
Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
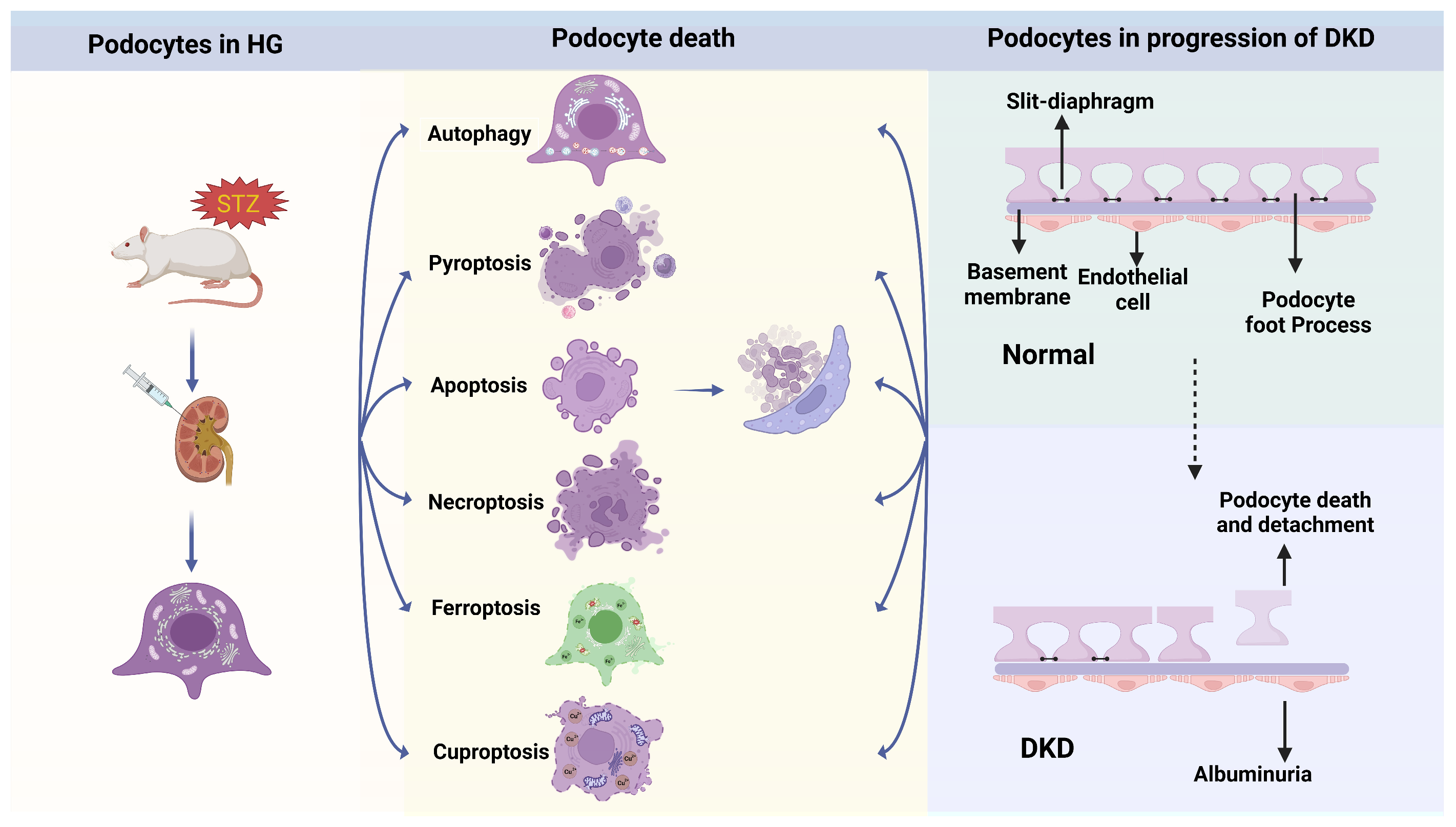
2. The Podocyte Deaths That Trigger the Progression of DKD
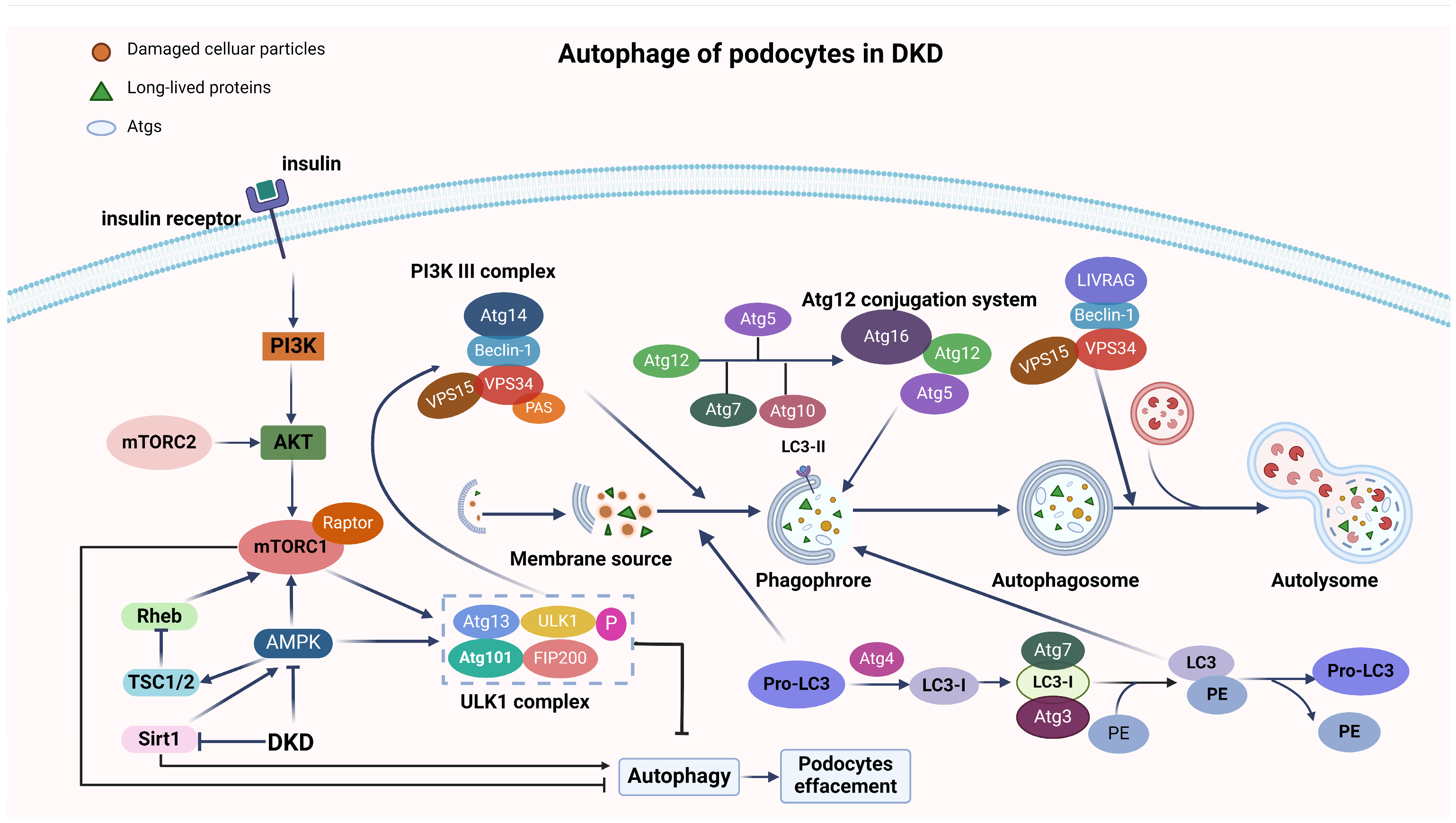
2.1. Autophagy
2.2. Pyroptosis
2.3. Apoptosis
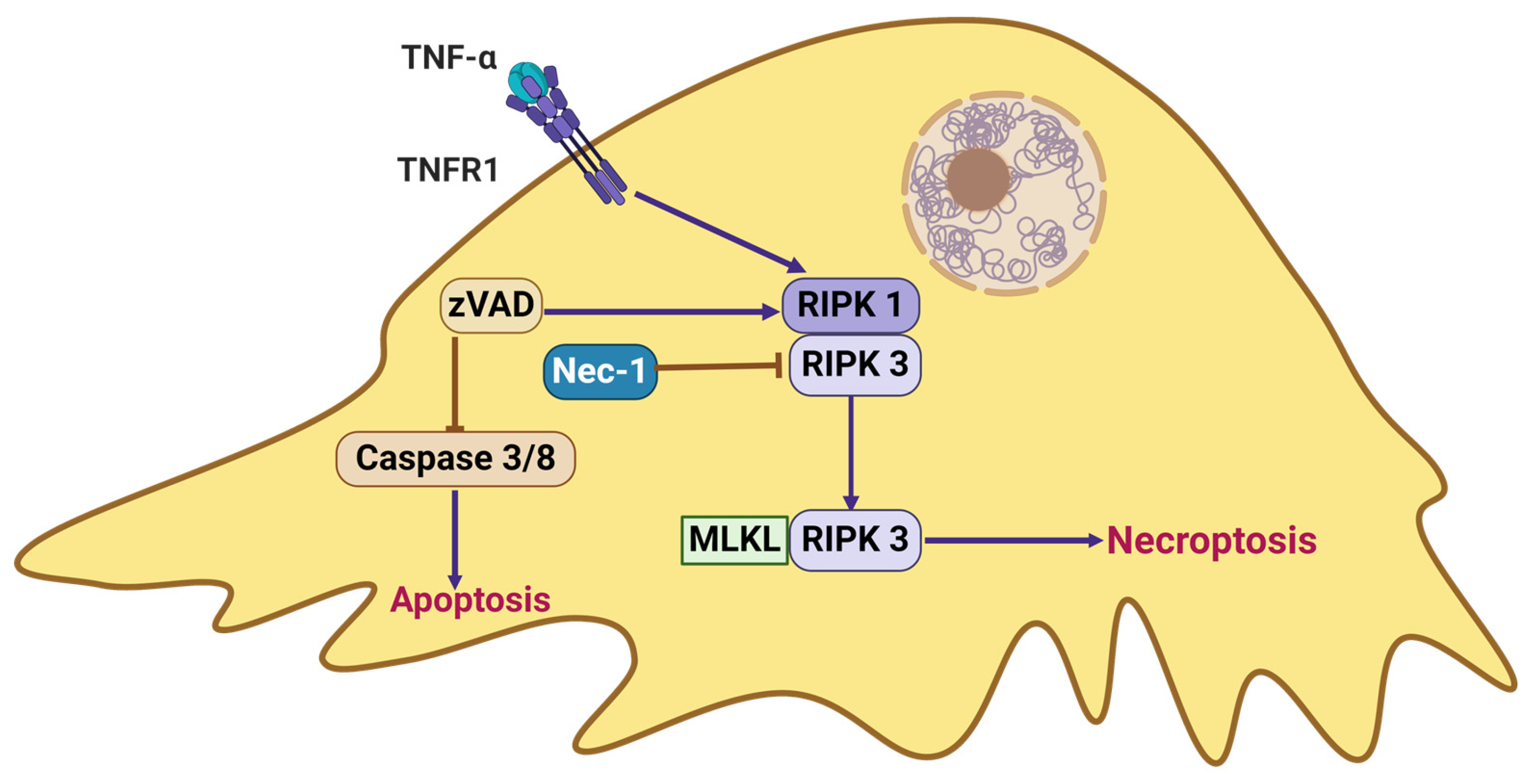
2.4. Necroptosis
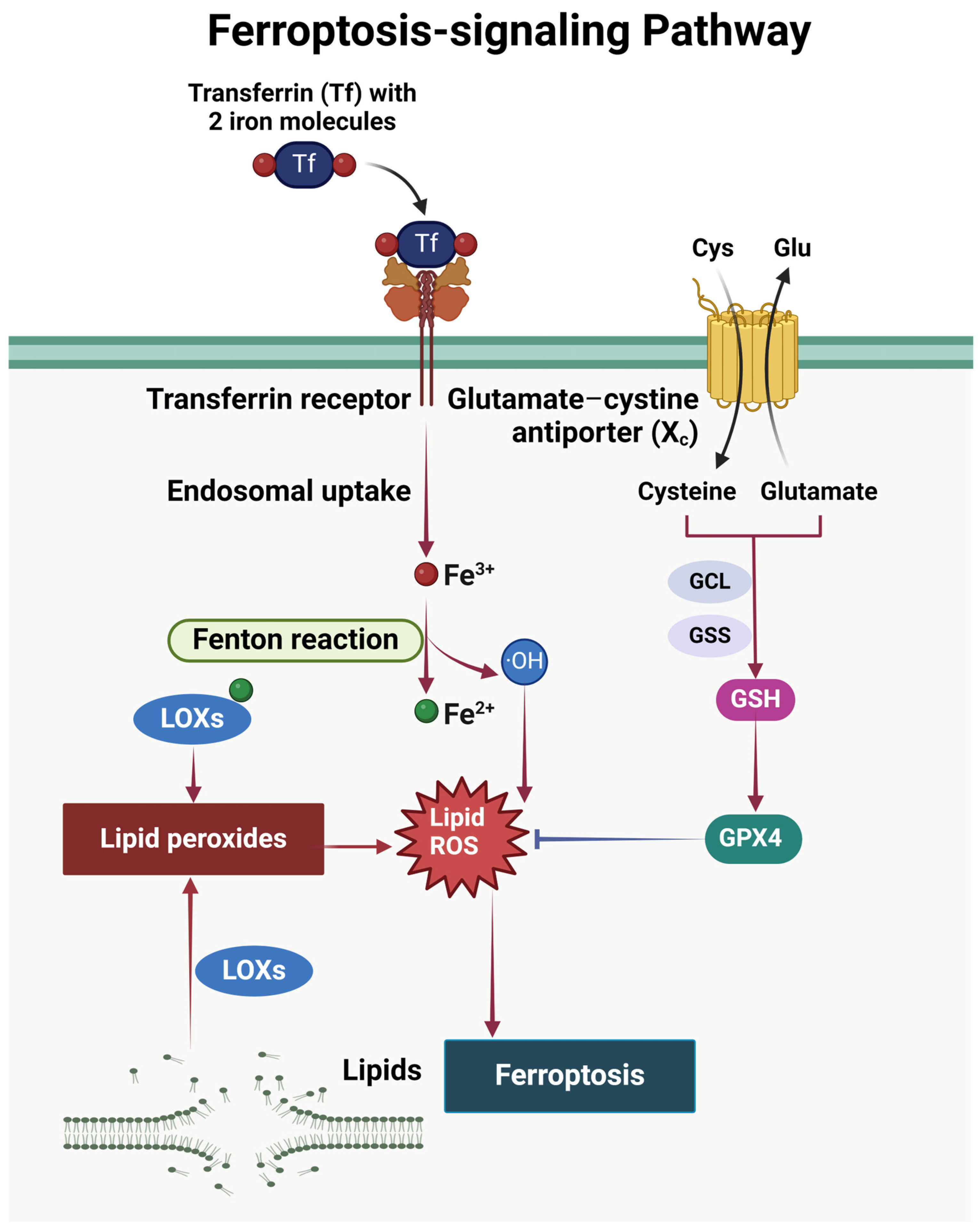
2.5. Ferroptosis
2.6. Others
3. Potential Therapeutic Targets for Preventing Podocyte Death in DKD
3.1. Potential Preclinical Therapeutic Approaches Targeting Podocyte Death
3.2. Clinical Study
4. Conclusions and Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bowe, B.; Xie, Y.; Li, T.; Mokdad, A.H.; Xian, H.; Yan, Y.; Maddukuri, G.; Al-Aly, Z. Changes in the US Burden of Chronic Kidney Disease from2002 to 2016: An Analysis of the Global Burden of Disease Study. JAMA Netw. Open 2018, 1, e184412. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; De Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic Kidney Disease: A Report froman ADA Consensus Conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, N.; Wu, Y.; Wang, M.; Yang, S.; Zheng, Y.; Deng, X.; Xiang, D.; Zhu, Y.; Xu, P.; et al. Global, Regional, and National Burden of Diabetes-Related Chronic Kidney Disease from1990 to 2019. Front. Endocrinol. 2021, 12, 672350. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B. The Global Burden of Diabetic Kidney Disease: Time Trends and Gender Gaps. Curr. Diab Rep. 2019, 19, 18. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Mallick, N.P.; Gokal, R. Haemodialysis. Lancet 1999, 353, 737–742. [Google Scholar] [CrossRef]
- Van Raalte, D.H.; Bjornstad, P.; Cherney, D.Z.I.; De Boer, I.H.; Fioretto, P.; Gordin, D.; Persson, F.; Rosas, S.E.; Rossing, P.; Schaub, J.A.; et al. Combination Therapy for Kidney Disease in People with Diabetes Mellitus. Nat. Rev. Nephrol. 2024, 20, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H.; et al. Global, Regional, and National Burden of Diabetes from 1990 to 2021, with Projections of Prevalence to 2050: A Systematic Analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef]
- IDF Diabetes Atlas. Available online: https://diabetesatlas.org/atlas/diabetes-and-kidney-disease/ (accessed on 4 June 2024).
