kim, S.Y.; Kim, H.; Kim, H.J.; Hwang, S.K.; Park, K.S. Potential Phytotherapy of DSS-Induced Colitis: Ameliorating Reactive Oxygen Species-Mediated Necroptosis and Gut Dysbiosis with a New Crataegus pinnatifida Bunge Variety-Daehong. Antioxidants 2024, 13, 340. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.T.; Vasconcelos, Q.; Aragão, G.F. Fructooligosaccharides on inflammation, immunomodulation, oxidative stress, and gut immune response: A systematic review. Nutr. Rev. 2022, 80, 709–722. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Wang, Y.; Xu, H.; Mei, X.; Yu, D.; Wang, Y.; Li, W. Antioxidant Properties of Probiotic Bacteria. Nutrients 2017, 9, 521. [Google Scholar] [CrossRef]
- Khalil, N.A.; NA, A.L.; JZ, A.L.; Mohamed Ahmed, I.A. Anti-inflammatory effects of bay laurel (Laurus nobilis L.) towards the gut microbiome in dextran sodium sulfate induced colitis animal models. Food Sci. Nutr. 2024, 12, 2650–2660. [Google Scholar] [CrossRef]
- Yu, F.; Hu, X.; Ren, H.; Wang, X.; Shi, R.; Guo, J.; Chang, J.; Zhou, X.; Jin, Y.; Li, Y.; et al. Protective effect of synbiotic combination of Lactobacillus plantarum SC-5 and olive oil extract tyrosol in a murine model of ulcerative colitis. J. Transl. Med. 2024, 22, 308. [Google Scholar] [CrossRef]
- Tan, L.T.; Chan, K.G.; Pusparajah, P.; Yin, W.F.; Khan, T.M.; Lee, L.H.; Goh, B.H. Mangrove derived Streptomyces sp. MUM265 as a potential source of antioxidant and anticolon-cancer agents. BMC Microbiol. 2019, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Torres-Maravilla, E.; Boucard, A.S.; Al Azzaz, J.; Gontier, S.; Kulakauskas, S.; Langella, P.; Bermúdez-Humarán, L.G. Assessment of the safety of Levilactobacillus brevis CNCM I-5321, a probiotic candidate strain isolated from pulque with anti-proliferative activities. Benef. Microbes 2023, 14, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Oh, N.S.; Joung, J.Y.; Lee, J.Y.; Kim, Y.J.; Kim, Y.; Kim, S.H. A synbiotic combination of Lactobacillus gasseri 505 and Cudrania tricuspidata leaf extract prevents hepatic toxicity induced by colorectal cancer in mice. J. Dairy. Sci. 2020, 103, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.S.; Kanmani, P.; Yuvaraj, N.; Paari, K.A.; Pattukumar, V.; Thirunavukkarasu, C.; Arul, V. Lactobacillus plantarum AS1 isolated from south Indian fermented food Kallappam suppress 1,2-dimethyl hydrazine (DMH)-induced colorectal cancer in male Wistar rats. Appl. Biochem. Biotechnol. 2012, 166, 620–631. [Google Scholar] [CrossRef]
- Desrouillères, K.; Millette, M.; Bagheri, L.; Maherani, B.; Jamshidian, M.; Lacroix, M. The synergistic effect of cell wall extracted from probiotic biomass containing Lactobacillus acidophilus CL1285, L. casei LBC80R, and L. rhamnosus CLR2 on the anticancer activity of cranberry juice-HPLC fractions. J. Food Biochem. 2020, 44, e13195. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; HA, A.L.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Pryczynicz, A.; Dymicka-Piekarska, V.; Kamińska, J.; Koper-Lenkiewicz, O.; Matowicka-Karna, J.; Kędra, B.; Zalewska, A.; et al. Pro-Oxidant Enzymes, Redox Balance and Oxidative Damage to Proteins, Lipids and DNA in Colorectal Cancer Tissue. Is Oxidative Stress Dependent on Tumour Budding and Inflammatory Infiltration? Cancers 2020, 12, 1636. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Carini, F.; Mazzola, M.; Rappa, F.; Jurjus, A.; Geagea, A.G.; Al Kattar, S.; Bou-Assi, T.; Jurjus, R.; Damiani, P.; Leone, A.; et al. Colorectal Carcinogenesis: Role of Oxidative Stress and Antioxidants. Anticancer. Res. 2017, 37, 4759–4766. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.Y.; Park, J.; Jang, E.S.; Chi, S.W. 8-Oxoguanine: From oxidative damage to epigenetic and epitranscriptional modification. Exp. Mol. Med. 2022, 54, 1626–1642. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Yang, J.; Zhang, J.; Zhang, G. The lipid peroxidation product EKODE exacerbates colonic inflammation and colon tumorigenesis. Redox Biol. 2021, 42, 101880. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hwang, I.; Kang, Y.N.; Choi, I.J.; Kim, D.K. Genetic characteristics of mitochondrial DNA was associated with colorectal carcinogenesis and its prognosis. PLoS ONE 2015, 10, e0118612. [Google Scholar] [CrossRef] [PubMed]
- López, I.; Oliveira, L.P.; Tucci, P.; Álvarez-Valín, F.; Coudry, R.A.; Marín, M. Different mutation profiles associated to P53 accumulation in colorectal cancer. Gene 2012, 499, 81–87. [Google Scholar] [CrossRef]
- Tomonaga, T.; Izumi, H.; Yoshiura, Y.; Nishida, C.; Yatera, K.; Morimoto, Y. Examination of Surfactant Protein D as a Biomarker for Evaluating Pulmonary Toxicity of Nanomaterials in Rat. Int. J. Mol. Sci. 2021, 22, 4635. [Google Scholar] [CrossRef]
- Obtulowicz, T.; Swoboda, M.; Speina, E.; Gackowski, D.; Rozalski, R.; Siomek, A.; Janik, J.; Janowska, B.; Ciesla, J.M.; Jawien, A.; et al. Oxidative stress and 8-oxoguanine repair are enhanced in colon adenoma and carcinoma patients. Mutagenesis 2010, 25, 463–471. [Google Scholar] [CrossRef]
- Lei, L.; Zhang, J.; Decker, E.A.; Zhang, G. Roles of Lipid Peroxidation-Derived Electrophiles in Pathogenesis of Colonic Inflammation and Colon Cancer. Front. Cell Dev. Biol. 2021, 9, 665591. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, J.; Edin, M.L.; Wang, Y.; Luo, Y.; Wan, D.; Yang, H.; Song, C.Q.; Xue, W.; Sanidad, K.Z.; et al. Targeted Metabolomics Identifies the Cytochrome P450 Monooxygenase Eicosanoid Pathway as a Novel Therapeutic Target of Colon Tumorigenesis. Cancer Res. 2019, 79, 1822–1830. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Yang, H.; Shao, D.; Zhao, X.; Zhang, G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic. Biol. Med. 2019, 131, 237–242. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, S.; Ahn, D.U. Protein oxidation: Basic principles and implications for meat quality. Crit. Rev. Food Sci. Nutr. 2013, 53, 1191–1201. [Google Scholar] [CrossRef]
- Yang, H.Y.; Chay, K.O.; Kwon, J.; Kwon, S.O.; Park, Y.K.; Lee, T.H. Comparative proteomic analysis of cysteine oxidation in colorectal cancer patients. Mol. Cells 2013, 35, 533–542. [Google Scholar] [CrossRef]
- Cui, Z.; Cong, M.; Yin, S.; Li, Y.; Ye, Y.; Liu, X.; Tang, J. Role of protein degradation systems in colorectal cancer. Cell Death Discov. 2024, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Gao, L.; Loveless, J.; Shay, C.; Teng, Y. Targeting ROS-Mediated Crosstalk between Autophagy and Apoptosis in Cancer. Adv. Exp. Med. Biol. 2020, 1260, 1–12. [Google Scholar] [CrossRef]
- Huang, Z.; Gan, S.; Zhuang, X.; Chen, Y.; Lu, L.; Wang, Y.; Qi, X.; Feng, Q.; Huang, Q.; Du, B.; et al. Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy. Cells 2022, 11, 2472. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.C.; Parish, S.L.; Williams, C.S. Healthcare expenditures for autism during times of school transition: Some vulnerable families fall behind. Matern. Child. Health J. 2014, 18, 1936–1944. [Google Scholar] [CrossRef]
