
Exploring the Molecular Mechanism of Skeletal Muscle Development in Ningxiang Pig by Weighted Gene Co-Expression Network Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Module Identification
2.2. Screening Skeletal Muscle Development-Related Co-Expression Gene Modules by WGCNA
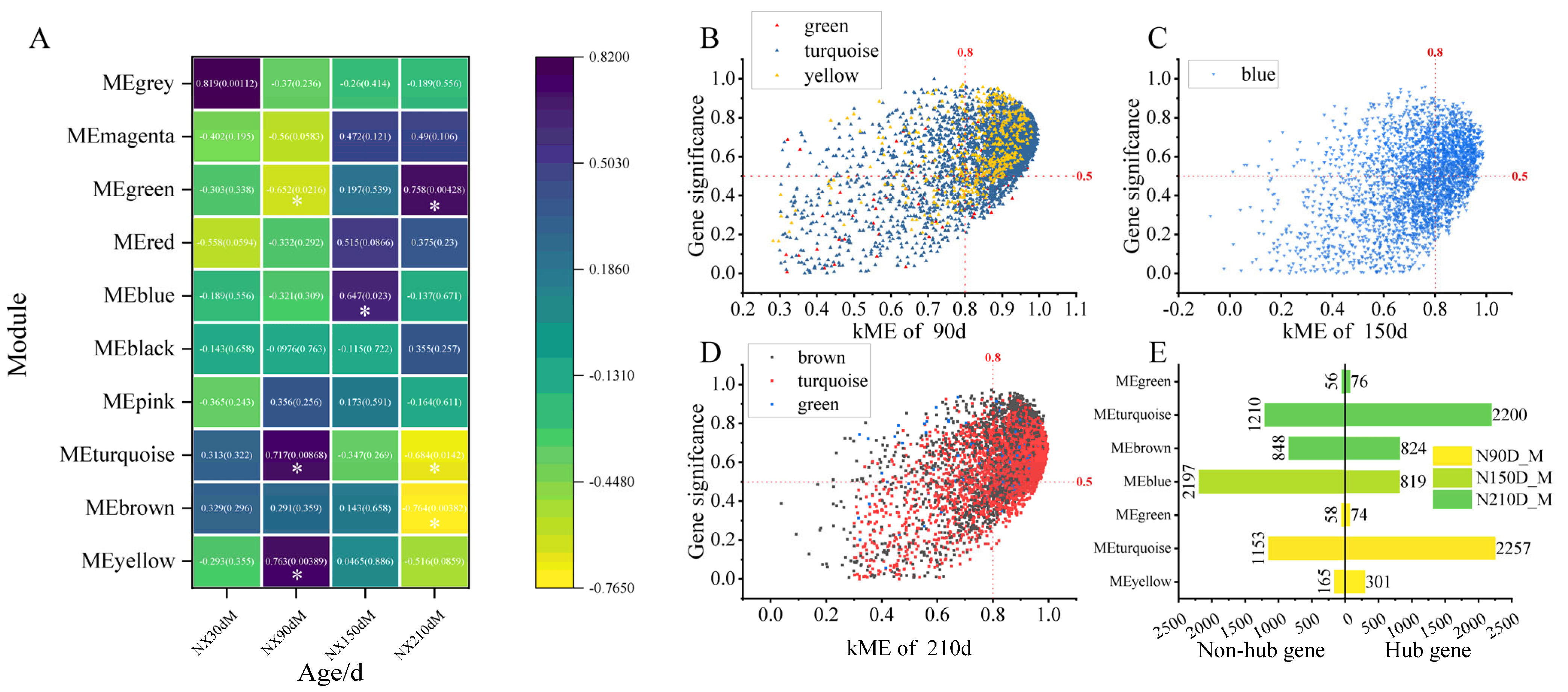