- Quist, S.W.; Van Schoonhoven, A.V.; Bakker, S.J.L.; Pochopień, M.; Postma, M.J.; Van Loon, J.M.T.; Paulissen, J.H.J. Cost-Effectiveness of Finerenone in Chronic Kidney Disease Associated with Type 2 Diabetes in The Netherlands. Cardiovasc. Diabetol. 2023, 22, 328. [Google Scholar] [CrossRef]
- Li, Y.; Ning, Y.; Shen, B.; Shi, Y.; Song, N.; Fang, Y.; Ding, X. Temporal Trends in Prevalence and Mortality for Chronic Kidney Disease in China from 1990 to 2019: An Analysis of the Global Burden of Disease Study 2019. Clin. Kidney J. 2023, 16, 312–321. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes Management in Chronic Kidney Disease: A Consensus Report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2022, 102, 974–989. [Google Scholar] [CrossRef]
- Conti, S.; Remuzzi, G.; Benigni, A.; Tomasoni, S. Imaging the Kidney with an Unconventional Scanning Electron Microscopy Technique: Analysis of the Subpodocyte Space in Diabetic Mice. Int. J. Mol. Sci. 2022, 23, 1699. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-Induced Reactive Oxygen Species Cause Apoptosis of Podocytes and Podocyte Depletion at the Onset of Diabetic Nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Meliambro, K.; He, J.C.; Campbell, K.N. Podocyte-Targeted Therapies—Progress and Future Directions. Nat. Rev. Nephrol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Márquez, E.; Riera, M.; Pascual, J.; Soler, M.J. Renin-Angiotensin System within the Diabetic Podocyte. Am. J. Physiol. Ren. Physiol. 2015, 308, F1–F10. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate Immunity in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. Epigenetics and Epigenomics in Diabetic Kidney Disease and Metabolic Memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Thomas, M.C. Targeting the Pathobiology of Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2021, 28, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, J.M.; Gifford, C.C.; Tang, J.; Higgins, P.J.; Samarakoon, R. Emerging Role of Tumor Suppressor P53 in Acute and Chronic Kidney Diseases. Cell Mol. Life Sci. 2022, 79, 474. [Google Scholar] [CrossRef]
- Guo, Q.; Zhong, W.; Duan, A.; Sun, G.; Cui, W.; Zhuang, X.; Liu, L. Protective or Deleterious Role of Wnt/Beta-Catenin Signaling in Diabetic Nephropathy: An Unresolved Issue. Pharmacol. Res. 2019, 144, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Erekat, N.S. Programmed Cell Death in Diabetic Nephropathy: A Review of Apoptosis, Autophagy, and Necroptosis. Med. Sci. Monit. 2022, 28, e937766. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, Y.; Deng, X.; Li, Y.; Ma, X.; Zeng, J.; Zhao, Y. Diabetic Nephropathy: Focusing on Pathological Signals, Clinical Treatment, and Dietary Regulation. Biomed. Pharmacother. 2023, 159, 114252. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, M.; Cheng, C.K.; Li, Q. Tubular Injury in Diabetic Kidney Disease: Molecular Mechanisms and Potential Therapeutic Perspectives. Front. Endocrinol. 2023, 14, 1238927. [Google Scholar] [CrossRef]
- Fu, J.; Wei, C.; Lee, K.; Zhang, W.; He, W.; Chuang, P.; Liu, Z.; He, J.C. Comparison of Glomerular and Podocyte mRNA Profiles in Streptozotocin-Induced Diabetes. J. Am. Soc. Nephrol. 2016, 27, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, A.; Pan, Y.; Cao, S.; Terker, A.S.; Wang, S.; Fan, X.; Toth, C.L.; Ramirez Solano, M.A.; Michell, D.L.; et al. Profile of Podocyte Translatome During Development of Type 2 and Type 1 Diabetic Nephropathy Using Podocyte-Specific TRAP mRNA RNA-Seq. Diabetes 2021, 70, 2377–2390. [Google Scholar] [CrossRef]
- Chen, S.-J.; Lv, L.-L.; Liu, B.-C.; Tang, R.-N. Crosstalk between Tubular Epithelial Cells and Glomerular Endothelial Cells in Diabetic Kidney Disease. Cell Prolif. 2020, 53, e12763. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The Renin-Angiotensin-Aldosterone System and Its Suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef]
- Kuang, Z.; Hou, N.; Kan, C.; Han, F.; Qiu, H.; Sun, X. The Protective Effects of SGLT-2 Inhibitors, GLP-1 Receptor Agonists, and RAAS Blockers against Renal Injury in Patients with Type 2 Diabetes. Int. Urol. Nephrol. 2023, 55, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Wilcox, C.S.; Arendshorst, W.J. Renal Autoregulation in Health and Disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chang, Y.-H.; Yang, S.-Y.; Wu, K.-D.; Chu, T.-S. Update of Pathophysiology and Management of Diabetic Kidney Disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef]
- Guedes, M.; Pecoits-Filho, R. Can We Cure Diabetic Kidney Disease? Present and Future Perspectives from a Nephrologist’s Point of View. J. Intern. Med. 2022, 291, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.-Y.; Yoo, T.-H. Pathophysiologic Mechanisms and Potential Biomarkers in Diabetic Kidney Disease. Diabetes Metab. J. 2022, 46, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zheng, H.J.; Zhang, W.; Lou, W.; Xia, C.; Han, X.T.; Huang, W.J.; Zhang, F.; Wang, Y.; Liu, W.J. Accelerated Kidney Aging in Diabetes Mellitus. Oxid. Med. Cell Longev. 2020, 2020, 1234059. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Nakamura, N.; Suematsu, M.; Kaseda, K.; Matsui, T. Advanced Glycation End Products: A Molecular Target for Vascular Complications in Diabetes. Mol. Med. 2015, 21 (Suppl. S1), S32–S40. [Google Scholar] [CrossRef]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular Endothelial Cell Injury and Cross Talk in Diabetic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Li, M.; Armelloni, S.; Mattinzoli, D.; Ikehata, M.; Chatziantoniou, C.; Alfieri, C.; Molinari, P.; Chadjichristos, C.E.; Malvica, S.; Castellano, G. Crosstalk Mechanisms between Glomerular Endothelial Cells and Podocytes in Renal Diseases and Kidney Transplantation. Kidney Res. Clin. Pr. 2024, 43, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, J.; Xuan, C.; Ma, H. A Review on the Physiological and Pathophysiological Role of Endothelial Glycocalyx. J. Biochem. Mol. Tox 2020, 34, e22571. [Google Scholar] [CrossRef] [PubMed]
- Gamez, M.; Elhegni, H.E.; Fawaz, S.; Ho, K.H.; Campbell, N.W.; Copland, D.A.; Onions, K.L.; Butler, M.J.; Wasson, E.J.; Crompton, M.; et al. Heparanase Inhibition as a Systemic Approach to Protect the Endothelial Glycocalyx and Prevent Microvascular Complications in Diabetes. Cardiovasc. Diabetol. 2024, 23, 50. [Google Scholar] [CrossRef]
- Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef]
- Finch, N.C.; Neal, C.R.; Welsh, G.I.; Foster, R.R.; Satchell, S.C. The Unique Structural and Functional Characteristics of Glomerular Endothelial Cell Fenestrations and Their Potential as a Therapeutic Target in Kidney Disease. Am. J. Physiol. Ren. Physiol. 2023, 325, F465–F478. [Google Scholar] [CrossRef]
- Hu, S.; Hang, X.; Wei, Y.; Wang, H.; Zhang, L.; Zhao, L. Crosstalk among Podocytes, Glomerular Endothelial Cells and Mesangial Cells in Diabetic Kidney Disease: An Updated Review. Cell Commun. Signal 2024, 22, 136. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z. Mechanistic Pathogenesis of Endothelial Dysfunction in Diabetic Nephropathy and Retinopathy. Front. Endocrinol. 2022, 13, 816400. [Google Scholar] [CrossRef]
- Abu Seman, N.; Othman, S.H. Recent Progress in Genetics and Epigenetics Research on Diabetic Nephropathy in Malaysia. J. Diabetes Res. 2023, 2023, 9053580. [Google Scholar] [CrossRef] [PubMed]
- Khurana, I.; Kaipananickal, H.; Maxwell, S.; Birkelund, S.; Syreeni, A.; Forsblom, C.; Okabe, J.; Ziemann, M.; Kaspi, A.; Rafehi, H.; et al. Reduced Methylation Correlates with Diabetic Nephropathy Risk in Type 1 Diabetes. J. Clin. Investig. 2023, 133, e160959. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, J.; Li, Y.; Rao, J.; Xu, G. Epigenetics and Endoplasmic Reticulum in Podocytopathy during Diabetic Nephropathy Progression. Front. Immunol. 2022, 13, 1090989. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Sun, Y.; Nie, L.; Cui, A.; Zhao, P.; Leung, W.K.; Wang, Q. Metabolic Memory: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2024, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Qi, F.; Guo, F.; Shao, M.; Song, Y.; Ren, G.; Linlin, Z.; Qin, G.; Zhao, Y. An Update on Chronic Complications of Diabetes Mellitus: From Molecular Mechanisms to Therapeutic Strategies with a Focus on Metabolic Memory. Mol. Med. 2024, 30, 71. [Google Scholar] [CrossRef]
- Reddy, M.A.; Park, J.T.; Natarajan, R. Epigenetic Modifications and Diabetic Nephropathy. Kidney Res. Clin. Pract. 2012, 31, 139–150. [Google Scholar] [CrossRef]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef]
- Tang, G.; Li, S.; Zhang, C.; Chen, H.; Wang, N.; Feng, Y. Clinical Efficacies, Underlying Mechanisms and Molecular Targets of Chinese Medicines for Diabetic Nephropathy Treatment and Management. Acta Pharm. Sin. B 2021, 11, 2749–2767. [Google Scholar] [CrossRef]
- Vartak, T.; Godson, C.; Brennan, E. Therapeutic Potential of Pro-Resolving Mediators in Diabetic Kidney Disease. Adv. Drug Deliv. Rev. 2021, 178, 113965. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef]
- Dama, A.; Shpati, K.; Daliu, P.; Dumur, S.; Gorica, E.; Santini, A. Targeting Metabolic Diseases: The Role of Nutraceuticals in Modulating Oxidative Stress and Inflammation. Nutrients 2024, 16, 507. [Google Scholar] [CrossRef]
- Mohandes, S.; Doke, T.; Hu, H.; Mukhi, D.; Dhillon, P.; Susztak, K. Molecular Pathways That Drive Diabetic Kidney Disease. J. Clin. Investig. 2023, 133, e165654. [Google Scholar] [CrossRef]
- Kruczkowska, W.; Gałęziewska, J.; Kciuk, M.; Gielecińska, A.; Płuciennik, E.; Pasieka, Z.; Zhao, L.-Y.; Yu, Y.-J.; Kołat, D.; Kałuzińska-Kołat, Ż. Senescent Adipocytes and Type 2 Diabetes—Current Knowledge and Perspective Concepts. Biomol. Concepts. 2024, 15, 20220046. [Google Scholar] [CrossRef]
- Gao, P.; Zou, X.; Sun, X.; Zhang, C. Cellular Senescence in Metabolic-Associated Kidney Disease: An Update. Cells 2022, 11, 3443. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R.; Ferrario, F.; Villaggio, B.; Gianiorio, F.; Tosetti, F.; Weiss, U.; et al. Accelerated Senescence in the Kidneys of Patients with Type 2 Diabetic Nephropathy. Am. J. Physiol.-Ren. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Iwano, M.; Suzuki, D.; Nakatani, K.; Kimura, K.; Harada, K.; Kubo, A.; Akai, Y.; Toyoda, M.; Kanauchi, M.; et al. Epithelial-Mesenchymal Transition as a Potential Explanation for Podocyte Depletion in Diabetic Nephropathy. Am. J. Kidney Dis. 2009, 54, 653–664. [Google Scholar] [CrossRef]
- Jiang, A.; Song, A.; Zhang, C. Modes of Podocyte Death in Diabetic Kidney Disease: An Update. J. Nephrol. 2022, 35, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Xing, X.; Li, M.; Liu, Y.; Xu, A.; Zhang, J. Podocyte Injury of Diabetic Nephropathy: Novel Mechanism Discovery and Therapeutic Prospects. Biomed. Pharmacother. 2023, 168, 115670. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The Mechanisms and Roles of Selective Autophagy in Mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Klionsky, D.J.; Shen, H.-M. The Emerging Mechanisms and Functions of Microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Tian, Y.; Li, Z.; Hu, W.; Ren, H.; Tian, E.; Zhao, Y.; Lu, Q.; Huang, X.; Yang, P.; Li, X.; et al. C. Elegans Screen Identifies Autophagy Genes Specific to Multicellular Organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef]
- Wang, B.; Qian, J.-Y.; Tang, T.-T.; Lin, L.-L.; Yu, N.; Guo, H.-L.; Ni, W.-J.; Lv, L.-L.; Wen, Y.; Li, Z.-L.; et al. VDR/Atg3 Axis Regulates Slit Diaphragm to Tight Junction Transition via P62-Mediated Autophagy Pathway in Diabetic Nephropathy. Diabetes 2021, 70, 2639–2651. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2020, 82, 297–322. [Google Scholar] [CrossRef]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.-I.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Yang, H.; Sun, J.; Sun, A.; Wei, Y.; Xie, W.; Xie, P.; Zhang, L.; Zhao, L.; Huang, Y. Podocyte Programmed Cell Death in Diabetic Kidney Disease: Molecular Mechanisms and Therapeutic Prospects. Biomed. Pharmacother. 2024, 177, 117140. [Google Scholar] [CrossRef]
- Njeim, R.; Merscher, S.; Fornoni, A. Mechanisms and Implications of Podocyte Autophagy in Chronic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2024, 326, F877–F893. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, Y.; Xue, J.; Jin, D.; Li, X.; Zhao, D.; Lian, F.; Qi, W.; Tong, X. The Critical Role of Dysregulated Autophagy in the Progression of Diabetic Kidney Disease. Front. Pharmacol. 2022, 13, 977410. [Google Scholar] [CrossRef]
