
Lignin Structural Characterization and Its Antioxidant Potential: A Comparative Evaluation by EPR, UV-Vis Spectroscopy, and DPPH Assays
 , , , , ,
, , , , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
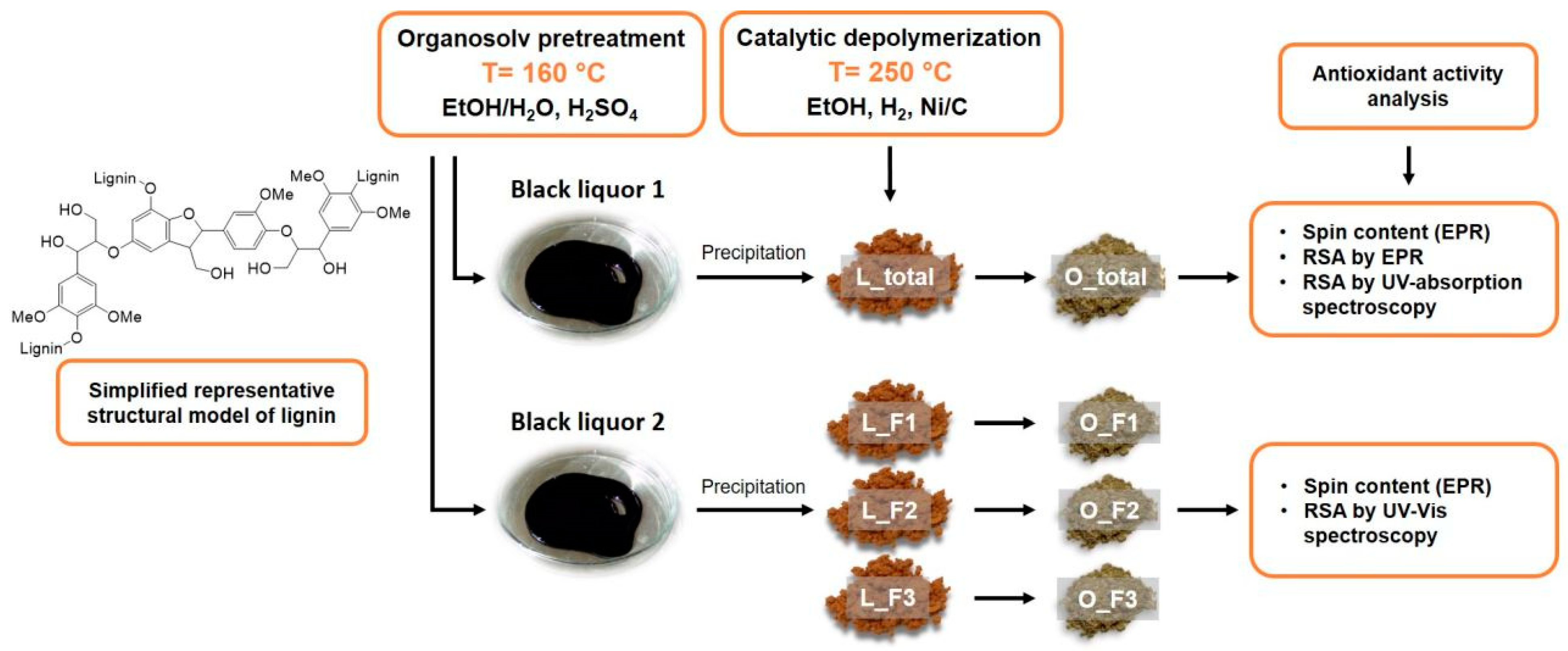
2.1. Variations in Lignin Molecular Weight and Functionality
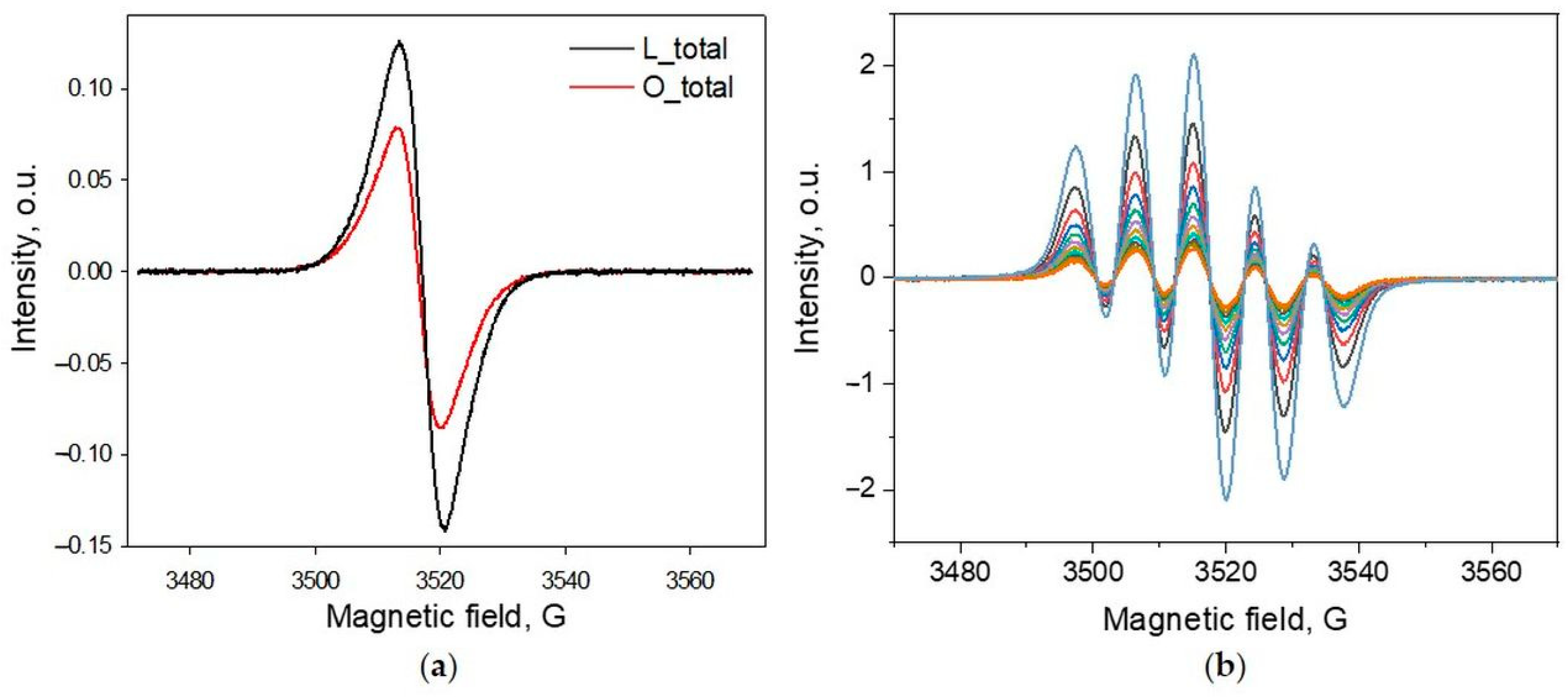
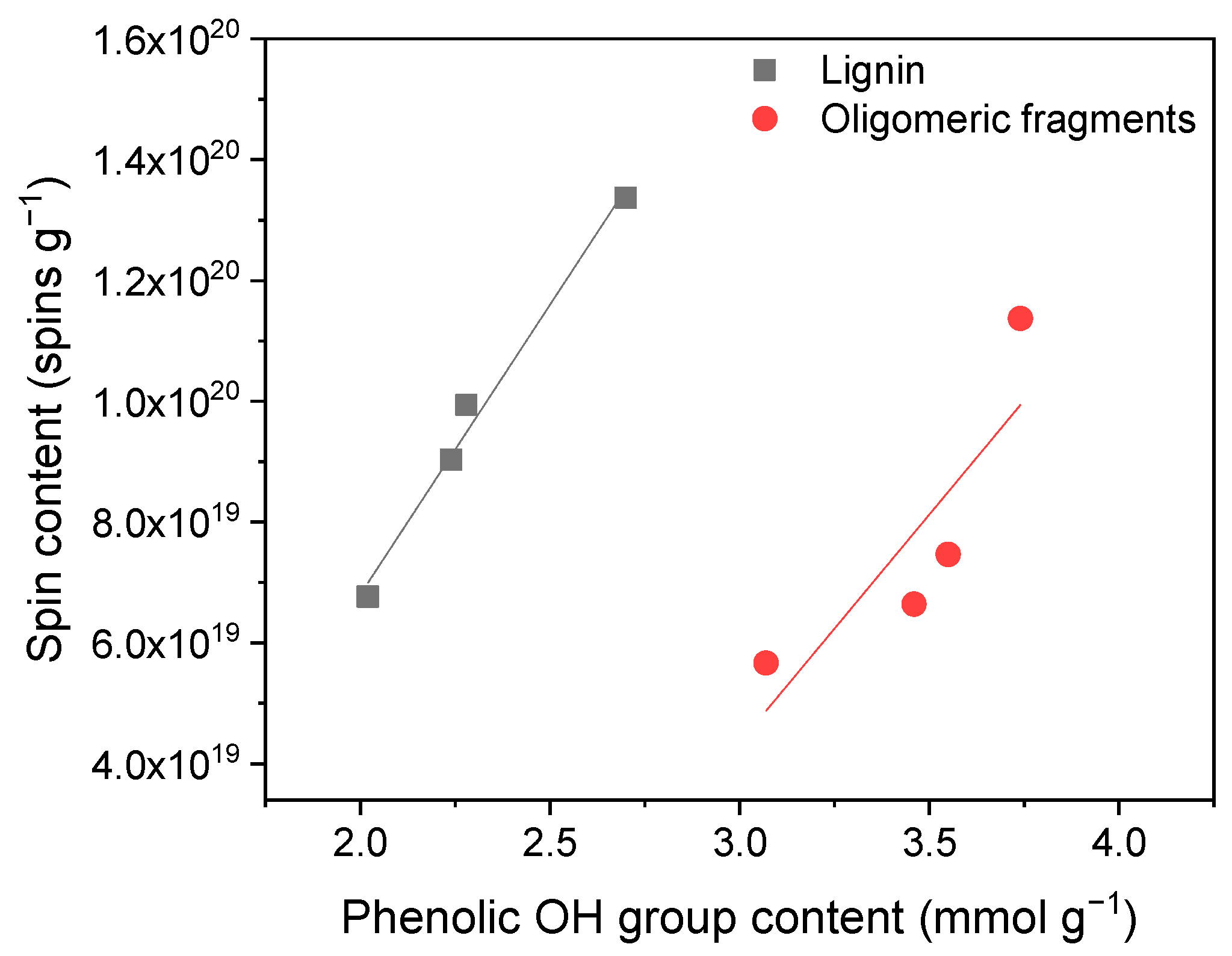
2.2. Measurement of Spin Content in Lignins by EPR
2.3. Radical Scavenging Activity by EPR
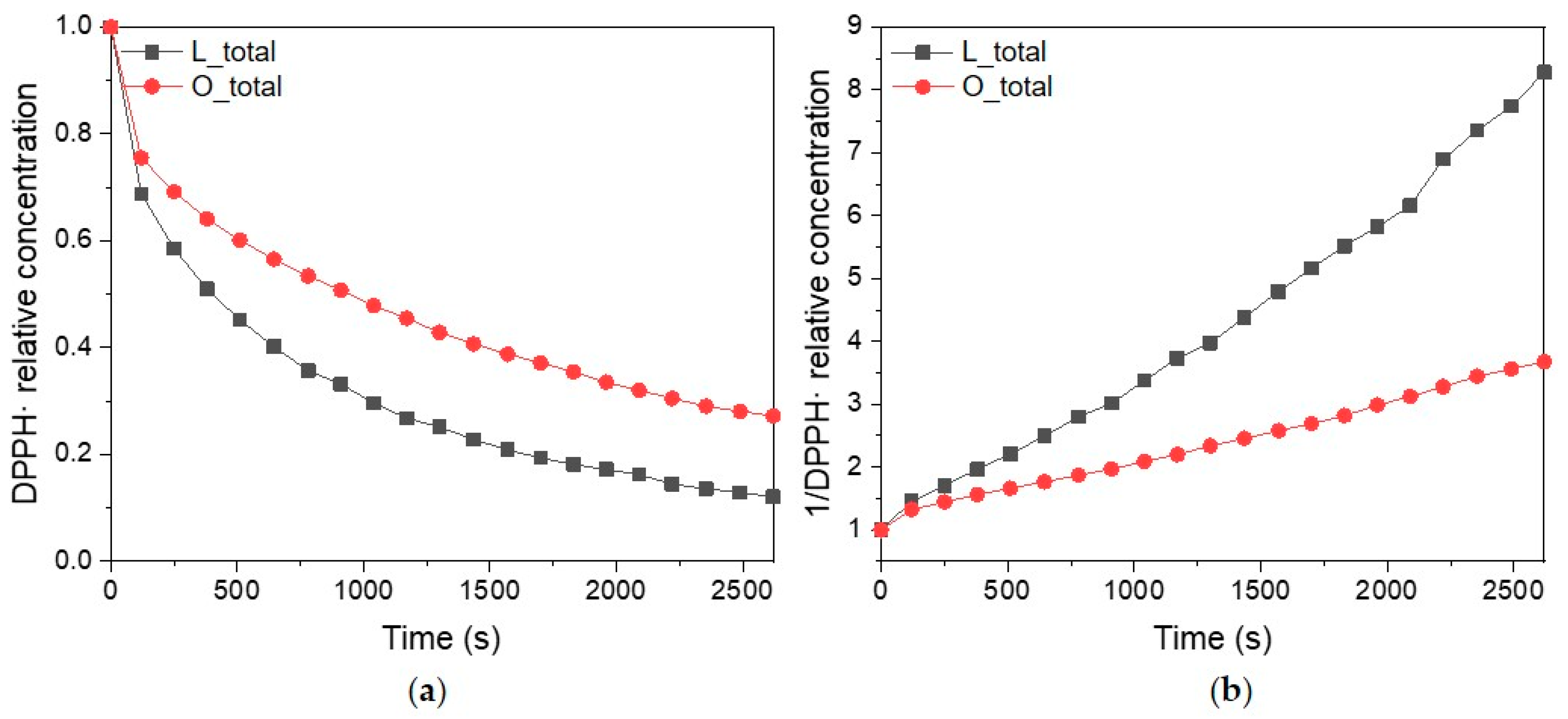
2.4. Radical Scavenging Activity by UV-Vis Spectroscopy
2.5. EPR vs. UV-Vis Spectroscopy
3. Materials and Methods
3.1. Lignin and Oligomeric Samples Preparation and Characterization
3.2. EPR Spin Counting and Radical Scavenging Activity
3.3. Radical Scavenging Activity by UV-Vis Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ullah, I.; Chen, Z.; Xie, Y.; Khan, S.S.; Singh, S.; Yu, C.; Cheng, G. Recent advances in biological activities of lignin and emerging biomedical applications: A short review. Int. J. Biol. Macromol. 2022, 208, 819–832. [Google Scholar] [CrossRef]
- Michalak, M. Plant-Derived Antioxidants: Significance in Skin Health and the Ageing Process. Int. J. Mol. Sci. 2022, 23, 8–12. [Google Scholar] [CrossRef]
- Kabir, A.S.; Yuan, Z.-S.; Kuboki, T.; Xu, C. Development of Lignin-Based Antioxidants for Polymers. In Production of Materials from Sustainable Biomass Resources. Biofuels and Biorefineries; Fang, Z., Smith, R., Jr., Tian, X.F., Eds.; Springer: Singapore, 2019; Volume 9, pp. 39–59. [Google Scholar] [CrossRef]
