Abstract
Nitric oxide (NO) is a highly versatile gasotransmitter that has first been shown to regulate cardiovascular function and then to exert tight control over a much broader range of processes, including neurotransmitter release, neuronal excitability, and synaptic plasticity. Endothelial NO synthase (eNOS) is usually far from the mind of synaptic neurophysiologists, who have focused most of their attention on neuronal NO synthase (nNOS) as the primary source of NO at the neurovascular unit (NVU). Nevertheless, the available evidence suggests that eNOS could also contribute to generating the burst of NO that, serving as volume intercellular messenger, is produced in response to neuronal activity in the brain parenchyma. Herein, we review the role of eNOS in both the regulation of cerebral blood flow and of synaptic plasticity and discuss the mechanisms by which cerebrovascular endothelial cells may transduce synaptic inputs into a NO signal. We further suggest that eNOS could play a critical role in vascular-to-neuronal communication by integrating signals converging onto cerebrovascular endothelial cells from both the streaming blood and active neurons.
1. Introduction
The assignment of the 1998 Nobel Prize in Physiology or Medicine to Robert F. Furchgott [1], Louis J. Ignarro [2], and Ferid Murad [3] “for their discoveries concerning nitric oxide (NO) as a signaling molecule in the cardiovascular system” sparked an avalanche of studies aiming at investigating the pleiotropic role of this versatile gasotransmitter [4]. It was rapidly established that NO is also critical in the central nervous system (CNS) by regulating multiple brain functions, including cerebral blood flow (CBF), blood-brain barrier (BBB) permeability, synaptic plasticity, and memory formation [5,6,7,8]. Endogenous NO is generated by the catalytic activity of three isoforms of the NO synthase (NOS) [6,7,8,9]: neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS), which are, respectively, encoded by the NOS1, NOS2, and NOS3 genes. Early studies suggested that nNOS was the main source of NO at the neurovascular unit (NVU), which represents the minimal functional unit in the brain and is composed of endothelial cells, mural cells, astrocyte end-feet, and neurons [10]. However, recent investigations based upon previous work in blood vessels unveiled the critical contribution of eNOS to NO signaling at the NVU [11]: eNOS can be recruited following synaptic activity to cause a local increase in CBF [12,13], thereby regulating neurovascular coupling (NVC), i.e., the mechanism by which an increase in neuronal activity is translated into an increase in local CBF [10]. Furthermore, eNOS-derived NO may modulate synaptic activity and long-term potentiation (LTP) by establishing an unexpected line of communication between microvascular endothelial cells and neurons [14,15,16,17], which falls within the broader concept of vascular-to-neuronal communication [18,19,20]. Therefore, endothelium-derived NO does not only serve as a volume intracellular messenger that supports ongoing neuronal function through NVC, but it can also participate in learning and memory formation [16,17,21].
Herein, we briefly summarize the anatomical organization of the brain microcirculation and the cellular organization of the NVU. Then, we survey the cellular localization of eNOS at the NVU, the molecular mechanisms of eNOS activation, and its downstream signaling pathways. Finally, we describe the mounting evidence that eNOS can be recruited by neuronal activity to regulate CBF and LTP. The unexpected implication of these novel pieces of information is that cerebrovascular endothelial cells, which are the main site of eNOS expression at the NVU [16,22,23,24], may fine-tune neuronal activity by both triggering NVC and directly regulating synaptic plasticity.
2. Cerebral Circulation and the NVU
An anastomotic ring at the base of the brain, known as the Willis circle, gives rise to the three main pairs of supplying arteries of the skull, known as the anterior, middle, and posterior cerebral arteries. The circle of Willis branches out into the main intracerebral arteries that ramify into progressively smaller arteries and arterioles that supply a large territory of the cerebral cortex en route to the cortical surface. Herein, a heavily interconnected network of pial arteries and arterioles runs on the surface of the brain within the subarachnoid space, thereby originating the penetrating arterioles, which dive into the brain parenchyma. The penetrating arterioles are lined by a single endothelial cell layer, which is covered by a thin basement membrane, enwrapped by 1–3 layers of vascular smooth muscle cells (VSMCs), and ensheathed by the pia mater. The outer limit of the perivascular space (or Virchow–Robin space) that surrounds the penetrating arterioles branching off the subarachnoid space and is filled with the cerebrospinal fluid (CSF) is formed by the astrocytic end-feet of the glia limitans. The vascular basement membrane and the glia limitans fuse together, thus obliterating the perivascular space, as the penetrating arterioles penetrate downward through the cerebral parenchyma and become intraparenchymal arterioles. At this level, cerebral microvessels present one single or discontinuous layer of VSMCs and are encased by the astrocytic end-feet (Figure 1) [5,10]. Intraparenchymal arterioles ramify into a heavily intercommunicating network of capillaries, which come in close contact with the neurons (<15 μm) and are therefore in the most suitable position to detect neuronal activity [25,26]. The capillary wall of the BBB consists of an endothelial monolayer that lacks VMSCs but is partially surrounded by contractile pericytes, which are also embedded into the vascular membrane basement and cover ≈30% of the capillary surface [27]. Astrocytic end-feet may cover the remaining 70% of the brain capillary surface and regulate the induction and maintenance of BBB properties (Figure 1) [10,27].
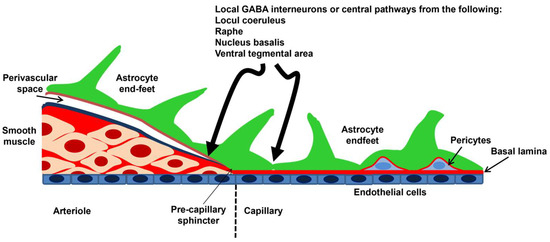
Figure 1.
Schematic representation of the NVU. The penetrating arterioles depart from the pial arteries and are surrounded by the perivascular space, also known as Virchow–Robin space, which may house perivascular macrophages and other cell types, including mast cells and Mato cells [5,10]. Their wall includes 1–3 layers of VSMCs that determine their resistance to blood flow: an increase in the contracting state reduces blood perfusion, while a decrease in the contracting state increases blood perfusion to downstream capillaries. The outer limit of the perivascular space is lined by the astrocyte end-feet, which give rise to the glia limitans membrane. When the basal lamina fuses with the glia limitans, the perivascular space is obliterated, and the penetrating arterioles become intraparenchymal arterioles. In the capillary vessels, VSMCs are replaced by pericytes, which are contractile cells closely embedded in the vascular wall, and the outer wall is still contacted by the astrocytic end-feet. Intraparenchymal arterioles and capillaries receive intrinsic innervation from local interneurons and subcortical pathways. Modified from [26] (https://creativecommons.org/licenses/by/4.0/ (accessed on 18 June 2024)).