- Chang, Y.; Li, F.; Wang, Z.; Zhao, Q.; Wang, Z.; Han, X.; Xu, Z.; Yu, C.; Liu, Y.; Chang, S.; et al. Oxidative balance score: A potential tool for reducing the risk of colorectal cancer and its subsites incidences. Front. Endocrinol. 2024, 15, 1397512. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Cheung, K.L.; Lee, J.H.; Khor, T.O.; Wu, T.Y.; Li, G.X.; Chan, J.; Yang, C.S.; Kong, A.N. Nrf2 knockout enhances intestinal tumorigenesis in Apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol. Carcinog. 2014, 53, 77–84. [Google Scholar] [CrossRef]
- Hammad, A.; Zheng, Z.H.; Gao, Y.; Namani, A.; Shi, H.F.; Tang, X. Identification of novel Nrf2 target genes as prognostic biomarkers in colitis-associated colorectal cancer in Nrf2-deficient mice. Life Sci. 2019, 238, 116968. [Google Scholar] [CrossRef] [PubMed]
- Medoro, A.; Saso, L.; Scapagnini, G.; Davinelli, S. NRF2 signaling pathway and telomere length in aging and age-related diseases. Mol. Cell. Biochem. 2023. [Google Scholar] [CrossRef]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, D.; Liu, Y.; Zhou, Y. Role of NRF2 in Colorectal Cancer Prevention and Treatment. Technol. Cancer Res. Treat. 2022, 21, 15330338221105736. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, Z.M.; Wang, K. Identification of ROS and KEAP1-related genes and verified targets of α-hederin induce cell death for CRC. Drug Dev. Res. 2024, 85, e22200. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Fan, C.W.; Tseng, W.K.; Chen, J.R.; Hua, C.C. The tumour/normal tissue ratio of Keap1 protein is a predictor for lymphovascular invasion in colorectal cancer: A correlation study between the Nrf2 and KRas pathways. Biomarkers 2022, 27, 701–707. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef]
- Pouremamali, F.; Pouremamali, A.; Dadashpour, M.; Soozangar, N.; Jeddi, F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun. Signal. 2022, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Song, X.; Yu, W.; Pan, Y.; Zhang, Y.; Jian, H.; He, B. The role and mechanism of cinnamaldehyde in cancer. J. Food Drug Anal. 2024, 32, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tsujinaka, S.; Miura, T.; Kitamura, Y.; Suzuki, H.; Shibata, C. Inflammatory Bowel Disease and Colorectal Cancer: Epidemiology, Etiology, Surveillance, and Management. Cancers 2023, 15, 4154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Veauthier, B.; Hornecker, J.R. Crohn’s Disease: Diagnosis and Management. Am. Fam. Physician 2018, 98, 661–669. [Google Scholar] [PubMed]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef]
- Abu-Freha, N.; Cohen, B.; Gordon, M.; Weissmann, S.; Kestenbaum, E.H.; Vosko, S.; Abu-Tailakh, M.; Ben-Shoshan, L.; Cohen, D.L.; Shirin, H. Colorectal cancer among inflammatory bowel disease patients: Risk factors and prevalence compared to the general population. Front. Med. 2023, 10, 1225616. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxid. Med. Cell. Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Kosaka, T.; Yoshino, J.; Inui, K.; Wakabayashi, T.; Kobayashi, T.; Watanabe, S.; Hayashi, S.; Hirokawa, Y.; Shiraishi, T.; Yamamoto, T.; et al. Involvement of NAD(P)H:quinone oxidoreductase 1 and superoxide dismutase polymorphisms in ulcerative colitis. DNA Cell Biol. 2009, 28, 625–631. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Zhao, B.L.; Qian, Z.; Xu, Y.; Ding, Y.Q. Association of Glutathione S-Transferase M1 null genotype with inflammatory bowel diseases: A systematic review and meta-analysis. Medicine 2019, 98, e17722. [Google Scholar] [CrossRef] [PubMed]
- Hertervig, E.; Nilsson, A.; Seidegård, J. The expression of glutathione transferase mu in patients with inflammatory bowel disease. Scand. J. Gastroenterol. 1994, 29, 729–735. [Google Scholar] [CrossRef]
- Varzari, A.; Deyneko, I.V.; Tudor, E.; Turcan, S. Polymorphisms of glutathione S-transferase and methylenetetrahydrofolate reductase genes in Moldavian patients with ulcerative colitis: Genotype-phenotype correlation. Meta Gene 2016, 7, 76–82. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, T.; Sun, D.; Xie, C.; Wang, T.; Liu, X.; Wang, J.; Wang, Q.; Luo, Y.; Wang, P.; et al. Rutaecarpine inhibits KEAP1-NRF2 interaction to activate NRF2 and ameliorate dextran sulfate sodium-induced colitis. Free Radic. Biol. Med. 2020, 148, 33–41. [Google Scholar] [CrossRef]
- Karban, A.; Hartman, C.; Eliakim, R.; Waterman, M.; Nesher, S.; Barnett-Griness, O.; Shamir, R. Paraoxonase (PON)1 192R allele carriage is associated with reduced risk of inflammatory bowel disease. Dig. Dis. Sci. 2007, 52, 2707–2715. [Google Scholar] [CrossRef]
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: Review of the evidence. Tech. Coloproctol. 2019, 23, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz Shahbaz, S.; Koushki, K.; Ayati, S.H.; Bland, A.R.; Bezsonov, E.E.; Sahebkar, A. Inflammasomes and Colorectal Cancer. Cells 2021, 10, 2172. [Google Scholar] [CrossRef]
- Patel, M.; Horgan, P.G.; McMillan, D.C.; Edwards, J. NF-κB pathways in the development and progression of colorectal cancer. Transl. Res. 2018, 197, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, Q.; Fan, H.; Wang, J.; Liu, X.; Wang, H.; Wang, Y.; Hu, R. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem. Pharmacol. 2016, 105, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.L.; Yin, H.R.; He, Q.Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef]
- Deng, Q.; Geng, Y.; Zhao, L.; Li, R.; Zhang, Z.; Li, K.; Liang, R.; Shao, X.; Huang, M.; Zuo, D.; et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett. 2019, 442, 21–30. [Google Scholar] [CrossRef]
- Hassanzadeh, P. Colorectal cancer and NF-κB signaling pathway. Gastroenterol. Hepatol. Bed Bench 2011, 4, 127–132. [Google Scholar]
- Karin, M. How NF-kappaB is activated: The role of the IkappaB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Berkovich, L.; Gerber, M.; Katzav, A.; Kidron, D.; Avital, S. NF-kappa B expression in resected specimen of colonic cancer is higher compared to its expression in inflammatory bowel diseases and polyps. Sci. Rep. 2022, 12, 16645. [Google Scholar] [CrossRef]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-κB signaling pathway in the pathogenesis of colorectal cancer. Gene 2020, 726, 144132. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Rajitha, B.; Belalcazar, A.; Nagaraju, G.P.; Shaib, W.L.; Snyder, J.P.; Shoji, M.; Pattnaik, S.; Alam, A.; El-Rayes, B.F. Inhibition of NF-κB translocation by curcumin analogs induces G0/G1 arrest and downregulates thymidylate synthase in colorectal cancer. Cancer Lett. 2016, 373, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, P.; Zhang, W.; Du, Q.; Tang, J.; Wang, H.; Lu, J.; Hu, R. GEN-27, a Newly Synthetic Isoflavonoid, Inhibits the Proliferation of Colon Cancer Cells in Inflammation Microenvironment by Suppressing NF-κB Pathway. Mediat. Inflamm. 2016, 2016, 2853040. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Sun, M.; Motolani, A.; Lu, T. The Pivotal Player: Components of NF-κB Pathway as Promising Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 7429. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-κB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2021, 9, 809952. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, M.J.; Vicente, A.M.; Araico, A.; Dominguez, J.N.; Terencio, M.C.; Ferrándiz, M.L. Role of nuclear factor-kappaB and heme oxygenase-1 in the mechanism of action of an anti-inflammatory chalcone derivative in RAW 264.7 cells. Br. J. Pharmacol. 2004, 142, 1191–1199. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, T.; Ye, Z.; Li, H.; Zhao, Y.; Chen, W.; Fu, Y.; Ye, Z.; Sun, A.; Li, Z. Carbon monoxide (CO) inhibits hydrogen peroxide (H(2)O(2))-induced oxidative stress and the activation of NF-κB signaling in lens epithelial cells. Exp. Eye Res. 2018, 166, 29–39. [Google Scholar] [CrossRef]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Therap. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef]