- Qian, Y.; Qiu, X.; Zhu, S. Sunscreen performance of lignin from different technical resources and their general synergistic effect with synthetic sunscreens. ACS Sustain. Chem. Eng. 2016, 4, 4029–4035. [Google Scholar] [CrossRef]
- Gordobil, O.; Olaizola, P.; Banales, J.M.; Labidi, J. Lignins from agroindustrial by-products as natural ingredients for cosmetics: Chemical structure and in vitro sunscreen and cytotoxic activities. Molecules 2020, 25, 1131. [Google Scholar] [CrossRef]
- Rácz, A.; Papp, N.; Balogh, E.; Fodor, M.; Héberger, K. Comparison of antioxidant capacity assays with chemometric methods. Anal. Methods 2015, 7, 4216–4224. [Google Scholar] [CrossRef]
- Rumpf, J.; Burger, R.; Schulze, M. Statistical evaluation of DPPH, ABTS, FRAP, and Folin-Ciocalteu assays to assess the antioxidant capacity of lignins. Int. J. Biol. Macromol. 2023, 233, 123470. [Google Scholar] [CrossRef]
- Takatsuka, M.; Goto, S.; Kobayashi, K.; Otsuka, Y.; Shimada, Y. Evaluation of pure antioxidative capacity of antioxidants: ESR spectroscopy of stable radicals by DPPH and ABTS assays with singular value decomposition. Food Biosci. 2022, 48, 101714. [Google Scholar] [CrossRef]
- Patil, S.V.; Argyropoulos, D.S. Stable Organic Radicals in Lignin: A Review. ChemSusChem 2017, 10, 3284–3303. [Google Scholar] [CrossRef]
- Davies, M.J. Detection and characterisation of radicals using electron paramagnetic resonance (EPR) spin trapping and related methods. Methods 2016, 109, 21–30. [Google Scholar] [CrossRef]
- Sanna, D.; Delogu, G.; Mulas, M.; Schirra, M.; Fadda, A. Determination of Free Radical Scavenging Activity of Plant Extracts Through DPPH Assay: An EPR and UV-Vis Study. Food Anal. Methods 2012, 5, 759–766. [Google Scholar] [CrossRef]