- Cho, W.; Oh, H.; Choi, S.W.; Abd El-Aty, A.M.; Birdal, O.; Jeong, J.H.; Song, J.-H.; Jung, T.W. CTRP4 Attenuates Apoptosis and Epithelial-Mesenchymal Transition Markers in Podocytes through an AMPK/Autophagy-Dependent Pathway. Biochem. Biophys. Res. Commun. 2023, 682, 104–110. [Google Scholar] [CrossRef]
- Orvieto, R.; Aizer, A.; Gleicher, N. Is There Still a Rationale for Non-Invasive PGT-A by Analysis of Cell-Free DNA Released by Human Embryos into Culture Medium? Hum. Reprod. 2021, 36, 1186–1190. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, L.; Liu, X.-Q.; Huang, Y.-B.; Wang, A.-L.; Zeng, H.-X.; Gao, L.; Zhu, Q.-J.; Xia, L.-L.; Wu, Y.-G. Paeoniflorin Binds to VEGFR2 to Restore Autophagy and Inhibit Apoptosis for Podocyte Protection in Diabetic Kidney Disease through PI3K-AKT Signaling Pathway. Phytomedicine 2022, 106, 154400. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of Cell Death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. β-Thujaplicin Induces Autophagic Cell Death, Apoptosis, and Cell Cycle Arrest through ROS-Mediated Akt and P38/ERK MAPK Signaling in Human Hepatocellular Carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef]
- Chen, D.; Liu, Y.; Chen, J.; Lin, H.; Guo, H.; Wu, Y.; Xu, Y.; Zhou, Y.; Zhou, W.; Lu, R.; et al. JAK/STAT Pathway Promotes the Progression of Diabetic Kidney Disease via Autophagy in Podocytes. Eur. J. Pharmacol. 2021, 902, 174121. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Liu, F.; Yu, M.; Zhao, Y.; Yan, G.; Xue, J.; Sun, Y.; Zhao, D.; Li, X.; Qi, W.; et al. Jiedu Tongluo Baoshen Formula Enhances Podocyte Autophagy and Reduces Proteinuria in Diabetic Kidney Disease by Inhibiting PI3K/Akt/mTOR Signaling Pathway. J. Ethnopharmacol. 2022, 293, 115246. [Google Scholar] [CrossRef]
- Heintz, L.; Meyer-Schwesinger, C. The Intertwining of Autophagy and the Ubiquitin Proteasome System in Podocyte (Patho)Physiology. Cell Physiol. Biochem. 2021, 55, 68–95. [Google Scholar] [PubMed]
- Cybulsky, A.V. Endoplasmic Reticulum Stress, the Unfolded Protein Response and Autophagy in Kidney Diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Li, X.-Z.; Jiang, H.; Xu, L.; Liu, Y.-Q.; Tang, J.-W.; Shi, J.-S.; Yu, X.-J.; Wang, X.; Du, L.; Lu, Q.; et al. Sarsasapogenin Restores Podocyte Autophagy in Diabetic Nephropathy by Targeting GSK3β Signaling Pathway. Biochem. Pharmacol. 2021, 192, 114675. [Google Scholar] [CrossRef]
- Su, P.-P.; Liu, D.-W.; Zhou, S.-J.; Chen, H.; Wu, X.-M.; Liu, Z.-S. Down-Regulation of Risa Improves Podocyte Injury by Enhancing Autophagy in Diabetic Nephropathy. Mil. Med. Res. 2022, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Ma, F.; Liu, T.; Yang, L.; Mao, H.; Wang, Y.; Peng, L.; Li, P.; Zhan, Y. Sirtuins in Kidney Diseases: Potential Mechanism and Therapeutic Targets. Cell Commun. Signal 2024, 22, 114. [Google Scholar] [CrossRef]
- Ala, M. Sestrin2 Signaling Pathway Regulates Podocyte Biology and Protects against Diabetic Nephropathy. J. Diabetes Res. 2023, 2023, 8776878. [Google Scholar] [CrossRef]
- Liu, M.; Liang, K.; Zhen, J.; Zhou, M.; Wang, X.; Wang, Z.; Wei, X.; Zhang, Y.; Sun, Y.; Zhou, Z.; et al. Sirt6 Deficiency Exacerbates Podocyte Injury and Proteinuria through Targeting Notch Signaling. Nat. Commun. 2017, 8, 413. [Google Scholar] [CrossRef]
- Liu, T.; Yang, L.; Mao, H.; Ma, F.; Wang, Y.; Li, S.; Li, P.; Zhan, Y. Sirtuins as Novel Pharmacological Targets in Podocyte Injury and Related Glomerular Diseases. Biomed. Pharmacother. 2022, 155, 113620. [Google Scholar] [CrossRef]
- Buetow, L.; Huang, D.T. Structural Insights into the Catalysis and Regulation of E3 Ubiquitin Ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef]
- Kulkarni, A.; Preeti, K.; Tryphena, K.P.; Srivastava, S.; Singh, S.B.; Khatri, D.K. Proteostasis in Parkinson’s Disease: Recent Development and Possible Implication in Diagnosis and Therapeutics. Ageing Res. Rev. 2023, 84, 101816. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.-H.; Chen, Y.-H.; Huang, T.-Y. Ubiquitin-Mediated Regulation of Autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and Autophagy-Related Pathways in Cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Liu, W.J.; Gan, Y.; Huang, W.F.; Wu, H.; Zhang, X.; Zheng, H.J.; Liu, H. Lysosome Restoration to Activate Podocyte Autophagy: A New Therapeutic Strategy for Diabetic Kidney Disease. Cell Death Dis. 2019, 10, 806. [Google Scholar] [CrossRef]
- Cui, Z.; Cong, M.; Yin, S.; Li, Y.; Ye, Y.; Liu, X.; Tang, J. Role of Protein Degradation Systems in Colorectal Cancer. Cell Death Discov. 2024, 10, 141. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [PubMed]
- Zhu, W.; Li, Y.-Y.; Zeng, H.-X.; Liu, X.-Q.; Sun, Y.-T.; Jiang, L.; Xia, L.-L.; Wu, Y.-G. Carnosine Alleviates Podocyte Injury in Diabetic Nephropathy by Targeting Caspase-1-Mediated Pyroptosis. Int. Immunopharmacol. 2021, 101, 108236. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic Cell Death Defends against Intracellular Pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in Pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, Z.; Li, Y. Relevance of the Pyroptosis-Related Inflammasome Pathway in the Pathogenesis of Diabetic Kidney Disease. Front. Immunol. 2021, 12, 603416. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Pan, J.; Zhou, Z.-L.; Yin, F.; Xie, H.-Y.; Chen, P.-P.; Li, J.-Y.; Zheng, P.-Q.; Zhou, L.; Zhang, W.; et al. Caspase-11/4 and Gasdermin D-Mediated Pyroptosis Contributes to Podocyte Injury in Mouse Diabetic Nephropathy. Acta Pharmacol. Sin. 2021, 42, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-W.; Tang, M.-Q.; Liu, W.; Song, Y.; Gao, M.-J.; Ni, P.; Zhang, D.-D.; Mo, Q.-G.; Zhao, B.-Q. Dapagliflozin Prevents Kidney Podocytes Pyroptosis via miR-155-5p/HO-1/NLRP3 Axis Modulation. Int. Immunopharmacol. 2024, 131, 111785. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, J.; Jiang, L.; Liu, X.; Ge, Q.; Wang, Q.; Qi, X.; Wu, Y. Rutaecarpine Protects Podocytes in Diabetic Kidney Disease by Targeting VEGFR2/NLRP3-Mediated Pyroptosis. Int. Immunopharmacol. 2024, 130, 111790. [Google Scholar] [CrossRef]