- Bull, M.J.; Plummer, N.T. Part 1: The Human Gut Microbiome in Health and Disease. Integr. Med. 2014, 13, 17–22. [Google Scholar]
- Afzaal, M.; Saeed, F.; Shah, Y.A.; Hussain, M.; Rabail, R.; Socol, C.T.; Hassoun, A.; Pateiro, M.; Lorenzo, J.M.; Rusu, A.V.; et al. Human gut microbiota in health and disease: Unveiling the relationship. Front. Microbiol. 2022, 13, 999001. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef]
- Hamamah, S.; Amin, A.; Al-Kassir, A.L.; Chuang, J.; Covasa, M. Dietary Fat Modulation of Gut Microbiota and Impact on Regulatory Pathways Controlling Food Intake. Nutrients 2023, 15, 3365. [Google Scholar] [CrossRef] [PubMed]
- Flaig, B.; Garza, R.; Singh, B.; Hamamah, S.; Covasa, M. Treatment of Dyslipidemia through Targeted Therapy of Gut Microbiota. Nutrients 2023, 15, 228. [Google Scholar] [CrossRef]
- Hamamah, S.; Hajnal, A.; Covasa, M. Impact of Nutrition, Microbiota Transplant and Weight Loss Surgery on Dopaminergic Alterations in Parkinson’s Disease and Obesity. Int. J. Mol. Sci. 2022, 23, 7503. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Bai, C.; Brown, T.D.; Hood, L.E.; Tian, Q. Human Gut Microbiota and Gastrointestinal Cancer. Genom. Proteom. Bioinform. 2018, 16, 33–49. [Google Scholar] [CrossRef]
- Peng, X.; Gao, R.; Ren, J.; Lu, J.; Ma, X.; Li, P. Specific network information gain for detecting the critical state of colorectal cancer based on gut microbiome. Brief. Bioinform. 2023, 25, bbad465. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Wang, X.; Jia, Y.; Wen, L.; Mu, W.; Wu, X.; Liu, T.; Liu, X.; Fang, J.; Luan, Y.; Chen, P.; et al. Porphyromonas gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2021, 81, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.H.; Gao, Q.Y.; Cai, G.X.; Sun, X.M.; Sun, X.M.; Zou, T.H.; Chen, H.M.; Yu, S.Y.; Qiu, Y.W.; Gu, W.Q.; et al. Fecal Clostridium symbiosum for Noninvasive Detection of Early and Advanced Colorectal Cancer: Test and Validation Studies. EBioMedicine 2017, 25, 32–40. [Google Scholar] [CrossRef]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674. [Google Scholar] [CrossRef]
- Dikeocha, I.J.; Al-Kabsi, A.M.; Chiu, H.T.; Alshawsh, M.A. Faecalibacterium prausnitzii Ameliorates Colorectal Tumorigenesis and Suppresses Proliferation of HCT116 Colorectal Cancer Cells. Biomedicines 2022, 10, 1128. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, W.; Liu, W.X.; Zhao, L.Y.; Huang, D.; Liu, X.D.; Chan, H.; Zhang, Y.; Zeng, J.D.; Coker, O.O.; et al. Streptococcus thermophilus Inhibits Colorectal Tumorigenesis Through Secreting β-Galactosidase. Gastroenterology 2021, 160, 1179–1193.e1114. [Google Scholar] [CrossRef]
- Qu, R.; Zhang, Y.; Ma, Y.; Zhou, X.; Sun, L.; Jiang, C.; Zhang, Z.; Fu, W. Role of the Gut Microbiota and Its Metabolites in Tumorigenesis or Development of Colorectal Cancer. Adv. Sci. 2023, 10, e2205563. [Google Scholar] [CrossRef]
- Prudhvi, K.; Jonnadula, J.; Kutti Sridharan, G.; Dominguez, M. Systemic sclerosis with renal crisis and pericardial effusion. Clin. Case Rep. 2020, 8, 3656–3657. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Fang, J.Y. Fusobacterium nucleatum, a key pathogenic factor and microbial biomarker for colorectal cancer. Trends Microbiol. 2023, 31, 159–172. [Google Scholar] [CrossRef]
- Bing, X.; Wang, Z.; Wei, F.; Gao, J.; Xu, D.; Zhang, L.; Wang, Y. Separation of m-Cresol from Coal Tar Model Oil Using Propylamine-Based Ionic Liquids: Extraction and Interaction Mechanism Exploration. ACS Omega 2020, 5, 23090–23098. [Google Scholar] [CrossRef]
- Bostanghadiri, N.; Razavi, S.; Shariati, A.; Talebi, M.; Mirkalantari, S.; Emami Razavi, A.; Darban-Sarokhalil, D. Exploring the interplay between Fusobacterium nucleatum with the expression of microRNA, and inflammatory mediators in colorectal cancer. Front. Microbiol. 2023, 14, 1302719. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Lessing, D.J.; Guo, M.; Chu, W. Fusobacterium nucleatum and its metabolite hydrogen sulfide alter gut microbiota composition and autophagy process and promote colorectal cancer progression. Microbiol. Spectr. 2023, 11, e0229223. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Salesse, L.; Garcia-Weber, D.; Marinelli, L.; Beguet-Crespel, F.; Brochard, V.; Le Gléau, C.; Jamet, A.; Doré, J.; Blottière, H.M.; et al. Fusobacterium nucleatum promotes inflammatory and anti-apoptotic responses in colorectal cancer cells via ADP-heptose release and ALPK1/TIFA axis activation. Gut Microbes 2024, 16, 2295384. [Google Scholar] [CrossRef]
- Xu, C.; Fan, L.; Lin, Y.; Shen, W.; Qi, Y.; Zhang, Y.; Chen, Z.; Wang, L.; Long, Y.; Hou, T.; et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes 2021, 13, 1980347. [Google Scholar] [CrossRef]
- Chen, S.; Su, T.; Zhang, Y.; Lee, A.; He, J.; Ge, Q.; Wang, L.; Si, J.; Zhuo, W.; Wang, L. Fusobacterium nucleatum promotes colorectal cancer metastasis by modulating KRT7-AS/KRT7. Gut Microbes 2020, 11, 511–525. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, L.; Li, M.; Zhang, Y.; Sun, M.; Wang, L.; Lin, J.; Cui, Y.; Chen, Q.; Jin, C.; et al. Fusobacterium nucleatum reduces METTL3-mediated m(6)A modification and contributes to colorectal cancer metastasis. Nat. Commun. 2022, 13, 1248. [Google Scholar] [CrossRef]
- Gao, Y.; Bi, D.; Xie, R.; Li, M.; Guo, J.; Liu, H.; Guo, X.; Fang, J.; Ding, T.; Zhu, H.; et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 398. [Google Scholar] [CrossRef]
- Li, B.; Wei, Z.; Wang, Z.; Xu, F.; Yang, J.; Lin, B.; Chen, Y.; Wenren, H.; Wu, L.; Guo, X.; et al. Fusobacterium nucleatum induces oxaliplatin resistance by inhibiting ferroptosis through E-cadherin/β-catenin/GPX4 axis in colorectal cancer. Free Radic. Biol. Med. 2024, 220, 125–138. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, L.; Leng, X.X.; Xie, Y.L.; Kang, Z.R.; Zhao, L.C.; Song, L.H.; Zhou, C.B.; Fang, J.Y. Fusobacterium nucleatum induces chemoresistance in colorectal cancer by inhibiting pyroptosis via the Hippo pathway. Gut Microbes 2024, 16, 2333790. [Google Scholar] [CrossRef]
- Zeng, W.; Pan, J.; Ye, G. miR-135b Aggravates Fusobacterium nucleatum-Induced Cisplatin Resistance in Colorectal Cancer by Targeting KLF13. J. Microbiol. 2024, 62, 63–73. [Google Scholar] [CrossRef]