- Celiz, G.; Renfige, M.; Finetti, M. Spectral analysis allows using the DPPH* UV–Vis assay to estimate antioxidant activity of colored compounds. Chem. Pap. 2020, 74, 3101–3109. [Google Scholar] [CrossRef]
- Zheng, L.; Lin, L.; Su, G.; Zhao, Q.; Zhao, M. Pitfalls of using 1,1-diphenyl-2-picrylhydrazyl (DPPH) assay to assess the radical scavenging activity of peptides: Its susceptibility to interference and low reactivity towards peptides. Food Res. Int. 2015, 76, 359–365. [Google Scholar] [CrossRef]
- de Menezes, B.B.; Frescura, L.M.; Duarte, R.; Villetti, M.A.; da Rosa, M.B. A critical examination of the DPPH method: Mistakes and inconsistencies in stoichiometry and IC50 determination by UV–Vis spectroscopy. Anal. Chim. Acta. 2021, 1157, 338398. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.; Shahidi, F. Revisiting DPPH (2,2-diphenyl-1-picrylhydrazyl) assay as a useful tool in antioxidant evaluation: A new IC100 concept to address its limitations. J. Food Bioact. 2019, 7, 36–42. [Google Scholar] [CrossRef]
- Yeo, J.D.; Shahidi, F. Critical Re-Evaluation of DPPH assay: Presence of pigments affects the results. J. Agric. Food Chem. 2019, 67, 7526–7529. [Google Scholar] [CrossRef]
- Lange, H.; Schiffels, P.; Sette, M.; Sevastyanova, O.; Crestini, C. Fractional Precipitation of Wheat Straw Organosolv Lignin: Macroscopic Properties and Structural Insights. ACS Sustain. Chem. Eng. 2016, 4, 5136–5151. [Google Scholar] [CrossRef]
- Smit, A.T.; Dezaire, T.; Riddell, L.A.; Bruijnincx, P.C.A. Reductive Partial Depolymerization of Acetone Organosolv Lignin to Tailor Lignin Molar Mass, Dispersity, and Reactivity for Polymer Applications. ACS Sustain. Chem. Eng. 2023, 11, 6070–6080. [Google Scholar] [CrossRef]