Neurons are obviously key cellular constituents of the NVU. The “extrinsic innervation” of the pial surface vasculature is brought about by sympathetic nerves from the superior cervical ganglion, parasympathetic nerves from the sphenopalatine and otic ganglia, and the trigeminal ganglion. The “extrinsic innervation” regulates the contractile tone of pial VSMCs by releasing multiple neurotransmitters and neuromodulators, including norepinephrine, acetylcholine, vasoactive intestinal peptide (VIP), neuropeptide Y, calcitonin gene-related peptide (CGRP), and NO itself, and is primarily responsible for shifting the upper limit of the myogenic response, also known as autoregulation, toward higher pressures [28,29]. Penetrating arterioles are barely innervated by extrinsic fibers, while intraparenchymal arterioles receive extensive innervation by local γ-aminobutyric acid (GABA) interneurons, often with the interposition of astrocytes. In addition, intraparenchymal arterioles receive neuronal inputs from sub-cortical neurons that are located in the basal forebrain, raphe nucleus, and basal forebrain (Figure 1) [28,29]. The primary role of this “intrinsic innervation” is to adjust the intrinsic vasomotor activity of intracerebral microvessels to neuronal activity, according to a mechanism known as NVC [28,29]. Therefore, brain capillary endothelial cells and local pyramidal neurons/interneurons can feed back onto one another, with the BBB regulating the supply of oxygen (O2) and nutrients to active neurons and the latter delivering signaling mediators (e.g., neurotransmitters and neuromodulators) to the vascular cells.
3. NOS Isoforms and NO Signaling at the NVU
The discovery of NO as a signaling messenger in the brain has taken advantage of the identification of NO as a vasorelaxing mediator in the cardiovascular system [30,31]. This observation supports the view, more broadly discussed below, that eNOS, i.e., solely responsible for NO production in blood vessels, could play a role that goes beyond the regulation of BBB permeability and NVC [11]. The endogenous synthesis of NO at the NVU is strictly controlled by the constitutive NOS isoforms (cNOSs), i.e., nNOS and eNOS, which are both activated upon an increase in intracellular Ca2+ concentration ([Ca2+]i) and the recruitment of the Ca2+/dependent calmodulin (CaM). Conversely, iNOS, which is constitutively bound to CaM and is therefore Ca2+-independent, is barely detectable in the CNS but can be expressed in astrocytes, microglia, and blood-derived macrophages in response to an immune challenge and will not be further addressed here [7,30,31].
3.1. nNOS and eNOS: Cellular Localization within the NVU and Mechanisms of Synaptic Activation
nNOS can be activated by glutamate release during synaptic activity. The expression pattern of nNOS is restricted to discrete neuronal subtypes, such as inhibitory interneurons and a small population of excitatory neurons in the cerebral cortex, striatum, hippocampus, cerebellum, and olfactory bulb [22,30,32,33,34]. Glutamate triggers Ca2+ influx through N-methyl-D-aspartate (NMDA) receptors (NMDARs) [32,35], thereby leading to a robust increase in dendritic Ca2+ concentration that engages CaM, which in turn binds to nNOS and accelerates NO production by facilitating interdomain electron transfer (Figure 2) [7,30,31]. The nNOS protein, via the scaffolding protein postsynaptic density-95 (PSD-95), is physically coupled with the GluN2B subunit of NMDARs; this ternary complex, nNOS–PSD95-NMDAR, ensures the efficiency and fidelity of the signal transduction pathway responsible for NO release in response to synaptic activation (Figure 2) [7,30,31].
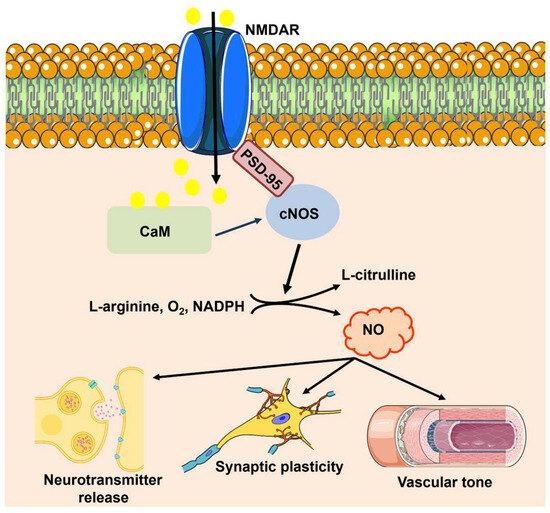
Figure 2.
NO signaling at the NVU. Synaptic activity leads to extracellular Ca2+ influx through NMDARs, thereby stimulating CaM activity. Then, CaM promotes the physical association of PSD-95 to the PDZ motif in the NMDAR protein, and PSD-95 and CaM assemble into a trimeric complex with nNOS. Ca2+ entry through NMDARs can also activate eNOS, but it is still unclear whether this mechanism requires the involvement of PSD-95. nNOS and cNOS catalyze the 5-electron oxidation of L-arginine into L-citrulline and NO by using the cofactors NADPH and BH4. Once synthetized, NO regulates neurotransmitter release, promotes LTP, and increases local CBF by promoting VSMC and pericyte relaxation.
Although early studies claimed that it could also be expressed in neurons [36,37,38], there is now no doubt that eNOS is primarily located within the endothelial component of the NVU [16,22,23,24]. Moreover, eNOS is also expressed in pericytes [39], although its functional role is yet to be unraveled [40]. An increase in [Ca2+]i is the main signal responsible for eNOS activation in endothelial cells throughout the vascular tree, including the brain microcirculation [11,41,42]. Interestingly, cerebrovascular endothelial cells also express NMDARs that can be stimulated by synaptically released glutamate to promote NO release (Figure 2) [11,43,44]. A recent study confirmed that Ca2+ entry through NMDARs may cause a spatially restricted sub-membrane Ca2+ domain that also elicits NO release in the human cerebrovascular endothelial cell line, hCMEC/D3 [45]. It should be noted that, in the other vascular districts, Ca2+ entry through store-operated Ca2+ channels (SOCs) is the primary stimulus responsible for eNOS activation in response to chemical stimulation [41,42,46]. However, an in vivo study revealed that synaptic activity stimulates Gq-coupled protein receptors (GqPCRs) that lead to endothelial Ca2+ oscillations and NO release by activating endoplasmic reticulum (ER)-dependent Ca2+ release through inositol-1,4,5-trisphosphate (InsP3) receptors (InsP3Rs) and extracellular Ca2+ entry through Transient Receptor Potential (TRP) Vanilloid 4 (TRPV4) [47]. It has been suggested that synaptically elicited endothelial Ca2+ oscillations could be driven by mGluR1 and mGluR5, which are GqPCRs widely expressed in cerebrovascular endothelial cells [48,49]. Subsequent investigations showed that other neurotransmitters and neuromodulators, including acetylcholine, GABA, and histamine, were also able to induce oscillatory or biphasic endothelial Ca2+ signals that led to NO release in vitro [48,50,51,52,53,54]. Finally, an additional source of Ca2+ for eNOS activation at the NVU could be provided by the mechanosensitive Piezo1 channels [55], which are activated by laminar shear stress and are coupled to eNOS in multiple vascular districts [56]. Therefore, it is likely that any increase in [Ca2+]i can recruit eNOS in cerebrovascular endothelial cells.
Throughout the peripheral circulation, eNOS is spatially segregated in caveolae [41,42], where it is tonically inhibited by the physical association with caveolin-1 (CaV-1) and is bound to the plasma membrane due to NH2-terminal fatty acylation by palmitic and myristic acid [21,57]. The increase in endothelial [Ca2+]i serves to stimulate the Ca2+/CaM to displace CaV-1 from the Ca2+-binding domain and activate eNOS [21]. However, a recent investigation showed that eNOS signaling and caveolae-mediated pathways may not overlap in the mouse brain microcirculation [12]. This finding prompts the search for alternative mechanisms of Ca2+/CaM-dependent eNOS activation in cerebrovascular endothelial cells. Interestingly, eNOS can also be activated upon phosphorylation by different signaling pathways, including phosphoinositide 3 kinase (PI3K)/protein kinase B (PKB/Akt), extracellular regulated protein kinases 1/2 (ERK1/2)/mitogen-activated protein kinase (MAPK), and cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) [21,57]. Furthermore, NO release from endothelial cells could also be elicited by Ca2+/CaM-dependent protein kinase II (CaMKII)-mediated eNOS phosphorylation [58,59]. This evidence suggests that synaptic activity or chemical stimulation could activate eNOS by causing an increase in CaMKII activity, which could overcome the lack of PSD95.