2.2.1. Development Stage Feature Module and Hub Gene Identification
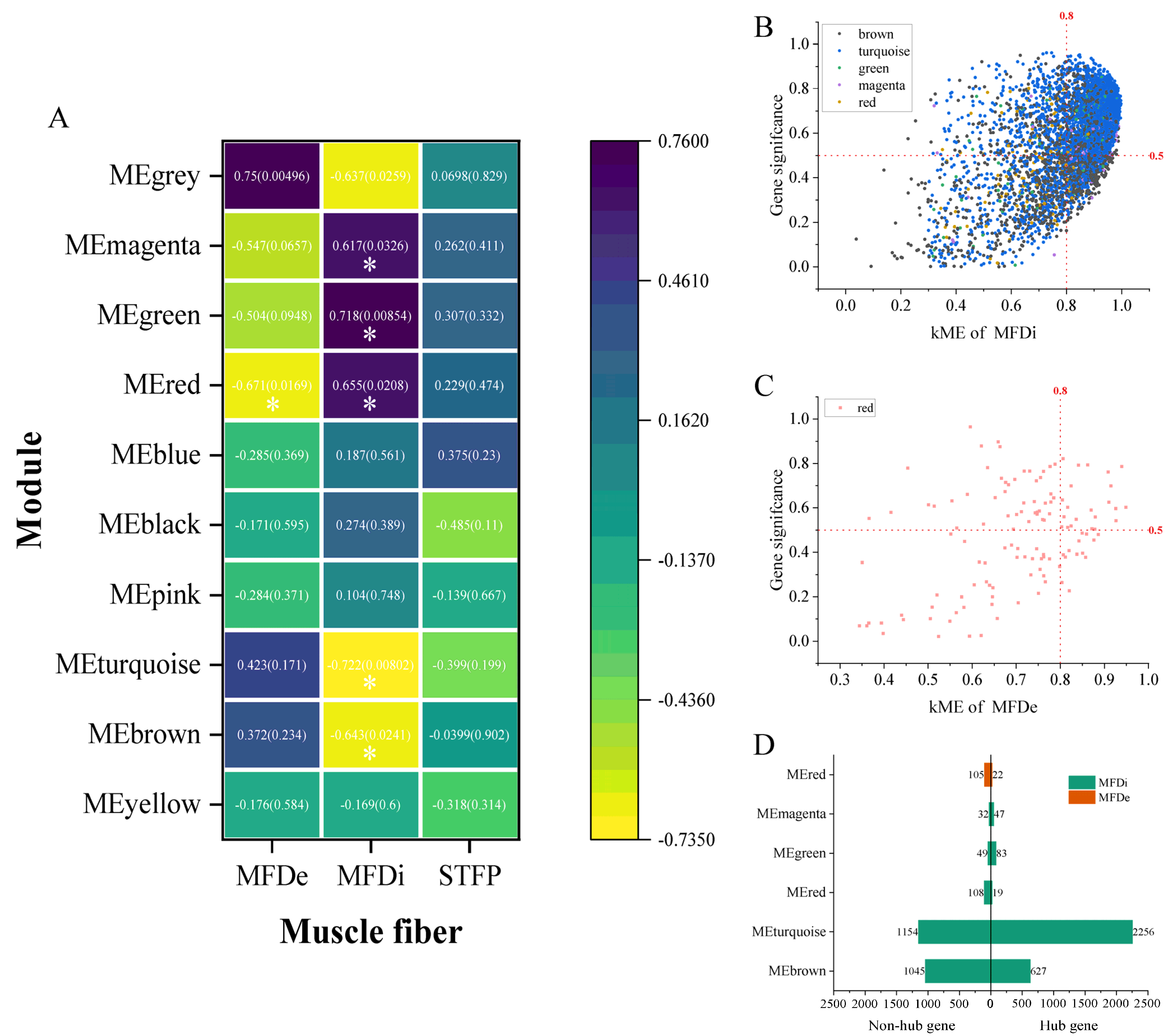
2.2.2. Key Modules of Muscle Fiber-Related Indexes of LD and