- Karunakaran, G.; Yang, Y.; Tremblay, V.; Ning, Z.; Martin, J.; Belaouad, A.; Figeys, D.; Brunzelle, J.S.; Giguere, P.M.; Stintzi, A.; et al. Structural analysis of Atopobium parvulum SufS cysteine desulfurase linked to Crohn’s disease. FEBS Lett. 2022, 596, 898–909. [Google Scholar] [CrossRef]
- Coker, O.O.; Liu, C.; Wu, W.K.K.; Wong, S.H.; Jia, W.; Sung, J.J.Y.; Yu, J. Altered gut metabolites and microbiota interactions are implicated in colorectal carcinogenesis and can be non-invasive diagnostic biomarkers. Microbiome 2022, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Bose, S.; Roy, R.; Paul, D.; Rawat, S.; Nilwe, P.; Chauhan, N.K.; Choudhury, S. Tumor tissue-specific bacterial biomarker panel for colorectal cancer: Bacteroides massiliensis, Alistipes species, Alistipes onderdonkii, Bifidobacterium pseudocatenulatum, Corynebacterium appendicis. Arch. Microbiol. 2022, 204, 348. [Google Scholar] [CrossRef]
- Kerdreux, M.; Edin, S.; Löwenmark, T.; Bronnec, V.; Löfgren-Burström, A.; Zingmark, C.; Ljuslinder, I.; Palmqvist, R.; Ling, A. Porphyromonas gingivalis in Colorectal Cancer and its Association to Patient Prognosis. J. Cancer 2023, 14, 1479–1485. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Hidalgo, I.; Sorolla, A.; Montal, R.; Pallisé, O.; Salud, A.; Parisi, E. Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities. Cancers 2021, 13, 37. [Google Scholar] [CrossRef]
- Irrazabal, T.; Thakur, B.K.; Kang, M.; Malaise, Y.; Streutker, C.; Wong, E.O.Y.; Copeland, J.; Gryfe, R.; Guttman, D.S.; Navarre, W.W.; et al. Limiting oxidative DNA damage reduces microbe-induced colitis-associated colorectal cancer. Nat. Commun. 2020, 11, 1802. [Google Scholar] [CrossRef]
- Dumitrescu, L.; Popescu-Olaru, I.; Cozma, L.; Tulbă, D.; Hinescu, M.E.; Ceafalan, L.C.; Gherghiceanu, M.; Popescu, B.O. Oxidative Stress and the Microbiota-Gut-Brain Axis. Oxid. Med. Cell. Longev. 2018, 2018, 2406594. [Google Scholar] [CrossRef]
- Aceto, G.M.; Catalano, T.; Curia, M.C. Molecular Aspects of Colorectal Adenomas: The Interplay among Microenvironment, Oxidative Stress, and Predisposition. Biomed. Res. Int. 2020, 2020, 1726309. [Google Scholar] [CrossRef]
- Schmitt, M.; Greten, F.R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- Allam-Ndoul, B.; Castonguay-Paradis, S.; Veilleux, A. Gut Microbiota and Intestinal Trans-Epithelial Permeability. Int. J. Mol. Sci. 2020, 21, 6402. [Google Scholar] [CrossRef]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef]
- Tsoi, H.; Chu, E.S.H.; Zhang, X.; Sheng, J.; Nakatsu, G.; Ng, S.C.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology 2017, 152, 1419–1433.e1415. [Google Scholar] [CrossRef]
- Huycke, M.M.; Moore, D.R. In vivo production of hydroxyl radical by Enterococcus faecalis colonizing the intestinal tract using aromatic hydroxylation. Free Radic. Biol. Med. 2002, 33, 818–826. [Google Scholar] [CrossRef]
- Sears, C.L.; Geis, A.L.; Housseau, F. Bacteroides fragilis subverts mucosal biology: From symbiont to colon carcinogenesis. J. Clin. Investig. 2014, 124, 4166–4172. [Google Scholar] [CrossRef]
- Rajkumar, S.D.; Gunabooshanam, B.; Joseph, L.D.; D’Cruze, L. Utility of immunohistochemical expression of E-cadherin in colorectal carcinoma. Prz. Gastroenterol. 2022, 17, 59–66. [Google Scholar] [CrossRef]
- Cervelli, M.; Amendola, R.; Polticelli, F.; Mariottini, P. Spermine oxidase: Ten years after. Amino Acids 2012, 42, 441–450. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Romero-Gallo, J.; Barry, D.P.; Hoge, S.; de Sablet, T.; Delgado, A.G.; Wroblewski, L.E.; Piazuelo, M.B.; Yan, F.; et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 2011, 141, 1696–1708.e1691–1692. [Google Scholar] [CrossRef]
- Uemura, T.; Tanaka, Y.; Higashi, K.; Miyamori, D.; Takasaka, T.; Nagano, T.; Toida, T.; Yoshimoto, K.; Igarashi, K.; Ikegaya, H. Acetaldehyde-induced cytotoxicity involves induction of spermine oxidase at the transcriptional level. Toxicology 2013, 310, 1–7. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellavia, G.; Fratini, E.; Amendola, R.; Polticelli, F.; Barba, M.; Federico, R.; Signore, F.; Gucciardo, G.; Grillo, R.; et al. Spermine oxidase (SMO) activity in breast tumor tissues and biochemical analysis of the anticancer spermine analogues BENSpm and CPENSpm. BMC Cancer 2010, 10, 555. [Google Scholar] [CrossRef]
- Sun, L.; Yang, J.; Qin, Y.; Wang, Y.; Wu, H.; Zhou, Y.; Cao, C. Discovery and antitumor evaluation of novel inhibitors of spermine oxidase. J. Enzyme Inhib. Med. Chem. 2019, 34, 1140–1151. [Google Scholar] [CrossRef]
- Wang, X.; Allen, T.D.; May, R.J.; Lightfoot, S.; Houchen, C.W.; Huycke, M.M. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res. 2008, 68, 9909–9917. [Google Scholar] [CrossRef] [PubMed]
- Scheible, M.; Nguyen, C.T.; Luong, T.T.; Lee, J.H.; Chen, Y.W.; Chang, C.; Wittchen, M.; Camacho, M.I.; Tiner, B.L.; Wu, C.; et al. The Fused Methionine Sulfoxide Reductase MsrAB Promotes Oxidative Stress Defense and Bacterial Virulence in Fusobacterium nucleatum. mBio 2022, 13, e0302221. [Google Scholar] [CrossRef]
- Silva, V.L.; Diniz, C.G.; Cara, D.C.; Santos, S.G.; Nicoli, J.R.; Carvalho, M.A.; Farias, L.M. Enhanced pathogenicity of Fusobacterium nucleatum adapted to oxidative stress. Microb. Pathog. 2005, 39, 131–138. [Google Scholar] [CrossRef]
- Frances Nakamya, M.; Ayoola, M.B.; Shack, L.A.; Mohamed, M.; Swiatlo, E.; Nanduri, B. Arginine Decarboxylase Is Essential for Pneumococcal Stress Responses. Pathogens 2021, 10, 286. [Google Scholar] [CrossRef]
- McDowell, R.; Perrott, S.; Murchie, P.; Cardwell, C.; Hughes, C.; Samuel, L. Oral antibiotic use and early-onset colorectal cancer: Findings from a case-control study using a national clinical database. Br. J. Cancer 2022, 126, 957–967. [Google Scholar] [CrossRef]
- Aneke-Nash, C.; Yoon, G.; Du, M.; Liang, P. Antibiotic use and colorectal neoplasia: A systematic review and meta-analysis. BMJ Open Gastroenterol. 2021, 8, e000601. [Google Scholar] [CrossRef]
- Voskarides, K. An evolutionary explanation for antibiotics’ association with increased colon cancer risk. Evol. Med. Public. Health 2022, 10, 214–220. [Google Scholar] [CrossRef]
- Kim, M.; Yun, S.Y.; Lee, Y.; Lee, H.; Yong, D.; Lee, K. Clinical Differences in Patients Infected with Fusobacterium and Antimicrobial Susceptibility of Fusobacterium Isolates Recovered at a Tertiary-Care Hospital in Korea. Ann. Lab. Med. 2022, 42, 188–195. [Google Scholar] [CrossRef]
- Ju, Y.; Liu, K.; Ma, G.; Zhu, B.; Wang, H.; Hu, Z.; Zhao, J.; Zhang, L.; Cui, K.; He, X.R.; et al. Bacterial antibiotic resistance among cancer inpatients in China: 2016-20. Qjm 2023, 116, 213–220. [Google Scholar] [CrossRef]