3.2. eNOS Signaling within the NVU
eNOS (as well as nNOS and iNOS) is an oxidoreductase that catalyzes the oxidation of L-arginine to NO and L-citrulline, with nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD) participating in the electron transfer chain and O2 serving as the final electron acceptor (Figure 2). The cofactor tetrahydrobiopterin (BH4) accelerates the rates and improves the efficiency of the electron transfer, thereby ensuring that L-arginine oxidation is coupled with NO synthesis [9,21,57]. Its physicochemical properties hint at NO as a candidate volume transmitter in the brain [30,60], being able to diffuse within a finite volume in all directions away from the site of production and integrate the activity of multiple cell types, i.e., neurons, astrocytes, endothelial cells, and mural cells, irrespective of their synaptic connectivity. Of course, most of the attention has been paid to nNOS- rather than eNOS-dependent NO signaling. For instance, the activation of NMDARs with NMDA induces a transient NO signal that spreads for ≈400 μm within a radius of 100 μm from the stimulation site [61]. These measurements have been carried out in the CA1 region of the hippocampus by means of an electrochemical sensor [61] that may detect the endogenous production of NO by both nNOS and eNOS, which is quite abundant in pyramidal neurons of the CA1 region [22]. Intriguingly, the NO signal triggered by the exogenous application of NMDA was suppressed by NG-nitro-l-arginine (l-NNA), which blocks both isoforms [61]. In 2005, the notion that cerebrovascular endothelial cells also express NMDARs and can respond to synaptic stimulation by producing NO was yet to come [11,43,44,45,62], and the NO signal was entirely ascribed to nNOS activation [61]. Nevertheless, we can now reinterpret those pioneering findings in light of the new information and conclude that the diffusional spread of NO from the NMDA ejection site is likely to be supported by multiple NOS sources [61], including both nNOS and eNOS.
In agreement with this hypothesis, Garthwaite has recalled that the prototypic example of volume transmission is provided by NO signaling in the blood vessels, where NO generated in endothelial cells diffuses to the overlying VSMCs to cause vasorelaxation [30,31,63]. In addition, Garthwaite has already pinpointed the potential role of eNOS in volume transmission, especially at the capillary level, where the density of the microvascular network is such as to boost NO production induced by synaptic activity [30]. Therefore, eNOS could integrate the endothelial Ca2+ signals deriving from the stimulation of both abluminal NMDARs and GqPCRs (which receive synaptic contacts and can be activated by neurotransmitters and neuromodulators) and luminal GqPCRs and mechanosensitive channels (which detect blood-borne chemical and physical signals). The ensuing NO signal generated by the simultaneous activation of a cluster of endothelial cells [42,64,65,66] could then spread from its site of synthesis to the other cellular constituents of the vascular wall and to the brain parenchyma. This would remarkably extend the spatial range of action of endothelial NO, which could not only regulate VSMC/pericyte contraction and endothelial permeability but also neuronal activity, axonal excitability, and synaptic plasticity [11,17,30]. As anticipated above, the capillary district of the bran microcirculation is ideally suitable for volume transmission by NO. Herein, virtually every neuron has its own capillary, and when synaptic stimulation coordinates the activity of a population of neurons and endothelial cells that are closely packed within the same volume of brain tissue, the recruitment of multiple NO sources (i.e., eNOS and nNOS) may result in the rapid buildup of NO in between those cells, thereby also influencing the activity of more distant cells (neurons, astrocytes, and vascular cells).
3.3. The Regulation of CBF by eNOS
It has long been known that NO plays a primary role in NVC, also known as functional hyperemia [10,26,32]. Glutamate is the most abundant excitatory neurotransmitter in the brain and, therefore, is the best-characterized stimulus for NVC [10]. For the reasons highlighted in Chapter 3.2, most studies focused on nNOS activation by neuronal NMDARs followed by NO-mediated vasorelaxation of the intraparenchymal arterioles [5,10,32]. NO diffuses to the overlying VSMCs to activate the stimulated soluble guanylyl cyclase (sGC) and induce the production of cyclic guanosine-3′,5′-monophosphate (cGMP) (Figure 3). cGMP, in turn, stimulates cGMP-dependent protein kinase (PKG), which causes VSMC relaxation by phosphorylating multiple targets (Figure 3) [26]. Furthermore, synaptically released NO could also relax precapillary sphincters and pericytes at first-order capillaries, thereby enhancing blood perfusion to downstream microvessels [67]. A non-classical mechanism by which NO may regulate functions other than the vascular tone, e.g., DNA repair, transcription, and cell growth, is achieved through post-translational modification of target proteins. Non-classical NO signaling includes S-nitrosylation of protein thiols, oxidative nitration, hydroxylation, S-glutathionylation, and metal nitrosylation of transition metals [68,69].
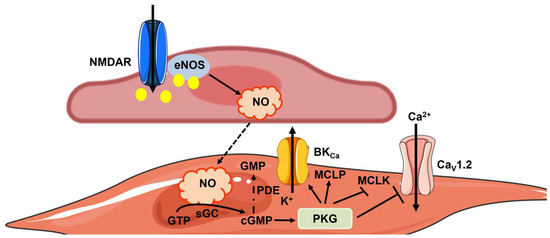
Figure 3.
The mechanisms of NO-mediated vasorelaxation. NO can be produced by eNOS upon synaptic activation of NMDARs, although it is likely that any increase in endothelial [Ca2+]i stimulates eNOS at the NVU [25,48]. Endothelium-derived NO may then diffuse to overlying VSMCs to induce relaxation by stimulating sGC activity, thereby resulting in GTP conversion into cGMP. In accordance, cGMP activates PKG, which in turn activates myosin light-chain phosphatase (MLCP) and inhibits myosin light-chain kinase (MLCK), thereby reducing the Ca2+-sensitivity of the contractile machinery. Moreover, PKG reduces VSMC excitability by activating large-conductance Ca2+-activated K+ channels (BKCa) and inhibiting voltage-gated CaV1.2 channels, thereby promoting VSMC hyperpolarization and preventing voltage-gated Ca2+ entry. The signal transduction pathway elicited by NO can be interrupted when cGMP is hydrolyzed into an inactive 5′-GMP metabolite by phosphodiesterase (PDE) activity.
Endothelium-derived NO was claimed to be primarily involved in the hemodynamic response to the stimulation of basal forebrain acetylcholine neurons, which broadly project onto the intraparenchymal arterioles and capillaries of the cortex [21,26,28]. In line with this, the acetylcholine-induced increase in CBF is impaired by the genetic deletion of eNOS [70] and M5 muscarinic receptors [71,72], which can activate eNOS via a long-lasting increase in endothelial [Ca2+]i [50]. Furthermore, eNOS has been involved in the hemodynamic response of the somatosensory cortex to whisker stimulation, which triggers ATP release from synaptically activated astrocytes [13]. Then, ATP binds to P2Y1 receptors on the adjacent endothelial cells, thereby promoting eNOS activation and the local increase in CBF [13] through the elevation in endothelial [Ca2+]i [73].