- Kim, D.; Ban, K.-Y.; Lee, G.-H.; Jun, H.-S. Lysophosphatidic Acid Induces Podocyte Pyroptosis in Diabetic Nephropathy by an Increase of Egr1 Expression via Downregulation of EzH2. Int. J. Mol. Sci. 2023, 24, 9968. [Google Scholar] [CrossRef]
- Liu, B.-H.; Tu, Y.; Ni, G.-X.; Yan, J.; Yue, L.; Li, Z.-L.; Wu, J.-J.; Cao, Y.-T.; Wan, Z.-Y.; Sun, W.; et al. Total Flavones of Abelmoschus Manihot Ameliorates Podocyte Pyroptosis and Injury in High Glucose Conditions by Targeting METTL3-Dependent m6A Modification-Mediated NLRP3-Inflammasome Activation and PTEN/PI3K/Akt Signaling. Front. Pharmacol. 2021, 12, 667644. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, Z.; Luo, Z.; Yang, D.; Hao, Y.; Hu, J.; Feng, J.; Zhu, Z.; Luo, Q.; Zhang, Z.; et al. STING Deletion Alleviates Podocyte Injury through Suppressing Inflammation by Targeting NLRP3 in Diabetic Kidney Disease. Cell Signal 2023, 109, 110777. [Google Scholar] [CrossRef]
- Gao, Y.; Ma, Y.; Xie, D.; Jiang, H. ManNAc Protects against Podocyte Pyroptosis via Inhibiting Mitochondrial Damage and ROS/NLRP3 Signaling Pathway in Diabetic Kidney Injury Model. Int. Immunopharmacol. 2022, 107, 108711. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.; Lv, Z.; Shu, A.; Du, Q.; Wang, W.; Chen, Y.; Xu, H. Study on the Inhibitive Effect of Catalpol on Diabetic Nephropathy. Life Sci. 2020, 257, 118120. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H.; Kerr, J.F.R.; Currie, A.R. Cell Death: The Significance of Apoptosis. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1980; Volume 68, pp. 251–306. ISBN 978-0-12-364468-8. [Google Scholar]
- Yang, S.; Hu, C.; Chen, X.; Tang, Y.; Li, J.; Yang, H.; Yang, Y.; Ying, B.; Xiao, X.; Li, S.-Z.; et al. Crosstalk between Metabolism and Cell Death in Tumorigenesis. Mol. Cancer 2024, 23, 71. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.-D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 Protein Family: Attractive Targets for Cancer Therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef]
- Xu, Y.; Ye, H. Progress in Understanding the Mechanisms of Resistance to BCL-2 Inhibitors. Exp. Hematol. Oncol. 2022, 11, 31. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The Role of Mitochondrial Factors in Apoptosis: A Russian Roulette with More than One Bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The Biochemistry of Apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular Mechanisms of Cell Death in Neurological Diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanisms of Caspase Activation and Inhibition during Apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs Less Travelled: TRAIL in Cancer Biology and Therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-Enzymatic Role as a Scaffold for Assembly of a Pro-Inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef]
- Mouasni, S.; Tourneur, L. FADD at the Crossroads between Cancer and Inflammation. Trends Immunol. 2018, 39, 1036–1053. [Google Scholar] [CrossRef]
- Mandal, R.; Barrón, J.C.; Kostova, I.; Becker, S.; Strebhardt, K. Caspase-8: The Double-Edged Sword. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188357. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, C.; Ye, Q.; Tong, L.; Jiang, H.; Zhu, X.; Huang, L.; Lin, W.; Fu, H.; Wang, J.; et al. Podocyte Apoptosis in Diabetic Nephropathy by BASP1 Activation of the P53 Pathway via WT1. Acta Physiol. 2021, 232, e13634. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, H.; Zhu, J.; Song, N.; Lu, Z.; Fang, Y.; Teng, J.; Dai, Y.; Ding, X. Inhibition of P53/miR-34a/SIRT1 Axis Ameliorates Podocyte Injury in Diabetic Nephropathy. Biochem. Biophys. Res. Commun. 2021, 559, 48–55. [Google Scholar] [CrossRef]
- Feng, Y.; Sun, Z.; Fu, J.; Zhong, F.; Zhang, W.; Wei, C.; Chen, A.; Liu, B.-C.; He, J.C.; Lee, K. Podocyte-Derived Soluble RARRES1 Drives Kidney Disease Progression through Direct Podocyte and Proximal Tubular Injury. Kidney Int. 2024, 106, 50–66. [Google Scholar] [CrossRef]
- Gong, M.; Guo, Y.; Dong, H.; Wu, F.; He, Q.; Gong, J.; Lu, F. Modified Hu-Lu-Ba-Wan Protects Diabetic Glomerular Podocytes via Promoting PKM2-Mediated Mitochondrial Dynamic Homeostasis. Phytomedicine 2024, 123, 155247. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yuan, Q.; Tang, B.; Xie, Y.; Cao, Y.; Qiu, Y.; Zeng, J.; Wang, Z.; Su, H.; Zhang, C. CPT1A Protects Podocytes fromLipotoxicity and Apoptosis In Vitro and Alleviates Diabetic Nephropathy In Vivo. Diabetes 2024, 73, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Chi, K.; Ji, Y.; Zhang, K.; Li, P.; Fu, Z.; Wang, X.; Cui, S.; Shen, W.; et al. IGFBP2 Induces Podocyte Apoptosis Promoted by Mitochondrial Damage via Integrin A5/FAK in Diabetic Kidney Disease. Apoptosis 2024, 29, 1109–1125. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, S.; Tian, X.; Yuan, L.; Gao, Y.; Liu, D.; Qi, H.; Wang, S.; Liu, Z.; Li, Y.; et al. miR-4645-3p Attenuates Podocyte Injury and Mitochondrial Dysfunction in Diabetic Kidney Disease by Targeting Cdk5. FASEB J. 2024, 38, e23668. [Google Scholar] [CrossRef]
- Chan, F.K.-M.; Luz, N.F.; Moriwaki, K. Programmed Necrosis in the Cross Talk of Cell Death and Inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, H.; Hu, Y.; Fang, Y.; Qi, C.; Huang, J.; Cai, X.; Wu, H.; Ding, X.; Zhang, Z. High Glucose-Induced Apoptosis and Necroptosis in Podocytes Is Regulated by UCHL1 via RIPK1/RIPK3 Pathway. Exp. Cell Res. 2019, 382, 111463. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-X.; Fang, Q.-J.; Wan, Y.-G.; Liu, Y.-L.; Wang, Y.; Wu, W.; Tu, Y.; Wang, M.-Z.; Wang, D.-G.; Ge, H.-T. Effects and mechanisms of total flavones of Abelmoschus manihot in inhibiting podocyte necroptosis and renal fibrosis in diabetic kidney disease. Zhongguo Zhong Yao Za Zhi 2023, 48, 4137–4146. [Google Scholar]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis Is a Type of Autophagy-Dependent Cell Death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Wu, Q.; Huang, F. Targeting Ferroptosis as a Prospective Therapeutic Approach for Diabetic Nephropathy. Ann. Med. 2024, 56, 2346543. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Tang, Z.; Luo, L. The Crosstalk between Oncogenic Signaling and Ferroptosis in Cancer. Crit. Rev. Oncol. Hematol. 2024, 197, 104349. [Google Scholar] [CrossRef]