- Oberc, A.M.; Fiebig-Comyn, A.A.; Tsai, C.N.; Elhenawy, W.; Coombes, B.K. Antibiotics Potentiate Adherent-Invasive E. coli Infection and Expansion. Inflamm. Bowel Dis. 2019, 25, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Hamoya, T.; Miyamoto, S.; Tomono, S.; Fujii, G.; Nakanishi, R.; Komiya, M.; Tamura, S.; Fujimoto, K.; Toshima, J.; Wakabayashi, K.; et al. Chemopreventive effects of a low-side-effect antibiotic drug, erythromycin, on mouse intestinal tumors. J. Clin. Biochem. Nutr. 2017, 60, 199–207. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Zheng, X.; Ren, L.; Yang, Y.; Li, W.; Fu, W.; Wang, J.; Du, G. Tumorigenic bacteria in colorectal cancer: Mechanisms and treatments. Cancer Biol. Med. 2021, 19, 147–162. [Google Scholar] [CrossRef]
- Heidari, A.; Emami, M.H.; Maghool, F.; Mohammadzadeh, S.; Kadkhodaei Elyaderani, P.; Safari, T.; Fahim, A.; Kamali Dolatabadi, R. Molecular epidemiology, antibiotic resistance profile and frequency of integron 1 and 2 in adherent-invasive Escherichia coli isolates of colorectal cancer patients. Front. Microbiol. 2024, 15, 1366719. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- DeStefano Shields, C.E.; Van Meerbeke, S.W.; Housseau, F.; Wang, H.; Huso, D.L.; Casero, R.A., Jr.; O’Hagan, H.M.; Sears, C.L. Reduction of Murine Colon Tumorigenesis Driven by Enterotoxigenic Bacteroides fragilis Using Cefoxitin Treatment. J. Infect. Dis. 2016, 214, 122–129. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, B.; Zheng, Q.; Li, H.; Meng, X.; Zhou, F.; Zhang, L. A Review of Gut Microbiota-Derived Metabolites in Tumor Progression and Cancer Therapy. Adv. Sci. 2023, 10, e2207366. [Google Scholar] [CrossRef]
- Avuthu, N.; Guda, C. Meta-Analysis of Altered Gut Microbiota Reveals Microbial and Metabolic Biomarkers for Colorectal Cancer. Microbiol. Spectr. 2022, 10, e0001322. [Google Scholar] [CrossRef]
- Wu, Y.; Jiao, N.; Zhu, R.; Zhang, Y.; Wu, D.; Wang, A.J.; Fang, S.; Tao, L.; Li, Y.; Cheng, S.; et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat. Commun. 2021, 12, 3063. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577–588. [Google Scholar] [CrossRef]
- Duizer, C.; de Zoete, M.R. The Role of Microbiota-Derived Metabolites in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 8024. [Google Scholar] [CrossRef]
- Xiong, R.G.; Zhou, D.D.; Wu, S.X.; Huang, S.Y.; Saimaiti, A.; Yang, Z.J.; Shang, A.; Zhao, C.N.; Gan, R.Y.; Li, H.B. Health Benefits and Side Effects of Short-Chain Fatty Acids. Foods 2022, 11, 2863. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Zhang, D.; Jian, Y.P.; Zhang, Y.N.; Li, Y.; Gu, L.T.; Sun, H.H.; Liu, M.D.; Zhou, H.L.; Wang, Y.S.; Xu, Z.X. Short-chain fatty acids in diseases. Cell Commun. Signal. 2023, 21, 212. [Google Scholar] [CrossRef]
- Alvandi, E.; Wong, W.K.M.; Joglekar, M.V.; Spring, K.J.; Hardikar, A.A. Short-chain fatty acid concentrations in the incidence and risk-stratification of colorectal cancer: A systematic review and meta-analysis. BMC Med. 2022, 20, 323. [Google Scholar] [CrossRef]
- Wu, Q.L.; Fang, X.T.; Wan, X.X.; Ding, Q.Y.; Zhang, Y.J.; Ji, L.; Lou, Y.L.; Li, X. Fusobacterium nucleatum-induced imbalance in microbiome-derived butyric acid levels promotes the occurrence and development of colorectal cancer. World J. Gastroenterol. 2024, 30, 2018–2037. [Google Scholar] [CrossRef]
- Son, M.Y.; Cho, H.S. Anticancer Effects of Gut Microbiota-Derived Short-Chain Fatty Acids in Cancers. J. Microbiol. Biotechnol. 2023, 33, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.M.; Bast, A.; Vanhoutvin, S.A.; Fischer, M.A.; Kodde, A.; Troost, F.J.; Venema, K.; Brummer, R.J. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin. Nutr. 2009, 28, 88–93. [Google Scholar] [CrossRef]
- Huang, W.; Guo, H.L.; Deng, X.; Zhu, T.T.; Xiong, J.F.; Xu, Y.H.; Xu, Y. Short-Chain Fatty Acids Inhibit Oxidative Stress and Inflammation in Mesangial Cells Induced by High Glucose and Lipopolysaccharide. Exp. Clin. Endocrinol. Diabetes 2017, 125, 98–105. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, W.; Xu, M.; Xie, H.; Pu, Z. GPR43 regulation of mitochondrial damage to alleviate inflammatory reaction in sepsis. Aging (Albany NY) 2021, 13, 22588–22610. [Google Scholar] [CrossRef]
- Masui, R.; Sasaki, M.; Funaki, Y.; Ogasawara, N.; Mizuno, M.; Iida, A.; Izawa, S.; Kondo, Y.; Ito, Y.; Tamura, Y.; et al. G protein-coupled receptor 43 moderates gut inflammation through cytokine regulation from mononuclear cells. Inflamm. Bowel Dis. 2013, 19, 2848–2856. [Google Scholar] [CrossRef]
- Hu, Y.; Le Leu, R.K.; Christophersen, C.T.; Somashekar, R.; Conlon, M.A.; Meng, X.Q.; Winter, J.M.; Woodman, R.J.; McKinnon, R.; Young, G.P. Manipulation of the gut microbiota using resistant starch is associated with protection against colitis-associated colorectal cancer in rats. Carcinogenesis 2016, 37, 366–375. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Fawad, J.A.; Luzader, D.H.; Hanson, G.F.; Moutinho, T.J., Jr.; McKinney, C.A.; Mitchell, P.G.; Brown-Steinke, K.; Kumar, A.; Park, M.; Lee, S.; et al. Histone Deacetylase Inhibition by Gut Microbe-Generated Short-Chain Fatty Acids Entrains Intestinal Epithelial Circadian Rhythms. Gastroenterology 2022, 163, 1377–1390.e1311. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Noonan, E.J.; Giardina, C. HDAC inhibition prevents NF-kappa B activation by suppressing proteasome activity: Down-regulation of proteasome subunit expression stabilizes I kappa B alpha. Biochem. Pharmacol. 2005, 70, 394–406. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Short-Chain Fatty Acids Manifest Stimulative and Protective Effects on Intestinal Barrier Function Through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell Physiol. Biochem. 2018, 49, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Scharlau, D.; Borowicki, A.; Habermann, N.; Hofmann, T.; Klenow, S.; Miene, C.; Munjal, U.; Stein, K.; Glei, M. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 2009, 682, 39–53. [Google Scholar] [CrossRef]
- Salvi, P.S.; Cowles, R.A. Butyrate and the Intestinal Epithelium: Modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef]
- Siavoshian, S.; Blottiere, H.M.; Cherbut, C.; Galmiche, J.P. Butyrate stimulates cyclin D and p21 and inhibits cyclin-dependent kinase 2 expression in HT-29 colonic epithelial cells. Biochem. Biophys. Res. Commun. 1997, 232, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shannar, A.A.F.; Wu, R.; Chou, P.; Sarwar, M.S.; Kuo, H.C.; Peter, R.M.; Wang, Y.; Su, X.; Kong, A.N. Butyrate Drives Metabolic Rewiring and Epigenetic Reprogramming in Human Colon Cancer Cells. Mol. Nutr. Food Res. 2022, 66, e2200028. [Google Scholar] [CrossRef]
- Guo, W.; Liu, J.; Sun, J.; Gong, Q.; Ma, H.; Kan, X.; Cao, Y.; Wang, J.; Fu, S. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic. Biol. Med. 2020, 152, 728–742. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, W.I.; Bae, J.H.; Cho, M.K.; Lee, S.H.; Nam, H.S.; Choi, I.H.; Cho, S.W. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann. Surg. Treat. Res. 2020, 98, 159–167. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Gonzalez-Donquiles, C.; Alonso-Molero, J.; Fernandez-Villa, T.; Vilorio-Marqués, L.; Molina, A.J.; Martín, V. The NRF2 transcription factor plays a dual role in colorectal cancer: A systematic review. PLoS ONE 2017, 12, e0177549. [Google Scholar] [CrossRef]