The discovery that NMDARs are also expressed in cerebrovascular endothelial cells led to a reappraisal of the cellular and molecular mechanisms of NVC at glutamatergic synapses [11,74,75]. In accordance, a series of studies showed that the exogenous administration of glutamate stimulated endothelial NMDARs to promote NO-mediated arteriolar relaxation in ex vivo mouse middle cerebral arteries [43,62,76]. A neuro-glial-endothelial axis provides endothelial NMDARs with the co-agonist D-serine in the mouse brain microcirculation [43], while the molecular make-up of human NMDARs does not support a critical role for either D-serine or glycine [11,45,75]. NMDARs are located in the abluminal membrane and, therefore, are ideally positioned to detect synaptic activity and recruit eNOS [43]. In line with these observations, the hemodynamic response to sensory stimulation is impaired by the genetic deletion of both endothelial NMDARs [44] and eNOS [12]. It follows that eNOS interacts with nNOS to regulate NVC, thereby complementing each other and providing redundancy to ensure that active neurons receive a sufficient amount of nutrients and O2 to maintain cerebral function [77]. Furthermore, studies carried out on healthy adult subjects suggest that both eNOS and nNOS contribute to generating the NO signal driving NVC in humans [78,79,80]. Interestingly, it has been proposed that basal CBF is not reduced in eNOS knockout mice due to autoregulation and a compensatory increase in perivascular nNOS expression or signaling [81,82,83]. The pathological implications of defective eNOS signaling fall beyond the scope of the present article [17,21,81]. However, eNOS-deficient mice, which are more prone to developing spontaneous hypertension and other defects in systemic circulation [84,85], also show an increase in infarct volume and reduced arteriogenesis after stroke [86]. Interestingly, genetic screening showed that polymorphisms in the NOS3 gene were associated with an increased risk of cerebral small vessel disease in patients, including silent brain infarction [87,88]. The physiological relevance of eNOS signaling at the NVU is, therefore, further highlighted by the pathological consequences of its impairment in both human patients and transgenic mouse models.
It should also be noted that eNOS-derived NO is not the sole vasorelaxing mediator by which cerebrovascular endothelial cells regulate CBF at the NVU. A complex ion signaling machinery, reviewed in [25,48,89,90], is set in motion by neuronal activity at the capillary level, thereby ensuring that both fast (i.e., EDH) and slow (i.e., intercellular Ca2+ waves) vasorelaxing signals travel back to up-stream arterioles and precapillary sphincters to steal blood from silent to active brain areas [47,91,92,93,94]. Altogether, this evidence re-evaluates the crucial role of endothelial cells in the regulation of blood flow in the brain microvasculature, although neuron-derived vasorelaxing pathways provide a backup mechanism to preserve (at least partially) NVC even during endothelial dysfunction or damage [10,32,77].
3.4. The Regulation of Synaptic Plasticity by eNOS
NO has been postulated to serve as a retrograde messenger at glutamatergic synapses, being produced in response to NMDARs-mediated Ca2+ entry and traveling back to the presynaptic terminal to increase glutamate release [31,95]. This mechanism explains the role of NO in synaptic plasticity, including LTP [63]. The source of NO during LTP induction is commonly claimed to be nNOS [6,96], but multiple pieces of evidence suggest that eNOS also generates NO during LTP in several brain regions. O’Dell and coworkers were the first to suggest that eNOS was responsible for LTP induction in the CA1 region of nNOS-deficient mice [36]. This pioneering finding was confirmed by three different research groups exploiting the adenoviral-mediated inhibition of eNOS activity [97], eNOS-deficient mice [98], and nNOS- and eNOS-deficient mice [99]. Subsequent studies showed that tetanic stimulation also requires eNOS to induce LTP in the cortex [100] and striatum [101]. It should again be noted that the interpretation of these studies was biased by the mislocalization of eNOS, which was wrongly claimed to also be expressed in dendritic spines [36]. The recognition that the immunocytochemical staining of eNOS in pyramidal neurons was artifactual, in association with more sensitive immunocytochemistry or in situ hybridization assays, unveiled the unexpected involvement of endothelium-derived NO in LTP [16,22,23,24]. Garthwaite and coworkers were the first to revise the view that cerebrovascular endothelial cells merely serve as a conduit of nutrients and O2 to active neurons [15]. They found that capillary endothelial cells tonically release NO to depolarize the optic nerve by activating sGC, thereby inducing the cyclic guanosin monophosphate (cGMP)-dependent activation of hyperpolarization-activated cyclic nucleotide-gated non-selective cation channels [15]. No other NOS isoforms, i.e., nNOS and iNOS, were involved in NO-dependent axonal depolarization. Furthermore, bradykinin, which is a widely employed endothelial agonist [42], boosted NO-dependent depolarization of the optic nerve [15]. A follow-up investigation shed further light on the role played by eNOS in hippocampal LTP [14]. This study revealed that LTP requires phasic bursts of NO release by nNOS, while eNOS provides the tonic NO signal (≈1 nM) that prevents LTP from rapidly decaying [14]. These findings help reconcile the double requirement for hippocampal LTP of both nNOS and eNOS that has been described in [99]. Furthermore, a recent investigation suggested that the hippocampal LTP may be supported by the neuro-glial-endothelial axis, which is indispensable for eNOS activation [102].
The concept of heart-brain axis (HBA) predicts that cardiovascular disorders, such as heart failure, atherosclerosis, and hypertension, may lead to vascular dementia by primarily causing cerebral hypoperfusion [103,104,105]. Similarly, aging and other cardiovascular risk factors, as well as environmental pollution, may cause early endothelial injury at the NVU, thereby accelerating the harmful consequences of amyloid-β (Aβ) peptide deposition and resulting in neurodegeneration and synaptic dysfunction in Alzheimer’s disease (AD) [5]. In addition, the amyloid hypothesis formulated by Selkoe [106] and Hardy [107] predicts that the excessive accumulation of the Aβ peptide in cerebral microvessels directly causes endothelial dysfunction (amyloid angiopathy) [17,108]. Intriguingly, transgenic mouse models of AD showed that cerebrovascular endothelial dysfunction appears long before the development of AD symptoms and the impairment of cognitive decline [17,21,108]. Endothelial dysfunction could certainly exacerbate cognitive decline by impairing CBF, dismantling BBB integrity, and promoting inflammation and astrogliosis [109,110]. In light of the unexpected role of eNOS in synaptic plasticity, we propose that endothelial dysfunction could impair the LTP induction mechanisms, thereby directly contributing to cognitive decline in AD disease [111]. In agreement with this hypothesis, the LTP/long-term depression (LTD) balance is shifted toward LTD in eNOS-deficient mice [101], which is a feature of AD [112]. Furthermore, the genetic deletion of eNOS has been shown to exacerbate spatial learning deficits in mouse models of vascular dementia [113] and AD [114], and in eNOS knockout mice after ischemic stroke [115].