- Miller, T.A.; Adams, R.N. Spin-Spin and Electron-Exchange Rates of Radicals and Radical Ions. J. Am. Chem. Soc. 1966, 88, 5713–5714. [Google Scholar] [CrossRef]
- Steelink, C. Free radical studies of lignin, lignin degradation products and soil humic acids. Geochim. Cosmochim. Acta 1964, 28, 1615–1622. [Google Scholar] [CrossRef]
- Nanassy, A.J.; Vyas, A. Electron Spin Resonance in Pyrolyzed Lignin. J. Macromol. Sci. Part B 1969, 3, 271–292. [Google Scholar] [CrossRef]
- Kocheva, L.S.; Karmanov, A.P.; Mironov, M.V.; Belyy, V.A.; Polina, I.N.; Pokryshkin, S.A. Characteristics of chemical structure of lignin biopolymer from Araucaria relict plant. Questions and answers of evolution. Int. J. Biol. Macromol. 2020, 159, 896–903. [Google Scholar] [CrossRef]
- Bährle, C.; Nick, T.U.; Bennati, M.; Jeschke, G.; Vogel, F. High-Field Electron Paramagnetic Resonance and Density Functional Theory Study of Stable Organic Radicals in Lignin: Influence of the Extraction Process, Botanical Origin, and Protonation Reactions on the Radical g Tensor. J. Phys. Chem. A 2015, 119, 6475–6482. [Google Scholar] [CrossRef]
- Chambon, C.L.; Fitriyanti, V.; Verdía, P.; Yang, S.M.; Hérou, S.; Titirici, M.M.; Brandt-Talbot, A.; Fennell, P.S.; Hallett, J.P. Fractionation by Sequential Antisolvent Precipitation of Grass, Softwood, and Hardwood Lignins Isolated Using Low-Cost Ionic Liquids and Water. ACS Sustain. Chem. Eng. 2020, 8, 3751–3761. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Sadeghifar, H.; Ragauskas, A.; Ragauskas, A.; Ragauskas, A.; Ragauskas, A. Perspective on Technical Lignin Fractionation. ACS Sustain. Chem. Eng. 2020, 8, 8086–8101. [Google Scholar] [CrossRef]