- Chen, R.; Zhu, S.; Zhao, R.; Liu, W.; Jin, L.; Ren, X.; He, H. Targeting Ferroptosis as a Potential Strategy to Overcome the Resistance of Cisplatin in Oral Squamous Cell Carcinoma. Front. Pharmacol. 2024, 15, 1402514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, J. Targeting Ferroptosis Regulators by Natural Products in Colorectal Cancer. Front. Pharmacol. 2024, 15, 1374722. [Google Scholar] [CrossRef]
- Nejadi Orang, F.; Abdoli Shadbad, M. Competing Endogenous RNA Networks and Ferroptosis in Cancer: Novel Therapeutic Targets. Cell Death Dis. 2024, 15, 357. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Kang, Z.; Zhang, F. Tanshinone IIA Suppresses Ferroptosis to Attenuate Renal Podocyte Injury in Diabetic Nephropathy through the Embryonic Lethal Abnormal Visual-like Protein 1 and Acyl-Coenzyme A Synthetase Long-Chain Family Member 4 Signaling Pathway. J. Diabetes Investig. 2024, 15, 1003–1016. [Google Scholar] [CrossRef]
- Jin, J.; Wang, Y.; Zheng, D.; Liang, M.; He, Q. A Novel Identified Circular RNA, mmu_mmu_circRNA_0000309, Involves in Germacrone-Mediated Improvement of Diabetic Nephropathy Through Regulating Ferroptosis by Targeting miR-188-3p/GPX4 Signaling Axis. Antioxid. Redox Signal 2022, 36, 740–759. [Google Scholar] [CrossRef]
- Jiao, Y.; Liu, X.; Shi, J.; An, J.; Yu, T.; Zou, G.; Li, W.; Zhuo, L. Unraveling the Interplay of Ferroptosis and Immune Dysregulation in Diabetic Kidney Disease: A Comprehensive Molecular Analysis. Diabetol. Metab. Syndr. 2024, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, Y.; Hu, J.-E.; Ding, Y.; Shen, Y.; Xu, H.; Chen, H.; Wu, N. Sp1-Mediated Upregulation of Prdx6 Expression Prevents Podocyte Injury in Diabetic Nephropathy via Mitigation of Oxidative Stress and Ferroptosis. Life Sci. 2021, 278, 119529. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, X.; Feng, M.; Yu, Y.; Hu, Y.; Jiang, W. Exosomal miR-223-3p from Bone Marrow Mesenchymal Stem Cells Targets HDAC2 to Downregulate STAT3 Phosphorylation to Alleviate HBx-Induced Ferroptosis in Podocytes. Front. Pharmacol. 2024, 15, 1327149. [Google Scholar] [CrossRef]
- Feng, J.; Chen, Z.; Ma, Y.; Yang, X.; Zhu, Z.; Zhang, Z.; Hu, J.; Liang, W.; Ding, G. AKAP1 Contributes to Impaired mtDNA Replication and Mitochondrial Dysfunction in Podocytes of Diabetic Kidney Disease. Int. J. Biol. Sci. 2022, 18, 4026–4042. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a Newly Characterized Form of Cell Death in Parkinson’s Disease That Is Regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Banerjee, S.; Lu, S.; Jain, A.; Wang, I.; Tao, H.; Srinivasan, S.; Nemeth, E.; He, P. Targeting PKCa Alleviates Iron Overload in Diabetes and Hemochromatosis through the Inhibition of Ferroportin. Blood J. 2024. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, J.; Wang, F. Copper Homeostasis and Cuproptosis in Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef]
- Al-Bayati, M.A.; Jamil, D.A.; Al-Aubaidy, H.A. Cardiovascular Effects of Copper Deficiency on Activity of Superoxide Dismutase in Diabetic Nephropathy. N. Am. J. Med. Sci. 2015, 7, 41–46. [Google Scholar]
- Chen, Y.; Liao, L.; Wang, B.; Wu, Z. Identification and Validation of Immune and Cuproptosis—Related Genes for Diabetic Nephropathy by WGCNA and Machine Learning. Front. Immunol. 2024, 15, 1332279. [Google Scholar]
- Thomasova, D.; Bruns, H.A.; Kretschmer, V.; Ebrahim, M.; Romoli, S.; Liapis, H.; Kotb, A.M.; Endlich, N.; Anders, H.-J. Murine Double Minute-2 Prevents P53-Overactivation-Related Cell Death (Podoptosis) of Podocytes. J. Am. Soc. Nephrol. 2015, 26, 1513–1523. [Google Scholar] [CrossRef]
- Lai, W.; Luo, D.; Li, Y.; Li, Y.; Wang, Q.; Hu, Z.; Ye, Z.; Peng, H. Irisin Ameliorates Diabetic Kidney Disease by Restoring Autophagy in Podocytes. FASEB J. 2023, 37, e23175. [Google Scholar] [CrossRef]
- Wang, L.; Tian, H.; Wang, H.; Mao, X.; Luo, J.; He, Q.; Wen, P.; Cao, H.; Fang, L.; Zhou, Y.; et al. Disrupting Circadian Control of Autophagy Induces Podocyte Injury and Proteinuria. Kidney Int. 2024, 105, 1020–1034. [Google Scholar] [CrossRef]
- Schelling, J.R. The Contribution of Lipotoxicity to Diabetic Kidney Disease. Cells 2022, 11, 3236. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D.; Chagnac, A.; De Vries, A.P.J.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-Related Glomerulopathy: Clinical and Pathologic Characteristics and Pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef]
- Murea, M.; Freedman, B.I.; Parks, J.S.; Antinozzi, P.A.; Elbein, S.C.; Ma, L. Lipotoxicity in Diabetic Nephropathy: The Potential Role of Fatty Acid Oxidation. Clin. J. Am. Soc. Nephrol. 2010, 5, 2373–2379. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Leung, V.T.M.; Chan, L.Y.Y.; Wong, S.S.H.; Chu, D.W.S.; Leung, J.C.K.; Ho, Y.W.; Lai, K.N.; Ma, L.; Elbein, S.C.; et al. The Acetyl-Coenzyme A Carboxylase Beta (ACACB) Gene Is Associated with Nephropathy in Chinese Patients with Type 2 Diabetes. Nephrol. Dial. Transplant. 2010, 25, 3931–3934. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.Q.; Chen, P.P.; Zhang, J.X.; Liu, L.; Wang, G.H.; Liu, X.Q.; Jiang, T.T.; Wang, M.Y.; Liu, W.T.; et al. Activation of Acetyl-CoA Synthetase 2 Mediates Kidney Injury in Diabetic Nephropathy. JCI Insight 2023, 8, e165817. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pan, Y.; Cao, S.; Sasaki, K.; Wang, Y.; Niu, A.; Fan, X.; Wang, S.; Zhang, M.-Z.; Harris, R.C. Podocyte EGFR Inhibits Autophagy Through Upregulation of Rubicon in Type 2 Diabetic Nephropathy. Diabetes 2021, 70, 562–576. [Google Scholar] [CrossRef]
- Shi, L.; Xiao, C.; Zhang, Y.; Xia, Y.; Zha, H.; Zhu, J.; Song, Z. Vitamin D/Vitamin D Receptor/Atg16L1 Axis Maintains Podocyte Autophagy and Survival in Diabetic Kidney Disease. Ren. Fail. 2022, 44, 694–705. [Google Scholar] [CrossRef]