Identification of Hub Gene
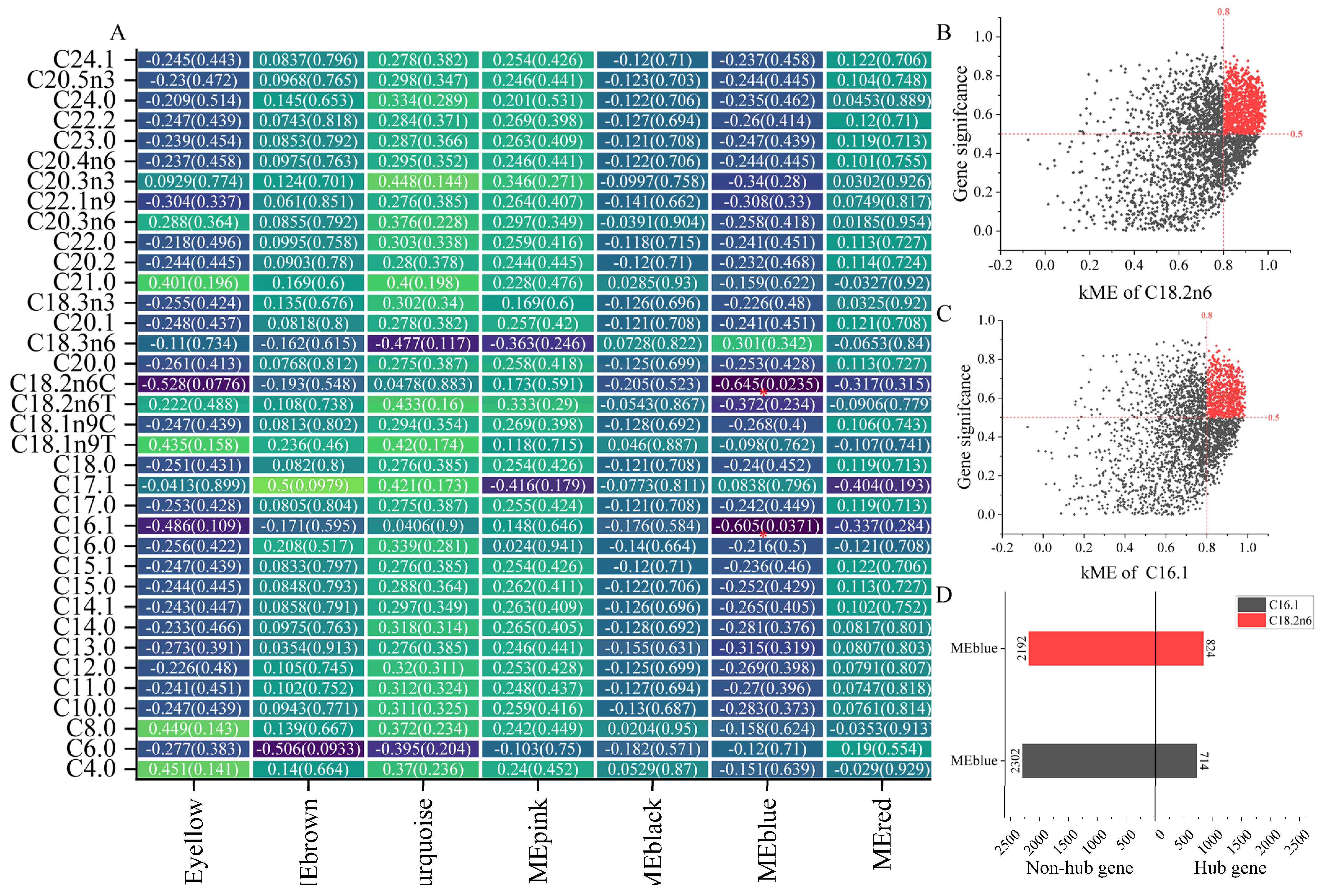
2.2.3. Key Module of Amino Acids and Fatty Acids in LD and Identification of Hub Gene