- Matos, M.; Claro, F.C.; Lima, T.A.M.; Avelino, F.; Hansel, F.A.; Maciel, G.M.; Lomonaco, D.; Magalhães, W.L.E. Acetone: Water fractionation of pyrolytic lignin improves its antioxidant and antibacterial activity. J. Anal. Appl. Pyrolysis 2021, 156, 105175. [Google Scholar] [CrossRef]
- An, L.; Wang, G.; Jia, H.; Liu, C.; Sui, W.; Si, C. Fractionation of enzymatic hydrolysis lignin by sequential extraction for enhancing antioxidant performance. Int. J. Biol. Macromol. 2017, 99, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP decolorization assay of antioxidant capacity reaction pathways. Int. J. Mol. Sci. 2020, 21, 1131. [Google Scholar] [CrossRef] [PubMed]
- Gulcin, İ.; Alwasel, S.H. DPPH Radical Scavenging Assay. Processes 2023, 11, 2248. [Google Scholar] [CrossRef]
- Pyrzynska, K.; Pȩkal, A. Application of free radical diphenylpicrylhydrazyl (DPPH) to estimate the antioxidant capacity of food samples. Anal. Methods 2013, 5, 4288–4295. [Google Scholar] [CrossRef]
- García, A.; Alriols, M.G.; Spigno, G.; Labidi, J. Lignin as natural radical scavenger. Effect of the obtaining and purification processes on the antioxidant behaviour of lignin. Biochem. Eng. J. 2012, 67, 173–185. [Google Scholar] [CrossRef]
- Gil-Chávez, J.; Gurikov, P.; Hu, X.; Meyer, R.; Reynolds, W.; Smirnova, I. Application of novel and technical lignins in food and pharmaceutical industries: Structure-function relationship and current challenges. Biomass Convers. Biorefinery 2021, 11, 2387–2403. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Dizhbite, T.; Lauberts, M.; Volperts, A.; Dobele, G.; Telysheva, G. Analytical pyrolysis—A tool for revealing of lignin structure-antioxidant activity relationship. J. Anal. Appl. Pyrolysis 2015, 113, 360–369. [Google Scholar] [CrossRef]