3.5. The Role of eNOS in Vascular-to-Neuronal Communication
In 2008, Moore and Cao introduced the concept of vascular-to-neuronal communication, according to which the mechanical stimulation of cerebrovascular endothelial cells during the hemodynamic response leads to a NO signal that may fine-tune neuronal activity [18]. The mechanosensitive channel activated by mechanical deformation and responsible for eNOS activation could be Piezo1 [55,56]. This concept has been reinforced by Filosa and coworkers, who showed that an increase in flow/pressure within the intraparenchymal arterioles of cortical brain slices caused an astrocyte-dependent decrease in the firing rate of pyramidal neurons [116,117]. It is unclear whether this signaling pathway requires the interposition of endothelial cells [20]; however, it lends strong support to the modulation of neuronal activity and synaptic processes by vascular inputs. On the one hand, assessing whether and how cerebrovascular endothelial cells generate NO in response to mechanical stimulation deriving from changes in the intraluminal pressure [56] or by the frictional force imposed by the train of red blood cells transiting through the capillaries in a single file [118] is a mandatory task. On the other hand, we propose to revise the concept of vascular-to-neuronal communication in light of the documented endothelial response to neuronal activity. Endothelium-derived NO may also be generated during synaptic activity, thereby serving as a volume intercellular messenger that contributes to induced and maintained LTP and targets mural cells to cause vasorelaxation and match the increased demand for blood supply (Figure 4). In addition, the burst of NO associated with eNOS activation could also modulate astrocyte activity, as envisaged by Moore and Cao [18]. It has long been known that the GqPCRs, mGluR1, and mGluR5, are involved in the astrocytic-dependent regulation of NVC, which occurs through the release of epoxyeicosatrienoic acids [119,120,121]. However, it is still debated whether astrocytes express mGluRs in vivo [10]. However, a series of independent studies showed that synaptic activity elicits Ca2+ signals in astrocytic end-feet and arteriolar vasodilation by promoting endothelial-dependent NO production (Figure 4), which could be driven by endothelial mGluR1 and mGluR5 [49,122]. Therefore, eNOS could play a critical role in integrating neuronal and vascular inputs, thereby ensuring a bidirectional mode of communication that guarantees the effective functioning of the NVU in the healthy brain microcirculation.
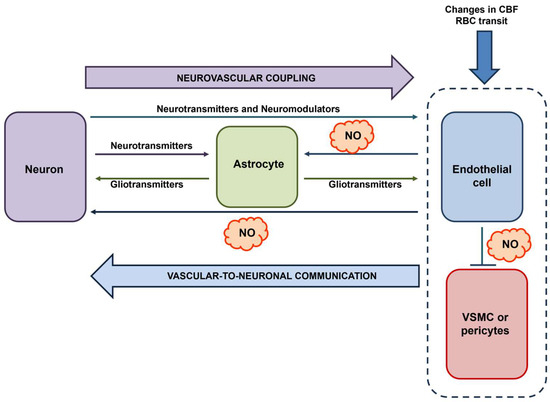
Figure 4.
The central role of eNOS in vascular-to-neuronal communication. Neuronal activity (possibly with the intermediation of astrocyte-derived signals) and blood-born mechanical signals (e.g., local changes in CBF evoked by the same neuronal activity or single-file transit of RBCs) lead to an increase in [Ca2+]i in cerebrovascular endothelial cells, thereby leading to eNOS activation and NO release. NO serves as a volume intercellular messenger that can target multiple cell types within the NVU: (1) neurons, thereby supporting LTP; (2) mural cells, i.e., VSMCs and pericytes, thereby promoting vasorelaxation; and (3) astrocytes, thereby eliciting intracellular Ca2+ signals. Inspired by [18,19].
4. Conclusions
The discovery of NO as a crucial regulator of cardiovascular function has paved the way for the identification of a highly versatile gasotransmitter with broader physiological implications [68], extending to immune responses, inflammation, osteogenesis, smooth muscle contraction, and neuronal signaling. The role of eNOS at the NVU has progressively gained momentum over the last decade, as nNOS was initially considered the primary source of NO within the brain parenchyma [16]. eNOS is unlikely to be the first thought of a neuroscientist at work to unlock the mechanisms by which NO is produced in response to neuronal activity to modulate CBF and synaptic plasticity. Nevertheless, it is now clear that the complex puzzle of NO signaling at the NVU misses crucial pieces without the addition of cerebrovascular endothelial cells and eNOS. Future work will have to assess whether endothelium-derived NO regulates CBF in brain areas other than the somatosensory cortex, e.g., in the hippocampus and cerebellum. It will also be mandatory to unravel whether eNOS supports LTP, as well as other modes of NO-dependent synaptic plasticity [96], throughout all glutamatergic synapses in the brain or at specific sites, e.g., the hippocampal CA1 region. Another indispensable step in filling the gap between hypotheses and knowledge would be to confirm whether the increased blood flow through brain capillaries during the hemodynamic response stimulates NO release. Therefore, we posit that the concept of vascular-to-neuronal communication should include endothelium-derived signals, including the NO burst, produced in response to neuronal activity upon the release of multiple neurotransmitters and neuromodulators. Finally, although pericytes have long been regarded as a target of NO signaling at the NVU, under both physiological [40] and pathological [123] conditions, emerging evidence indicates that they can also express eNOS [39]. Intriguingly, brain pericytes express a wide range of GqPCRs that are potentially involved in vasoconstriction, such as α1-adrenoreceptors and endothelin receptor type B [40]. Excessive eNOS signaling in brain pericytes during cerebral ischemia could play a pivotal role in triggering α-smooth muscle actin phenotype transformation [39] and pericyte contraction due to oxidative-nitrosative stress [123], thereby leading to the no-reflow phenomenon and patient death. Understanding the role of eNOS in the regulation of neurovascular interactions is predicted to shed light on alternative strategies to prevent or interfere with vascular cognitive decline by targeting the NO-sGC-cGMP signaling pathway [124].