- Salemkour, Y.; Yildiz, D.; Dionet, L.; ’t Hart, D.C.; Verheijden, K.A.T.; Saito, R.; Mahtal, N.; Delbet, J.-D.; Letavernier, E.; Rabant, M.; et al. Podocyte Injury in Diabetic Kidney Disease in Mouse Models Involves TRPC6-Mediated Calpain Activation Impairing Autophagy. J. Am. Soc. Nephrol. 2023, 34, 1823–1842. [Google Scholar] [CrossRef]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in Db/Db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, C.; Liu, L.; Xu, L.; Yao, L. Senolytic Combination of Dasatinib and Quercetin Protects against Diabetic Kidney Disease by Activating Autophagy to Alleviate Podocyte Dedifferentiation via the Notch Pathway. Int. J. Mol. Med. 2024, 53, 26. [Google Scholar] [CrossRef]
- Xiao, T.; Guan, X.; Nie, L.; Wang, S.; Sun, L.; He, T.; Huang, Y.; Zhang, J.; Yang, K.; Wang, J.; et al. Rapamycin Promotes Podocyte Autophagy and Ameliorates Renal Injury in Diabetic Mice. Mol. Cell Biochem. 2014, 394, 145–154. [Google Scholar] [CrossRef]
- Dong, D.; Fan, T.-T.; Ji, Y.-S.; Yu, J.-Y.; Wu, S.; Zhang, L. Spironolactone Alleviates Diabetic Nephropathy through Promoting Autophagy in Podocytes. Int. Urol. Nephrol. 2019, 51, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Toyonaga, J.; Tsuruya, K.; Ikeda, H.; Noguchi, H.; Yotsueda, H.; Fujisaki, K.; Hirakawa, M.; Taniguchi, M.; Masutani, K.; Iida, M. Spironolactone Inhibits Hyperglycemia-Induced Podocyte Injury by Attenuating ROS Production. Nephrol. Dial. Transplant. 2011, 26, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Q.; Jiang, L.; Li, Y.-Y.; Huang, Y.-B.; Hu, X.-R.; Zhu, W.; Wang, X.; Wu, Y.-G.; Meng, X.-M.; Qi, X.-M. Wogonin Protects Glomerular Podocytes by Targeting Bcl-2-Mediated Autophagy and Apoptosis in Diabetic Kidney Disease. Acta Pharmacol. Sin. 2022, 43, 96–110. [Google Scholar] [CrossRef]
- Barutta, F.; Bellini, S.; Kimura, S.; Hase, K.; Corbetta, B.; Corbelli, A.; Fiordaliso, F.; Bruno, S.; Biancone, L.; Barreca, A.; et al. Protective Effect of the Tunneling Nanotube-TNFAIP2/M-Sec System on Podocyte Autophagy in Diabetic Nephropathy. Autophagy 2023, 19, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Liu, X.; Tan, Z.; Tian, X.; Li, M.; Wei, J. Ferroptosis: A New Strategy for Chinese Herbal Medicine Treatment of Diabetic Nephropathy. Front. Endocrinol. 2023, 14, 1188003. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wang, R.; Wang, X.; Liu, J.; Han, Z.; Li, Q.; Jin, Y.; Liao, H. Research Progress of Natural Active Compounds on Improving Podocyte Function to Reduce Proteinuria in Diabetic Kidney Disease. Ren. Fail. 2023, 45, 2290930. [Google Scholar] [CrossRef]
- Chen, J.; Ou, Z.; Gao, T.; Yang, Y.; Shu, A.; Xu, H.; Chen, Y.; Lv, Z. Ginkgolide B Alleviates Oxidative Stress and Ferroptosis by Inhibiting GPX4 Ubiquitination to Improve Diabetic Nephropathy. Biomed. Pharmacother. 2022, 156, 113953. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Hu, W.; Han, X.; Cai, Y. Rhein Inhibited Ferroptosis and EMT to Attenuate Diabetic Nephropathy by Regulating the Rac1/NOX1/β-Catenin Axis. Front. Biosci. 2023, 28, 100. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-Y.; Wang, Z.-X.; Li, T.-S.; Ding, X.-Q.; Liu, Z.-H.; Yang, J.; Fang, L.; Kong, L.-D. SSBP1 Drives High Fructose-Induced Glomerular Podocyte Ferroptosis via Activating DNA-PK/P53 Pathway. Redox Biol. 2022, 52, 102303. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Qu, C.; Xiao, X.; Zhang, W.; Jiang, Y.; Wu, Z.; Song, D.; Peng, X.; Ma, X.; Zhao, Y. Flavonoids on Diabetic Nephropathy: Advances and Therapeutic Opportunities. Chin. Med. 2021, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Shi, C.; Bian, Y.; Yang, Z.; Mu, L.; Wu, H.; Duan, H.; Shi, Y. Sestrin2 Remedies Podocyte Injury via Orchestrating TSP-1/TGF-Β1/Smad3 Axis in Diabetic Kidney Disease. Cell Death Dis. 2022, 13, 663. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Han, S.; Zhu, Y.; Hu, F.; Wei, Y.; Wang, G. Kidney-Targeted Drug Delivery via Rhein-Loaded Polyethyleneglycol-Co-Polycaprolactone-Co-Polyethylenimine Nanoparticles for Diabetic Nephropathy Therapy. Int. J. Nanomed. 2018, 13, 3507–3527. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.-Q.; Tang, L.; Gao, Y.-B.; Wang, Y.-F.; Meng, Y.; Shen, C.; Shen, Z.-L.; Liu, Z.-Q.; Zhao, W.-J.; Liu, W.J. Effect of Baoshenfang Formula on Podocyte Injury via Inhibiting the NOX-4/ROS/P38 Pathway in Diabetic Nephropathy. J. Diabetes Res. 2019, 2019, 2981705. [Google Scholar] [CrossRef] [PubMed]
- Borges, C.M.; Papadimitriou, A.; Duarte, D.A.; Lopes de Faria, J.M.; Lopes de Faria, J.B. The Use of Green Tea Polyphenols for Treating Residual Albuminuria in Diabetic Nephropathy: A Double-Blind Randomised Clinical Trial. Sci. Rep. 2016, 6, 28282. [Google Scholar] [CrossRef]
- Packham, D.K.; Fraser, I.R.; Kerr, P.G.; Segal, K.R. Allogeneic Mesenchymal Precursor Cells (MPC) in Diabetic Nephropathy: A Randomized, Placebo-Controlled, Dose Escalation Study. EBioMedicine 2016, 12, 263–269. [Google Scholar] [CrossRef]
- He, J.; Liu, B.; Du, X.; Wei, Y.; Kong, D.; Feng, B.; Guo, R.; Asiamah, E.A.; Griffin, M.D.; Hynes, S.O.; et al. Amelioration of Diabetic Nephropathy in Mice by a Single Intravenous Injection of Human Mesenchymal Stromal Cells at Early and Later Disease Stages Is Associated with Restoration of Autophagy. Stem Cell Res. Ther. 2024, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, J.; Yue, G.; Xu, J. Placenta-Derived Mesenchymal Stem Cells Protect against Diabetic Kidney Disease by Upregulating Autophagy-Mediated SIRT1/FOXO1 Pathway. Ren. Fail. 2024, 46, 2303396. [Google Scholar] [CrossRef] [PubMed]
- Glastras, S.J.; Pollock, C.A. Targeted Identification of Risk and Treatment of Diabetic Kidney Disease. Nat. Rev. Nephrol. 2024, 20, 75–76. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, S.; Wang, N.; Zhang, C. Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 9035. https://doi.org/10.3390/ijms25169035
Zhong S, Wang N, Zhang C. Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets. International Journal of Molecular Sciences. 2024; 25(16):9035. https://doi.org/10.3390/ijms25169035
Chicago/Turabian StyleZhong, Suye, Na Wang, and Chun Zhang. 2024. "Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets" International Journal of Molecular Sciences 25, no. 16: 9035. https://doi.org/10.3390/ijms25169035
APA StyleZhong, S., Wang, N., & Zhang, C. (2024). Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets. International Journal of Molecular Sciences, 25(16), 9035. https://doi.org/10.3390/ijms25169035