- Sun, S.N.; Cao, X.F.; Xu, F.; Sun, R.C.; Jones, G.L. Structural features and antioxidant activities of lignins from steam-exploded bamboo (Phyllostachys pubescens). J. Agric. Food Chem. 2014, 62, 5939–5947. [Google Scholar] [CrossRef]
- Gong, W.; Xiang, Z.; Ye, F.; Zhao, G. Composition and structure of an antioxidant acetic acid lignin isolated from shoot shell of bamboo (Dendrocalamus latiforus). Ind. Crop. Prod. 2016, 91, 340–349. [Google Scholar] [CrossRef]
- Verrillo, M.; Savy, D.; Cangemi, S.; Savarese, C.; Cozzolino, V.; Piccolo, A. Valorization of lignins from energy crops and agro-industrial byproducts as antioxidant and antibacterial materials. J. Sci. Food Agric. 2022, 102, 2885–2892. [Google Scholar] [CrossRef]
- Polak, J.; Bartoszek, M.; Chora, M. Antioxidant Capacity: Experimental Determination by EPR Spectroscopy and Mathematical Modeling. J. Agric. Food Chem. 2015, 63, 6319–6324. [Google Scholar] [CrossRef]
- Jasiukaitytė-Grojzdek, E.; Huš, M.; Grilc, M.; Likozar, B. Acid-Catalyzed α-O-4 Aryl-Ether Cleavage Mechanisms in (Aqueous) γ-Valerolactone: Catalytic Depolymerization Reactions of Lignin Model Compound during Organosolv Pretreatment. ACS Sustain. Chem. Eng. 2020, 8, 17475–17486. [Google Scholar] [CrossRef] [PubMed]
- Kozmelj, T.R.; Bartolomei, E.; Dufour, A.; Leclerc, S.; Arnoux, P.; Likozar, B.; Jasiukaitytė-Grojzdek, E.; Grilc, M.; Le Brech, Y. Oligomeric fragments distribution, structure and functionalities upon ruthenium-catalyzed technical lignin depolymerization. Biomass Bioenergy 2024, 181, 107056. [Google Scholar] [CrossRef]