Author Contributions
Conceptualization, F.M.; investigation, F.M.; writing—original draft preparation, F.M.; writing—review and editing, F.M.; visualization, G.S., V.B., R.B.-R., G.D.S., G.G. and T.S.; supervision, F.M.; project administration, F.M.; funding acquisition, G.D.S. and F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research has been supported by #NEXTGENERATIONEU (NGEU) and funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006)—a multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022), and the European Commission’s FESR FSE 2014–2020 and POR CALABRIA FESR AZIONE 1.5.1 “Support for research infrastructures considered critical/crucial for regional systems” Nuova Piattaforma di Farmacologia Integrata e Tecnologie Avanzate. R.B.R. has been supported by the National Council of Science and Technology (CONACYT), identification number CVU121216.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, S.; Arnold, W.; Mittal, C.; Murad, F. Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J. Cycl. Nucleotide Res. 1977, 3, 23–35. [Google Scholar]
- Ignarro, L.J. Nitric oxide is not just blowing in the wind. Br. J. Pharmacol. 2019, 176, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Kourosh-Arami, M.; Hosseini, N.; Mohsenzadegan, M.; Komaki, A.; Joghataei, M.T. Neurophysiologic implications of neuronal nitric oxide synthase. Rev. Neurosci. 2020, 31, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Z.; Xiong, Y.; Zhou, Q.; Zhu, L.Q.; Liu, D. The emerging role of nitric oxide in the synaptic dysfunction of vascular dementia. Neural Regen. Res. 2025, 20, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Iova, O.M.; Marin, G.E.; Lazar, I.; Stanescu, I.; Dogaru, G.; Nicula, C.A.; Bulboaca, A.E. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders-An Overview. Antioxidants 2023, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Pla, A.F.; Moccia, F.; Tanzi, F.; Munaron, L. Old and new gasotransmitters in the cardiovascular system: Focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Curr. Pharm. Biotechnol. 2011, 12, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Brunetti, V.; Berra-Romani, R.; Moccia, F. The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 3914. [Google Scholar] [CrossRef]
- Chow, B.W.; Nunez, V.; Kaplan, L.; Granger, A.J.; Bistrong, K.; Zucker, H.L.; Kumar, P.; Sabatini, B.L.; Gu, C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 2020, 579, 106–110. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Davila, A.; Valcarcel-Ares, M.N.; Tucsek, Z.; Varamini, B.; Ballabh, P.; Sonntag, W.E.; Baur, J.A.; Csiszar, A.; et al. Purinergic glio-endothelial coupling during neuronal activity: Role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1837–H1845. [Google Scholar] [CrossRef]
- Hopper, R.A.; Garthwaite, J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J. Neurosci. 2006, 26, 11513–11521. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, G.; Bartus, K.; Malcolm, D.; Goodwin, D.; Kollb-Sielecka, M.; Dooldeniya, C.; Garthwaite, J. Signaling from blood vessels to CNS axons through nitric oxide. J. Neurosci. 2006, 26, 7730–7740. [Google Scholar] [CrossRef]
- Garthwaite, J. Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 2008, 27, 2783–2802. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; Austin, S.A. Endothelial nitric oxide: Protector of a healthy mind. Eur. Heart J. 2014, 35, 888–894. [Google Scholar] [CrossRef]
- Moore, C.I.; Cao, R. The hemo-neural hypothesis: On the role of blood flow in information processing. J. Neurophysiol. 2008, 99, 2035–2047. [Google Scholar] [CrossRef]
- Mishra, A. Binaural blood flow control by astrocytes: Listening to synapses and the vasculature. J. Physiol. 2017, 595, 1885–1902. [Google Scholar] [CrossRef]
- Presa, J.L.; Saravia, F.; Bagi, Z.; Filosa, J.A. Vasculo-Neuronal Coupling and Neurovascular Coupling at the Neurovascular Unit: Impact of Hypertension. Front. Physiol. 2020, 11, 584135. [Google Scholar] [CrossRef]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef]
- Blackshaw, S.; Eliasson, M.J.; Sawa, A.; Watkins, C.C.; Krug, D.; Gupta, A.; Arai, T.; Ferrante, R.J.; Snyder, S.H. Species, strain and developmental variations in hippocampal neuronal and endothelial nitric oxide synthase clarify discrepancies in nitric oxide-dependent synaptic plasticity. Neuroscience 2003, 119, 979–990. [Google Scholar] [CrossRef]
- Demas, G.E.; Kriegsfeld, L.J.; Blackshaw, S.; Huang, P.; Gammie, S.C.; Nelson, R.J.; Snyder, S.H. Elimination of aggressive behavior in male mice lacking endothelial nitric oxide synthase. J. Neurosci. 1999, 19, RC30. [Google Scholar] [CrossRef] [PubMed]
- Seidel, B.; Stanarius, A.; Wolf, G. Differential expression of neuronal and endothelial nitric oxide synthase in blood vessels of the rat brain. Neurosci. Lett. 1997, 239, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K(+) (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Hamel, E. Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 2006, 100, 1059–1064. [Google Scholar] [CrossRef]
- Agarwal, N.; Carare, R.O. Cerebral Vessels: An Overview of Anatomy, Physiology, and Role in the Drainage of Fluids and Solutes. Front. Neurol. 2020, 11, 611485. [Google Scholar] [CrossRef]
- Garthwaite, J. From synaptically localized to volume transmission by nitric oxide. J. Physiol. 2016, 594, 9–18. [Google Scholar] [CrossRef]
- Garthwaite, J. Nitric oxide as a multimodal brain transmitter. Brain Neurosci. Adv. 2018, 2, 2398212818810683. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.F.; Ledo, A.; Barbosa, R.M.; Laranjinha, J. Neurovascular-neuroenergetic coupling axis in the brain: Master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic. Biol. Med. 2017, 108, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Gotti, S.; Sica, M.; Viglietti-Panzica, C.; Panzica, G. Distribution of nitric oxide synthase immunoreactivity in the mouse brain. Microsc. Res. Tech. 2005, 68, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Chachlaki, K.; Garthwaite, J.; Prevot, V. The gentle art of saying NO: How nitric oxide gets things done in the hypothalamus. Nat. Rev. Endocrinol. 2017, 13, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, T.J.; Huang, P.L.; Dawson, T.M.; Dinerman, J.L.; Snyder, S.H.; Kandel, E.R.; Fishman, M.C. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science 1994, 265, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Dinerman, J.L.; Dawson, T.M.; Schell, M.J.; Snowman, A.; Snyder, S.H. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: Implications for synaptic plasticity. Proc. Natl. Acad. Sci. USA 1994, 91, 4214–4218. [Google Scholar] [CrossRef] [PubMed]
- Doyle, C.A.; Slater, P. Localization of neuronal and endothelial nitric oxide synthase isoforms in human hippocampus. Neuroscience 1997, 76, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, Y.; Li, B.; Luo, C.; Zuo, S.; Liu, X.; Zhang, J.H.; Ruan, H.; Feng, H. Hemoglobin induced NO/cGMP suppression Deteriorate Microcirculation via Pericyte Phenotype Transformation after Subarachnoid Hemorrhage in Rats. Sci. Rep. 2016, 6, 22070. [Google Scholar] [CrossRef]
- Longden, T.A.; Zhao, G.; Hariharan, A.; Lederer, W.J. Pericytes and the Control of Blood Flow in Brain and Heart. Annu. Rev. Physiol. 2023, 85, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Brunetti, V.; Perna, A.; Guerra, G.; Soda, T.; Berra-Romani, R. The Molecular Heterogeneity of Store-Operated Ca2+ Entry in Vascular Endothelial Cells: The Different roles of Orai1 and TRPC1/TRPC4 Channels in the Transition from Ca2+-Selective to Non-Selective Cation Currents. Int. J. Mol. Sci. 2023, 24, 3259. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Soda, T.; Berra-Romani, R.; Scarpellino, G. Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing. Int. J. Mol. Sci. 2023, 24, 16765. [Google Scholar] [CrossRef]