- Hodgkinson, K.; El Abbar, F.; Dobranowski, P.; Manoogian, J.; Butcher, J.; Figeys, D.; Mack, D.; Stintzi, A. Butyrate’s role in human health and the current progress towards its clinical application to treat gastrointestinal disease. Clin. Nutr. 2023, 42, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Eslami, M.; Sadrifar, S.; Karbalaei, M.; Keikha, M.; Kobyliak, N.M.; Yousefi, B. Importance of the Microbiota Inhibitory Mechanism on the Warburg Effect in Colorectal Cancer Cells. J. Gastrointest. Cancer 2020, 51, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Deng, W.; Xu, H.Z.; Zhou, C.; Zhang, F.; Chen, J.; Bao, Q.; Zhou, X.; Liu, M.; Li, J.; et al. Short-chain fatty acids reprogram metabolic profiles with the induction of reactive oxygen species production in human colorectal adenocarcinoma cells. Comput. Struct. Biotechnol. J. 2023, 21, 1606–1620. [Google Scholar] [CrossRef]
- Tailor, D.; Hahm, E.R.; Kale, R.K.; Singh, S.V.; Singh, R.P. Sodium butyrate induces DRP1-mediated mitochondrial fusion and apoptosis in human colorectal cancer cells. Mitochondrion 2014, 16, 55–64. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- di Gregorio, M.C.; Cautela, J.; Galantini, L. Physiology and Physical Chemistry of Bile Acids. Int. J. Mol. Sci. 2021, 22, 1780. [Google Scholar] [CrossRef]
- Durník, R.; Šindlerová, L.; Babica, P.; Jurček, O. Bile Acids Transporters of Enterohepatic Circulation for Targeted Drug Delivery. Molecules 2022, 27, 2961. [Google Scholar] [CrossRef]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Yntema, T.; Koonen, D.P.Y.; Kuipers, F. Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases. Nutrients 2023, 15, 1850. [Google Scholar] [CrossRef]
- Harris, S.C.; Devendran, S.; Méndez-García, C.; Mythen, S.M.; Wright, C.L.; Fields, C.J.; Hernandez, A.G.; Cann, I.; Hylemon, P.B.; Ridlon, J.M. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243(T). Gut Microbes 2018, 9, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Doden, H.; Sallam, L.A.; Devendran, S.; Ly, L.; Doden, G.; Daniel, S.L.; Alves, J.M.P.; Ridlon, J.M. Metabolism of Oxo-Bile Acids and Characterization of Recombinant 12α-Hydroxysteroid Dehydrogenases from Bile Acid 7α-Dehydroxylating Human Gut Bacteria. Appl. Environ. Microbiol. 2018, 84, e00235-18. [Google Scholar] [CrossRef]
- Dvorak, K.; Payne, C.M.; Chavarria, M.; Ramsey, L.; Dvorakova, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; Guy, N.; Condon, A.; et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett’s oesophagus. Gut 2007, 56, 763–771. [Google Scholar] [CrossRef]
- Payne, C.M.; Weber, C.; Crowley-Skillicorn, C.; Dvorak, K.; Bernstein, H.; Bernstein, C.; Holubec, H.; Dvorakova, B.; Garewal, H. Deoxycholate induces mitochondrial oxidative stress and activates NF-kappaB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis 2007, 28, 215–222. [Google Scholar] [CrossRef]
- Ignacio Barrasa, J.; Olmo, N.; Pérez-Ramos, P.; Santiago-Gómez, A.; Lecona, E.; Turnay, J.; Antonia Lizarbe, M. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis 2011, 16, 1054–1067. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Glinghammar, B.; Inoue, H.; Rafter, J.J. Deoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and the transcription factors NF-kB and AP-1. Carcinogenesis 2002, 23, 839–845. [Google Scholar] [CrossRef]
- Kundu, S.; Kumar, S.; Bajaj, A. Cross-talk between bile acids and gastrointestinal tract for progression and development of cancer and its therapeutic implications. IUBMB Life 2015, 67, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Aguilar, J.; Simon, F.; Cabello-Verrugio, C. Redox-Dependent Effects in the Physiopathological Role of Bile Acids. Oxid. Med. Cell. Longev. 2021, 2021, 4847941. [Google Scholar] [CrossRef]
- Lee, H.Y.; Crawley, S.; Hokari, R.; Kwon, S.; Kim, Y.S. Bile acid regulates MUC2 transcription in colon cancer cells via positive EGFR/PKC/Ras/ERK/CREB, PI3K/Akt/IkappaB/NF-kappaB and p38/MSK1/CREB pathways and negative JNK/c-Jun/AP-1 pathway. Int. J. Oncol. 2010, 36, 941–953. [Google Scholar] [CrossRef]
- Crowley-Weber, C.L.; Payne, C.M.; Gleason-Guzman, M.; Watts, G.S.; Futscher, B.; Waltmire, C.N.; Crowley, C.; Dvorakova, K.; Bernstein, C.; Craven, M.; et al. Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis 2002, 23, 2063–2080. [Google Scholar] [CrossRef]
- Huang, W.K.; Hsu, H.C.; Liu, J.R.; Yang, T.S.; Chen, J.S.; Chang, J.W.; Lin, Y.C.; Yu, K.H.; Kuo, C.F.; See, L.C. The Association of Ursodeoxycholic Acid Use With Colorectal Cancer Risk: A Nationwide Cohort Study. Medicine 2016, 95, e2980. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Zhang, C.; Tang, Q.; Bi, F. Ursodeoxycholic acid suppresses the malignant progression of colorectal cancer through TGR5-YAP axis. Cell Death Discov. 2021, 7, 207. [Google Scholar] [CrossRef]
- Kim, E.K.; Cho, J.H.; Kim, E.; Kim, Y.J. Ursodeoxycholic acid inhibits the proliferation of colon cancer cells by regulating oxidative stress and cancer stem-like cell growth. PLoS ONE 2017, 12, e0181183. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Caporaso, J.G.; Yellowhair, M.; Bokulich, N.A.; Padi, M.; Roe, D.J.; Wertheim, B.C.; Linhart, M.; Martinez, J.A.; Bilagody, C.; et al. Effects of ursodeoxycholic acid on the gut microbiome and colorectal adenoma development. Cancer Med. 2019, 8, 617–628. [Google Scholar] [CrossRef]
- He, Q.; Wu, J.; Ke, J.; Zhang, Q.; Zeng, W.; Luo, Z.; Gong, J.; Chen, Y.; He, Z.; Lan, P. Therapeutic role of ursodeoxycholic acid in colitis-associated cancer via gut microbiota modulation. Mol. Ther. 2023, 31, 585–598. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, R.; Zheng, X.; Lei, S.; Huang, F.; Xie, G.; Kwee, S.; Yu, H.; Farrar, C.; Sun, B.; et al. Ursodeoxycholic acid accelerates bile acid enterohepatic circulation. Br. J. Pharmacol. 2019, 176, 2848–2863. [Google Scholar] [CrossRef]
- Amaral, J.D.; Viana, R.J.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Okpara, E.S.; Hu, W.; Yan, C.; Wang, Y.; Liang, Q.; Chiang, J.Y.L.; Han, S. Interactive Relationships between Intestinal Flora and Bile Acids. Int. J. Mol. Sci. 2022, 23, 8343. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.L.; Zhang, L.Y.; Jiang, X.M.; Xue, C.H.; Chi, N.; Zhang, T.T.; Wang, Y.M. Effects of dietary choline, betaine, and L-carnitine on the generation of trimethylamine-N-oxide in healthy mice. J. Food Sci. 2020, 85, 2207–2215. [Google Scholar] [CrossRef]
- Rath, S.; Rud, T.; Pieper, D.H.; Vital, M. Potential TMA-Producing Bacteria Are Ubiquitously Found in Mammalia. Front. Microbiol. 2019, 10, 2966. [Google Scholar] [CrossRef]
- Zhen, J.; Zhou, Z.; He, M.; Han, H.X.; Lv, E.H.; Wen, P.B.; Liu, X.; Wang, Y.T.; Cai, X.C.; Tian, J.Q.; et al. The gut microbial metabolite trimethylamine N-oxide and cardiovascular diseases. Front. Endocrinol. 2023, 14, 1085041. [Google Scholar] [CrossRef]
- Shih, D.M.; Wang, Z.; Lee, R.; Meng, Y.; Che, N.; Charugundla, S.; Qi, H.; Wu, J.; Pan, C.; Brown, J.M.; et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 2015, 56, 22–37. [Google Scholar] [CrossRef]