- Meng, X.; Crestini, C.; Ben, H.; Hao, N.; Pu, Y.; Ragauskas, A.J.; Argyropoulos, D.S. Determination of hydroxyl groups in biorefinery resources via quantitative 31P NMR spectroscopy. Nat. Protoc. 2019, 14, 2627–2647. [Google Scholar] [CrossRef]
- Burghaus, O.; Rohrer, M.; Gotzinger, T.; Plato, M.; Mobius, K. A novel high-field/high-frequency EPR and ENDOR spectrometer operating at 3 mm wavelength. Meas. Sci. Technol. 1992, 3, 765–774. [Google Scholar] [CrossRef]
- Dizhbite, T.; Telysheva, G.; Jurkjane, V.; Viesturs, U. Characterization of the radical scavenging activity of lignins—Natural antioxidants. Bioresour. Technol. 2004, 95, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Alzagameem, A.; El Khaldi-Hansen, B.; Büchner, D.; Larkins, M.; Kamm, B.; Witzleben, S.; Schulze, M. Lignocellulosic biomass as source for lignin-based environmentally benign antioxidants. Molecules 2018, 23, 2664. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin Sample | SEC Results (g mol−1) | 31P-NMR Results (mmol g−1) | ||||
|---|---|---|---|---|---|---|
| Mw | Mn | Đ 1 | Aliphatic OH | Phenolic OH | Total OH | |
| L_F1 | 6950 | 2400 | 2.9 | 1.82 | 1.99 | 3.81 |
| L_F2 | 3450 | 1800 | 1.9 | 1.81 | 2.24 | 4.07 |
| L_F3 | 1850 | 1150 | 1.6 | 1.84 | 2.70 | 4.57 |
| L_total | 3400 | 1450 | 2.3 | 1.94 | 2.28 | 4.25 |
| O_F1 | 2150 | 1200 | 1.8 | 0.51 | 3.07 | 3.58 |
| O_F2 | 2100 | 1150 | 1.8 | 0.62 | 3.55 | 4.28 |
| O_F3 | 1600 | 1000 | 1.6 | 0.63 | 3.74 | 4.39 |
| O_total | 2000 | 1150 | 1.7 | 0.67 | 3.36 | 4.14 |