- Stobart, J.L.; Lu, L.; Anderson, H.D.; Mori, H.; Anderson, C.M. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2013, 110, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Lu, P.; Anderson, C.M. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10229–10231. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium. 2021, 99, 102454. [Google Scholar] [CrossRef]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.A.; Mughal, A.; Hennig, G.W.; Harraz, O.F.; Shui, B.; Lee, F.K.; Lee, J.C.; Reining, S.; Kotlikoff, M.I.; Konig, G.M.; et al. Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Sci. Adv. 2021, 7, eabh0101. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vascul. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABA(A) and GABA(B) Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Brunetti, V.; Pellavio, G.; Soda, T.; Laforenza, U.; Scarpellino, G.; Moccia, F. Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3. Cells 2023, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Shipsky, M.M.; Yan, G.; Abood, M.E.; Brailoiu, G.C. Mechanisms of modulation of brain microvascular endothelial cells function by thrombin. Brain Res. 2017, 1657, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Barlow, C.L.; Ramirez, S.H.; Abood, M.E.; Brailoiu, G.C. Effects of Platelet-Activating Factor on Brain Microvascular Endothelial Cells. Neuroscience 2018, 377, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Klug, N.R.; Senatore, A.J.; Hill-Eubanks, D.C.; Nelson, M.T. Piezo1 Is a Mechanosensor Channel in Central Nervous System Capillaries. Circ. Res. 2022, 130, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.R.; Harraz, O.F. Mechanosensing by Vascular Endothelium. Annu. Rev. Physiol. 2024, 86, 71–97. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and recent developments. Br. J. Pharmacol. 2019, 176, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.C.; Kou, Y.R.; Shyue, S.K.; Su, K.H.; Wei, J.; Cheng, L.C.; Yu, Y.B.; Pan, C.C.; Lee, T.S. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 2011, 91, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Kim, C.Y.; Zheng, C.; Jin, S.W.; Kim, J.Y.; Lee, S.Y.; Kim, M.Y.; Han, E.H.; Hwang, Y.P.; Jeong, H.G. Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKbeta/AMPK Signaling Pathway in Human Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 9407. [Google Scholar] [CrossRef]
- Steinert, J.R.; Kopp-Scheinpflug, C.; Baker, C.; Challiss, R.A.; Mistry, R.; Haustein, M.D.; Griffin, S.J.; Tong, H.; Graham, B.P.; Forsythe, I.D. Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron 2008, 60, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Ledo, A.; Barbosa, R.M.; Gerhardt, G.A.; Cadenas, E.; Laranjinha, J. Concentration dynamics of nitric oxide in rat hippocampal subregions evoked by stimulation of the NMDA glutamate receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 17483–17488. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2019, 39, 481–496. [Google Scholar] [CrossRef]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Clusters of specialized detector cells provide sensitive and high fidelity receptor signaling in the intact endothelium. FASEB J. 2016, 30, 2000–2013. [Google Scholar] [CrossRef]
- Lee, M.D.; Wilson, C.; Saunter, C.D.; Kennedy, C.; Girkin, J.M.; McCarron, J.G. Spatially structured cell populations process multiple sensory signals in parallel in intact vascular endothelium. Sci. Signal. 2018, 11, eaar4411. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D.; Buckley, C.; Zhang, X.; Louhivuori, L.; Uhlen, P.; Wilson, C.; McCarron, J.G. Small-world connectivity dictates collective endothelial cell signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2118927119. [Google Scholar] [CrossRef]
- Zambach, S.A.; Cai, C.; Helms, H.C.C.; Hald, B.O.; Dong, Y.; Fordsmann, J.C.; Nielsen, R.M.; Hu, J.; Lonstrup, M.; Brodin, B.; et al. Precapillary sphincters and pericytes at first-order capillaries as key regulators for brain capillary perfusion. Proc. Natl. Acad. Sci. USA 2021, 118, e2023749118. [Google Scholar] [CrossRef]
- Andrabi, S.M.; Sharma, N.S.; Karan, A.; Shahriar, S.M.S.; Cordon, B.; Ma, B.; Xie, J. Nitric Oxide: Physiological Functions, Delivery, and Biomedical Applications. Adv. Sci. 2023, 10, e2303259. [Google Scholar] [CrossRef]
- Martinez-Ruiz, A.; Cadenas, S.; Lamas, S. Nitric oxide signaling: Classical, less classical, and nonclassical mechanisms. Free Radic. Biol. Med. 2011, 51, 17–29. [Google Scholar] [CrossRef]
- Atochin, D.N.; Demchenko, I.T.; Astern, J.; Boso, A.E.; Piantadosi, C.A.; Huang, P.L. Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J. Cereb. Blood Flow Metab. 2003, 23, 1219–1226. [Google Scholar] [CrossRef]
- Yamada, M.; Lamping, K.G.; Duttaroy, A.; Zhang, W.; Cui, Y.; Bymaster, F.P.; McKinzie, D.L.; Felder, C.C.; Deng, C.X.; Faraci, F.M.; et al. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 14096–14101. [Google Scholar] [CrossRef] [PubMed]
- Araya, R.; Noguchi, T.; Yuhki, M.; Kitamura, N.; Higuchi, M.; Saido, T.C.; Seki, K.; Itohara, S.; Kawano, M.; Tanemura, K.; et al. Loss of M5 muscarinic acetylcholine receptors leads to cerebrovascular and neuronal abnormalities and cognitive deficits in mice. Neurobiol. Dis. 2006, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Bintig, W.; Begandt, D.; Schlingmann, B.; Gerhard, L.; Pangalos, M.; Dreyer, L.; Hohnjec, N.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; et al. Purine receptors and Ca2+ signalling in the human blood-brain barrier endothelial cell line hCMEC/D3. Purinergic Signal. 2012, 8, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, V.; Soda, T.; Berra-Romani, R.; De Sarro, G.; Guerra, G.; Scarpellino, G.; Moccia, F. Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age. Biomedicines 2024, 12, 880. [Google Scholar] [CrossRef] [PubMed]
- LeMaistre, J.L.; Sanders, S.A.; Stobart, M.J.; Lu, L.; Knox, J.D.; Anderson, H.D.; Anderson, C.M. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J. Cereb. Blood Flow Metab. 2012, 32, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Nippert, A.R.; Biesecker, K.R.; Newman, E.A. Mechanisms Mediating Functional Hyperemia in the Brain. Neuroscientist 2018, 24, 73–83. [Google Scholar] [CrossRef] [PubMed]
- O’Gallagher, K.; Puledda, F.; O’Daly, O.; Ryan, M.; Dancy, L.; Chowienczyk, P.J.; Zelaya, F.; Goadsby, P.J.; Shah, A.M. Neuronal nitric oxide synthase regulates regional brain perfusion in healthy humans. Cardiovasc. Res. 2022, 118, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Hoiland, R.L.; Caldwell, H.G.; Howe, C.A.; Nowak-Fluck, D.; Stacey, B.S.; Bailey, D.M.; Paton, J.F.R.; Green, D.J.; Sekhon, M.S.; Macleod, D.B.; et al. Nitric oxide is fundamental to neurovascular coupling in humans. J. Physiol. 2020, 598, 4927–4939. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.P.; Caldwell, H.G.; Gliemann, L. How do we kNOw the individual contribution of eNOS and nNOS for cerebral blood flow regulation? J. Physiol. 2022, 600, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.F.; Lin, G.; Chen, X.; Chen, L.; Zheng, W.; Raghow, R.; Zhou, F.M.; Shih, A.Y.; Tan, X.L. Endothelial Nitric Oxide Synthase-Deficient Mice: A Model of Spontaneous Cerebral Small-Vessel Disease. Am. J. Pathol. 2021, 191, 1932–1945. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, A.; Jing, Y.; Collie, N.D.; Zhang, H.; Liu, P. Altered neurovascular coupling and brain arginine metabolism in endothelial nitric oxide synthase deficient mice. Nitric Oxide 2019, 87, 60–72. [Google Scholar] [CrossRef]