- Jalandra, R.; Dalal, N.; Yadav, A.K.; Verma, D.; Sharma, M.; Singh, R.; Khosla, A.; Kumar, A.; Solanki, P.R. Emerging role of trimethylamine-N-oxide (TMAO) in colorectal cancer. Appl. Microbiol. Biotechnol. 2021, 105, 7651–7660. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Vajdi, M. Novel findings of the association between gut microbiota-derived metabolite trimethylamine N-oxide and inflammation: Results from a systematic review and dose-response meta-analysis. Crit. Rev. Food Sci. Nutr. 2020, 60, 2801–2823. [Google Scholar] [CrossRef]
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rong, X.; Pan, M.; Wang, T.; Yang, H.; Chen, X.; Xiao, Z.; Zhao, C. Integrated Analysis Reveals the Gut Microbial Metabolite TMAO Promotes Inflammatory Hepatocellular Carcinoma by Upregulating POSTN. Front. Cell Dev. Biol. 2022, 10, 840171. [Google Scholar] [CrossRef] [PubMed]
- Mirji, G.; Worth, A.; Bhat, S.A.; El Sayed, M.; Kannan, T.; Goldman, A.R.; Tang, H.Y.; Liu, Q.; Auslander, N.; Dang, C.V.; et al. The microbiome-derived metabolite TMAO drives immune activation and boosts responses to immune checkpoint blockade in pancreatic cancer. Sci. Immunol. 2022, 7, eabn0704. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front. Pharmacol. 2019, 10, 1360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, F.; Yuan, J.; Liu, H.; Wang, Y. Gut microbiota metabolites, redox status, and the related regulatory effects of probiotics. Heliyon 2023, 9, e21431. [Google Scholar] [CrossRef]
- Ding, S.; Hu, C.; Fang, J.; Liu, G. The Protective Role of Probiotics against Colorectal Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 8884583. [Google Scholar] [CrossRef]
- Ballini, A.; Santacroce, L.; Cantore, S.; Bottalico, L.; Dipalma, G.; Topi, S.; Saini, R.; De Vito, D.; Inchingolo, F. Probiotics Efficacy on Oxidative Stress Values in Inflammatory Bowel Disease: A Randomized Double-Blinded Placebo-Controlled Pilot Study. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.C.S.; Paulino, D.S.; Brambilla, S.R.; Camargo, J.A.; Persinoti, G.F.; Carvalheira, J.B.C. Microbiota modification by probiotic supplementation reduces colitis associated colon cancer in mice. World J. Gastroenterol. 2018, 24, 1995–2008. [Google Scholar] [CrossRef]
- Feng, T.; Wang, J. Oxidative stress tolerance and antioxidant capacity of lactic acid bacteria as probiotic: A systematic review. Gut Microbes 2020, 12, 1801944. [Google Scholar] [CrossRef]
- Bell, H.N.; Rebernick, R.J.; Goyert, J.; Singhal, R.; Kuljanin, M.; Kerk, S.A.; Huang, W.; Das, N.K.; Andren, A.; Solanki, S.; et al. Reuterin in the healthy gut microbiome suppresses colorectal cancer growth through altering redox balance. Cancer Cell 2022, 40, 185–200.e186. [Google Scholar] [CrossRef] [PubMed]
- Aghamohammad, S.; Sepehr, A.; Miri, S.T.; Najafi, S.; Rohani, M.; Pourshafiea, M.R. The effects of the probiotic cocktail on modulation of the NF-kB and JAK/STAT signaling pathways involved in the inflammatory response in bowel disease model. BMC Immunol. 2022, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Olejar, K.J.; On, S.L.W.; Chelikani, V. The Potential of Lactobacillus spp. for Modulating Oxidative Stress in the Gastrointestinal Tract. Antioxidants 2020, 9, 610. [Google Scholar] [CrossRef]
- Rocha-Ramírez, L.M.; Pérez-Solano, R.A.; Castañón-Alonso, S.L.; Moreno Guerrero, S.S.; Ramírez Pacheco, A.; García Garibay, M.; Eslava, C. Probiotic Lactobacillus Strains Stimulate the Inflammatory Response and Activate Human Macrophages. J. Immunol. Res. 2017, 2017, 4607491. [Google Scholar] [CrossRef]
- Finamore, A.; Ambra, R.; Nobili, F.; Garaguso, I.; Raguzzini, A.; Serafini, M. Redox Role of Lactobacillus casei Shirota Against the Cellular Damage Induced by 2,2′-Azobis (2-Amidinopropane) Dihydrochloride-Induced Oxidative and Inflammatory Stress in Enterocytes-Like Epithelial Cells. Front. Immunol. 2018, 9, 1131. [Google Scholar] [CrossRef]
- Riaz Rajoka, M.S.; Thirumdas, R.; Mehwish, H.M.; Umair, M.; Khurshid, M.; Hayat, H.F.; Phimolsiripol, Y.; Pallarés, N.; Martí-Quijal, F.J.; Barba, F.J. Role of Food Antioxidants in Modulating Gut Microbial Communities: Novel Understandings in Intestinal Oxidative Stress Damage and Their Impact on Host Health. Antioxidants 2021, 10, 1563. [Google Scholar] [CrossRef]
- Al-Numair, K.S.; Waly, M.I.; Ali, A.; Essa, M.M.; Farhat, M.F.; Alsaif, M.A. Dietary folate protects against azoxymethane-induced aberrant crypt foci development and oxidative stress in rat colon. Exp. Biol. Med. 2011, 236, 1005–1011. [Google Scholar] [CrossRef]
- Kang, X.; Liu, C.; Ding, Y.; Ni, Y.; Ji, F.; Lau, H.C.H.; Jiang, L.; Sung, J.J.; Wong, S.H.; Yu, J. Roseburia intestinalis generated butyrate boosts anti-PD-1 efficacy in colorectal cancer by activating cytotoxic CD8(+) T cells. Gut 2023, 72, 2112–2122. [Google Scholar] [CrossRef]
- Talwalkar, A.; Kailasapathy, K. The role of oxygen in the viability of probiotic bacteria with reference to L. acidophilus and Bifidobacterium spp. Curr. Issues Intest. Microbiol. 2004, 5, 1–8. [Google Scholar] [PubMed]
- Goffin, P.; Muscariello, L.; Lorquet, F.; Stukkens, A.; Prozzi, D.; Sacco, M.; Kleerebezem, M.; Hols, P. Involvement of pyruvate oxidase activity and acetate production in the survival of Lactobacillus plantarum during the stationary phase of aerobic growth. Appl. Environ. Microbiol. 2006, 72, 7933–7940. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.I.; Soliman, A.A.; Ahmed, H.A.; Gewaily, M.S.; Amer, A.A.; Shukry, M.; Abdel-Latif, H.M.R. Evaluation of different probiotics on growth, body composition, antioxidant capacity, and histoarchitecture of Mugil capito. Sci. Rep. 2024, 14, 7379. [Google Scholar] [CrossRef]
- de Moreno de LeBlanc, A.; LeBlanc, J.G.; Perdigón, G.; Miyoshi, A.; Langella, P.; Azevedo, V.; Sesma, F. Oral administration of a catalase-producing Lactococcus lactis can prevent a chemically induced colon cancer in mice. J. Med. Microbiol. 2008, 57, 100–105. [Google Scholar] [CrossRef]
- Deepak, V.; Sundar, W.A.; Pandian, S.R.K.; Sivasubramaniam, S.D.; Hariharan, N.; Sundar, K. Exopolysaccharides from Lactobacillus acidophilus modulates the antioxidant status of 1,2-dimethyl hydrazine-induced colon cancer rat model. 3 Biotech 2021, 11, 225. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Vinayagam, R.; Arokia Vijaya Anand, M.; Isa, N.M.; Ponnaiyan, R. Biochemical and molecular aspects of 1,2-dimethylhydrazine (DMH)-induced colon carcinogenesis: A review. Toxicol. Res. 2020, 9, 2–18. [Google Scholar] [CrossRef]
- Sengül, N.; Işık, S.; Aslım, B.; Uçar, G.; Demirbağ, A.E. The effect of exopolysaccharide-producing probiotic strains on gut oxidative damage in experimental colitis. Dig. Dis. Sci. 2011, 56, 707–714. [Google Scholar] [CrossRef]