| Lignin Sample | EPR Results | DPPH Assay 2 | |||
|---|---|---|---|---|---|
| Spin Content 1 (spins g−1) | g-Factor | ΔHpp (G) | RSA (%) | TEAC (μmol mg−1) | |
| L_F1 | 6.8 × 1019 | 2.0040 | 6.45 | 29.6 | 0.47 |
| L_F2 | 9.0 × 1019 | 2.0042 | 6.94 | 34.1 | 0.55 |
| L_F3 | 13.4 × 1019 | 2.0043 | 7.56 | 41.0 | 0.67 |
| L_total | 9.9 × 1019 | 2.0042 | 7.10 | 36.6 | 0.60 |
| O_F1 | 5.7 × 1019 | 2.0040 | 6.51 | 62.9 | 1.09 |
| O_F2 | 7.5 × 1019 | 2.0041 | 6.88 | 64.6 | 1.12 |
| O_F3 | 11.4 × 1019 | 2.0042 | 6.99 | 66.7 | 1.17 |
| O_total | 6.6 × 1019 | 2.0041 | 6.93 | 64.6 | 1.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozmelj, T.R.; Voinov, M.A.; Grilc, M.; Smirnov, A.I.; Jasiukaitytė-Grojzdek, E.; Lucia, L.; Likozar, B. Lignin Structural Characterization and Its Antioxidant Potential: A Comparative Evaluation by EPR, UV-Vis Spectroscopy, and DPPH Assays. Int. J. Mol. Sci. 2024, 25, 9044. https://doi.org/10.3390/ijms25169044
Kozmelj TR, Voinov MA, Grilc M, Smirnov AI, Jasiukaitytė-Grojzdek E, Lucia L, Likozar B. Lignin Structural Characterization and Its Antioxidant Potential: A Comparative Evaluation by EPR, UV-Vis Spectroscopy, and DPPH Assays. International Journal of Molecular Sciences. 2024; 25(16):9044. https://doi.org/10.3390/ijms25169044
Chicago/Turabian StyleKozmelj, Tina Ročnik, Maxim A. Voinov, Miha Grilc, Alex I. Smirnov, Edita Jasiukaitytė-Grojzdek, Lucian Lucia, and Blaž Likozar. 2024. "Lignin Structural Characterization and Its Antioxidant Potential: A Comparative Evaluation by EPR, UV-Vis Spectroscopy, and DPPH Assays" International Journal of Molecular Sciences 25, no. 16: 9044. https://doi.org/10.3390/ijms25169044