- Atochin, D.N.; Huang, P.L. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch. 2010, 460, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef]
- Cui, X.; Chopp, M.; Zacharek, A.; Zhang, C.; Roberts, C.; Chen, J. Role of endothelial nitric oxide synthetase in arteriogenesis after stroke in mice. Neuroscience 2009, 159, 744–750. [Google Scholar] [CrossRef]
- Song, J.; Kim, O.J.; Kim, H.S.; Bae, S.J.; Hong, S.P.; Oh, D.; Kim, N.K. Endothelial nitric oxide synthase gene polymorphisms and the risk of silent brain infarction. Int. J. Mol. Med. 2010, 25, 819–823. [Google Scholar] [CrossRef][Green Version]
- Hassan, A.; Gormley, K.; O’Sullivan, M.; Knight, J.; Sham, P.; Vallance, P.; Bamford, J.; Markus, H. Endothelial nitric oxide gene haplotypes and risk of cerebral small-vessel disease. Stroke 2004, 35, 654–659. [Google Scholar] [CrossRef]
- Jackson, W.F. Calcium-Dependent Ion Channels and the Regulation of Arteriolar Myogenic Tone. Front. Physiol. 2021, 12, 770450. [Google Scholar] [CrossRef]
- Jackson, W.F. Endothelial Ion Channels and Cell-Cell Communication in the Microcirculation. Front. Physiol. 2022, 13, 805149. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef]
- Harraz, O.F.; Longden, T.A.; Dabertrand, F.; Hill-Eubanks, D.; Nelson, M.T. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. USA 2018, 115, E3569–E3577. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Klug, N.R.; Mughal, A.; Koide, M.; Huerta de la Cruz, S.; Heppner, T.J.; Bonev, A.D.; Hill-Eubanks, D.; Nelson, M.T. Adenosine signaling activates ATP-sensitive K(+) channels in endothelial cells and pericytes in CNS capillaries. Sci. Signal. 2022, 15, eabl5405. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Garthwaite, J. NO as a multimodal transmitter in the brain: Discovery and current status. Br. J. Pharmacol. 2019, 176, 197–211. [Google Scholar] [CrossRef]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7, 190. [Google Scholar] [CrossRef]
- Kantor, D.B.; Lanzrein, M.; Stary, S.J.; Sandoval, G.M.; Smith, W.B.; Sullivan, B.M.; Davidson, N.; Schuman, E.M. A role for endothelial NO synthase in LTP revealed by adenovirus-mediated inhibition and rescue. Science 1996, 274, 1744–1748. [Google Scholar] [CrossRef]
- Wilson, R.I.; Yanovsky, J.; Godecke, A.; Stevens, D.R.; Schrader, J.; Haas, H.L. Endothelial nitric oxide synthase and LTP. Nature 1997, 386, 338. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Hawkins, R.D.; Martin, K.; Kiebler, M.; Huang, P.L.; Fishman, M.C.; Kandel, E.R. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 1996, 87, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Haul, S.; Godecke, A.; Schrader, J.; Haas, H.L.; Luhmann, H.J. Impairment of neocortical long-term potentiation in mice deficient of endothelial nitric oxide synthase. J. Neurophysiol. 1999, 81, 494–497. [Google Scholar] [CrossRef]
- Doreulee, N.; Sergeeva, O.A.; Yanovsky, Y.; Chepkova, A.N.; Selbach, O.; Godecke, A.; Schrader, J.; Haas, H.L. Cortico-striatal synaptic plasticity in endothelial nitric oxide synthase deficient mice. Brain Res. 2003, 964, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Takagi, H.; Stoney, P.N.; Echeverria, A.; Kuhn, B.; Hsu, K.S.; Takahashi, T. Anoxia-induced hippocampal LTP is regeneratively produced by glutamate and nitric oxide from the neuro-glial-endothelial axis. iScience 2024, 27, 109515. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Sun, D.; Wang, Y.; Yan, M.; Zheng, J.; Ren, J. Cognitive Impairment in Heart Failure: Landscape, Challenges, and Future Directions. Front. Cardiovasc. Med. 2021, 8, 831734. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, G.; Liang, Y.; He, S.; Lyu, M.; Zhu, Y. Heart-brain interaction in cardiogenic dementia: Pathophysiology and therapeutic potential. Front. Cardiovasc. Med. 2024, 11, 1304864. [Google Scholar] [CrossRef]
- Iadecola, C.; Gottesman, R.F. Neurovascular and Cognitive Dysfunction in Hypertension. Circ. Res. 2019, 124, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Duering, M.; Hachinski, V.; Joutel, A.; Pendlebury, S.T.; Schneider, J.A.; Dichgans, M. Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2019, 73, 3326–3344. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Santhanam, A.V.; Hinton, D.J.; Choi, D.S.; Katusic, Z.S. Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J. Neurochem. 2013, 127, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, X.; Ma, L.; Wei, W.; Li, Z.; Chang, S.; Wen, J.; Sun, J.; Li, H. Role of Abeta in Alzheimer’s-related synaptic dysfunction. Front. Cell Dev. Biol. 2022, 10, 964075. [Google Scholar] [CrossRef]
- An, L.; Shen, Y.; Chopp, M.; Zacharek, A.; Venkat, P.; Chen, Z.; Li, W.; Qian, Y.; Landschoot-Ward, J.; Chen, J. Deficiency of Endothelial Nitric Oxide Synthase (eNOS) Exacerbates Brain Damage and Cognitive Deficit in A Mouse Model of Vascular Dementia. Aging Dis. 2021, 12, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Jing, Y.; Mockett, B.G.; Zhang, H.; Abraham, W.C.; Liu, P. Partial Endothelial Nitric Oxide Synthase Deficiency Exacerbates Cognitive Deficit and Amyloid Pathology in the APPswe/PS1DeltaE9 Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 7316. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Jiang, Z.; Huai, Y.; Liao, J.K.; Lynch, K.A.; Zafonte, R.; Wood, L.J.; Wang, Q.M. Impaired Cognitive Performance in Endothelial Nitric Oxide Synthase Knockout Mice After Ischemic Stroke: A Pilot Study. Am. J. Phys. Med. Rehabil. 2018, 97, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Iddings, J.A.; Stern, J.E.; Blanco, V.M.; Croom, D.; Kirov, S.A.; Filosa, J.A. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J. Neurosci. 2015, 35, 8245–8257. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Ramiro Diaz, J.; Iddings, J.A.; Filosa, J.A. Vasculo-Neuronal Coupling: Retrograde Vascular Communication to Brain Neurons. J. Neurosci. 2016, 36, 12624–12639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Giannetto, M.; DeCourcey, J.; Kang, H.; Kang, N.; Li, Y.; Zheng, S.; Zhao, H.; Simmons, W.R.; Wei, H.S.; et al. Oxygen tension-mediated erythrocyte membrane interactions regulate cerebral capillary hyperemia. Sci. Adv. 2019, 5, eaaw4466. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Chaigneau, E.; Osmanski, B.F.; Charpak, S. Vascular Compartmentalization of Functional Hyperemia from the Synapse to the Pia. Neuron 2018, 99, 362–375.e4. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, X.; Gebremedhin, D.; Falck, J.R.; Harder, D.R.; Koehler, R.C. Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J. Cereb. Blood Flow Metab. 2008, 28, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Falck, J.R.; Roman, R.J.; Harder, D.R.; Koehler, R.C. Interaction of nitric oxide, 20-HETE, and EETs during functional hyperemia in whisker barrel cortex. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H619–H631. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.H.T.; Peringod, G.; Gordon, G.R. Astrocytes Integrate Behavioral State and Vascular Signals during Functional Hyperemia. Neuron 2018, 100, 1133–1148.e3. [Google Scholar] [CrossRef] [PubMed]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, E.; Schepers, M.; Ponsaerts, L.; Foulquier, S.; Bronckaers, A.; Vanmierlo, T.; Sandner, P.; Prickaerts, J. Soluble guanylyl cyclase: A novel target for the treatment of vascular cognitive impairment? Pharmacol. Res. 2023, 197, 106970. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).