- Kwun, S.Y.; Yoon, J.A.; Kim, G.Y.; Bae, Y.W.; Park, E.H.; Kim, M.D. Isolation of a Potential Probiotic Levilactobacillus brevis and Evaluation of Its Exopolysaccharide for Antioxidant and α-Glucosidase Inhibitory Activities. J. Microbiol. Biotechnol. 2024, 34, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Haghshenas, B.; Nami, Y. Bifidobacterium exopolysaccharides: New insights into engineering strategies, physicochemical functions, and immunomodulatory effects on host health. Front. Microbiol. 2024, 15, 1396308. [Google Scholar] [CrossRef]
- de Oliveira, C.S.; Baptistella, M.M.; Siqueira, A.P.; Carvalho, M.O.; Ramos, L.F.; Souto, B.S.; de Almeida, L.A.; Dos Santos, E.G.; Novaes, R.D.; Nogueira, E.S.C.; et al. Combination of vitamin D and probiotics inhibits chemically induced colorectal carcinogenesis in Wistar rats. Life Sci. 2023, 322, 121617. [Google Scholar] [CrossRef]
- Liu, J.; Wang, S.; Yi, R.; Long, X.; Zhao, X. Effect of Lactobacillus fermentum ZS40 on the NF-κB signaling pathway in an azomethane-dextran sulfate sodium-induced colon cancer mouse model. Front. Microbiol. 2022, 13, 953905. [Google Scholar] [CrossRef]
- Ma, F.; Song, Y.; Sun, M.; Wang, A.; Jiang, S.; Mu, G.; Tuo, Y. Exopolysaccharide Produced by Lactiplantibacillus plantarum-12 Alleviates Intestinal Inflammation and Colon Cancer Symptoms by Modulating the Gut Microbiome and Metabolites of C57BL/6 Mice Treated by Azoxymethane/Dextran Sulfate Sodium Salt. Foods 2021, 10, 3060. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xie, W.; Wan, X.; Deng, T. Clostridium butyricum modulates gut microbiota and reduces colitis associated colon cancer in mice. Int. Immunopharmacol. 2020, 88, 106862. [Google Scholar] [CrossRef]
- Fahmy, C.A.; Gamal-Eldeen, A.M.; El-Hussieny, E.A.; Raafat, B.M.; Mehanna, N.S.; Talaat, R.M.; Shaaban, M.T. Bifidobacterium longum Suppresses Murine Colorectal Cancer through the Modulation of oncomiRs and Tumor Suppressor miRNAs. Nutr. Cancer 2019, 71, 688–700. [Google Scholar] [CrossRef]
- Talero, E.; Bolivar, S.; Ávila-Román, J.; Alcaide, A.; Fiorucci, S.; Motilva, V. Inhibition of chronic ulcerative colitis-associated adenocarcinoma development in mice by VSL#3. Inflamm. Bowel Dis. 2015, 21, 1027–1037. [Google Scholar] [CrossRef]
- Kumar, M.; Kissoon-Singh, V.; Coria, A.L.; Moreau, F.; Chadee, K. Probiotic mixture VSL#3 reduces colonic inflammation and improves intestinal barrier function in Muc2 mucin-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G34–G45. [Google Scholar] [CrossRef] [PubMed]
- Cruz, B.; Conceição, L.L.D.; Mendes, T.A.O.; Ferreira, C.; Gonçalves, R.V.; Peluzio, M. Use of the synbiotic VSL#3 and yacon-based concentrate attenuates intestinal damage and reduces the abundance of Candidatus Saccharimonas in a colitis-associated carcinogenesis model. Food Res. Int. 2020, 137, 109721. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, B.; Zhang, S.; Yin, H.; Sun, L.; Pu, Y.; Zhou, B.; Sun, Y.; Li, X.; Fang, Y.; et al. Design of a self-driven probiotic-CRISPR/Cas9 nanosystem for sono-immunometabolic cancer therapy. Nat. Commun. 2022, 13, 7903. [Google Scholar] [CrossRef]
- Grenda, A.; Grenda, T.; Domaradzki, P.; Kwiatek, K. Enterococci-Involvement in Pathogenesis and Therapeutic Potential in Cancer Treatment: A Mini-Review. Pathogens 2022, 11, 687. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, P.; Rengaswamy, R.; Karunagaran, D.; Suraishkumar, G.K.; Sahoo, S. Metabolic modeling of host-microbe interactions for therapeutics in colorectal cancer. NPJ Syst. Biol. Appl. 2022, 8, 1. [Google Scholar] [CrossRef]
- Dong, J.; Wang, B.; Xiao, Y.; Liu, J.; Wang, Q.; Xiao, H.; Jin, Y.; Liu, Z.; Chen, Z.; Li, Y.; et al. Roseburia intestinalis sensitizes colorectal cancer to radiotherapy through the butyrate/OR51E1/RALB axis. Cell Rep. 2024, 43, 114407. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Chandrashekharappa, S.; Bodduluri, S.R.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun. 2019, 10, 89. [Google Scholar] [CrossRef]
- Rezaie, N.; Aghamohammad, S.; Haj Agha Gholizadeh Khiavi, E.; Khatami, S.; Sohrabi, A.; Rohani, M. The comparative anti-oxidant and anti-inflammatory efficacy of postbiotics and probiotics through Nrf-2 and NF-kB pathways in DSS-induced colitis model. Sci. Rep. 2024, 14, 11560. [Google Scholar] [CrossRef] [PubMed]
- Aboulgheit, A.; Karbasiafshar, C.; Zhang, Z.; Sabra, M.; Shi, G.; Tucker, A.; Sodha, N.; Abid, M.R.; Sellke, F.W. Lactobacillus plantarum probiotic induces Nrf2-mediated antioxidant signaling and eNOS expression resulting in improvement of myocardial diastolic function. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H839–H849. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Liu, K.H.; Owens, J.A.; Hunter-Chang, S.; Camacho, M.C.; Eboka, R.U.; Chandrasekharan, B.; Baker, N.F.; Darby, T.M.; Robinson, B.S.; et al. Gut-Resident Lactobacilli Activate Hepatic Nrf2 and Protect Against Oxidative Liver Injury. Cell Metab. 2020, 31, 956–968.e955. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Zhao, Y.; Liu, S.; Xu, H. Lactiplantibacillus plantarum P101 Attenuated Cyclophosphamide-Induced Liver Injury in Mice by Regulating the Nrf2/ARE Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 13424. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Sahu, N.; Saxena, S.; Pradhan, B.; Nayak, S.K.; Roychowdhury, A. Effects of Probiotics at the Interface of Metabolism and Immunity to Prevent Colorectal Cancer-Associated Gut Inflammation: A Systematic Network and Meta-Analysis With Molecular Docking Studies. Front. Microbiol. 2022, 13, 878297. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. Methods Mol. Biol. 2016, 1422, 297–307. [Google Scholar] [CrossRef]
- Do, E.J.; Hwang, S.W.; Kim, S.Y.; Ryu, Y.M.; Cho, E.A.; Chung, E.J.; Park, S.; Lee, H.J.; Byeon, J.S.; Ye, B.D.; et al. Suppression of colitis-associated carcinogenesis through modulation of IL-6/STAT3 pathway by balsalazide and VSL#3. J. Gastroenterol. Hepatol. 2016, 31, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, Z.; Zhu, L.; Ma, S.; Luo, Y.; Liang, H.; Liu, Q.; Chen, J.; Guli, S.; Chen, X. Orchestration of MUC2—The key regulatory target of gut barrier and homeostasis: A review. Int. J. Biol. Macromol. 2023, 236, 123862. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Huycke, M.M.; Abrams, V.; Moore, D.R. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 2002, 23, 529–536. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamamah, S.; Lobiuc, A.; Covasa, M. Antioxidant Role of Probiotics in Inflammation-Induced Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 9026. https://doi.org/10.3390/ijms25169026
Hamamah S, Lobiuc A, Covasa M. Antioxidant Role of Probiotics in Inflammation-Induced Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(16):9026. https://doi.org/10.3390/ijms25169026
Chicago/Turabian StyleHamamah, Sevag, Andrei Lobiuc, and Mihai Covasa. 2024. "Antioxidant Role of Probiotics in Inflammation-Induced Colorectal Cancer" International Journal of Molecular Sciences 25, no. 16: 9026. https://doi.org/10.3390/ijms25169026
APA StyleHamamah, S., Lobiuc, A., & Covasa, M. (2024). Antioxidant Role of Probiotics in Inflammation-Induced Colorectal Cancer. International Journal of Molecular Sciences, 25(16), 9026. https://doi.org/10.3390/ijms25169026