2.3. Functional Enrichment Analysis of Characteristic Genes of Muscle Phenotype Related Modules
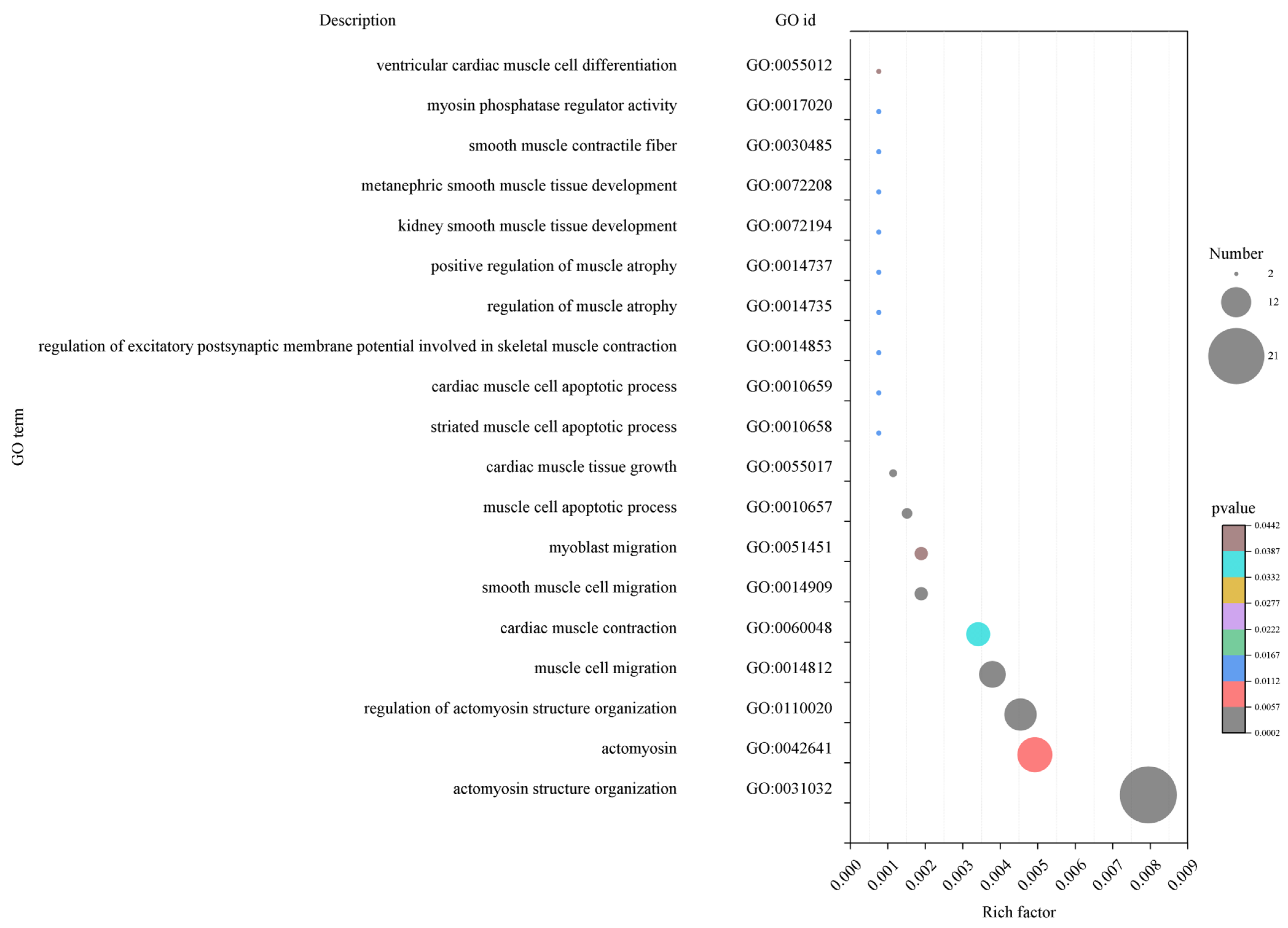
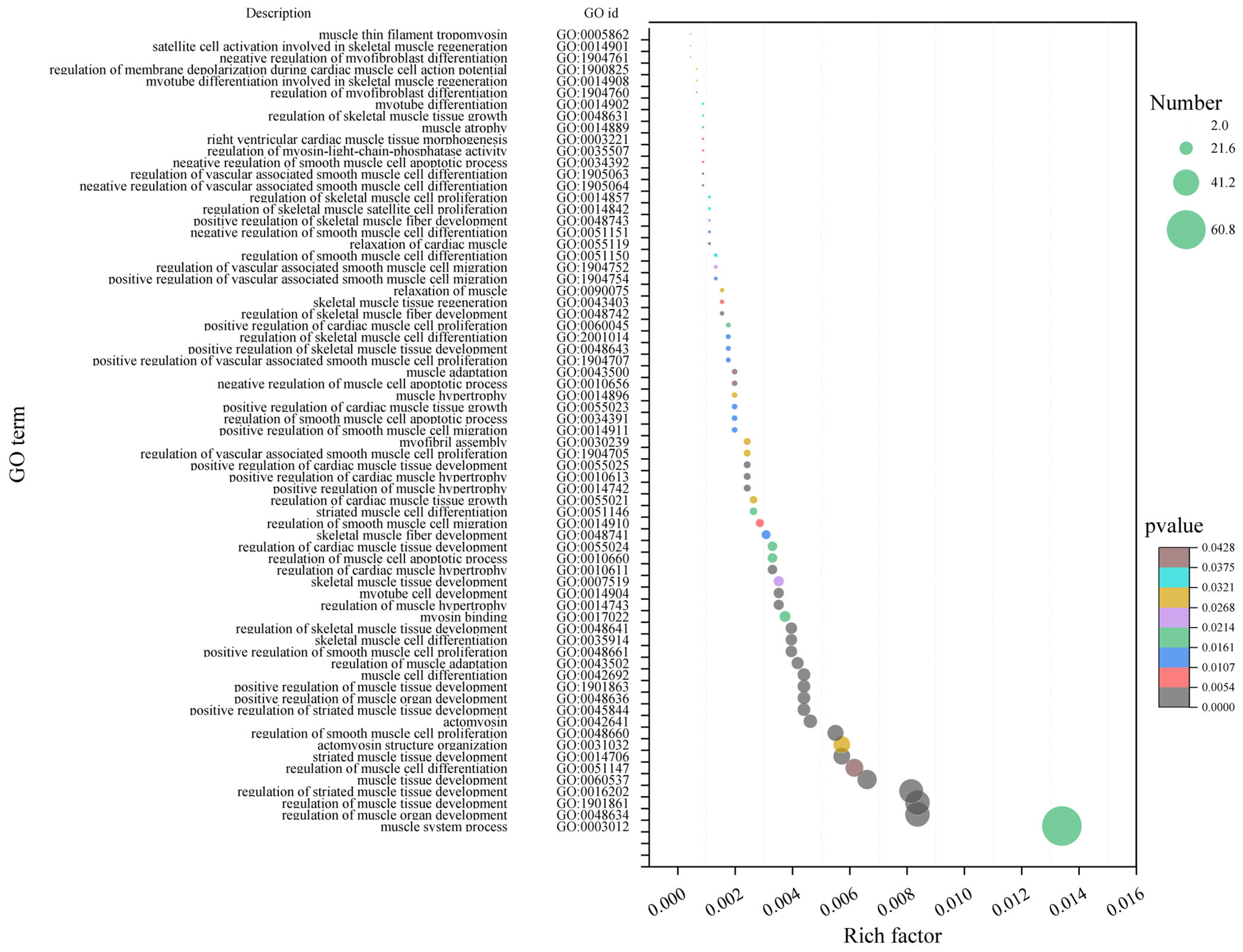
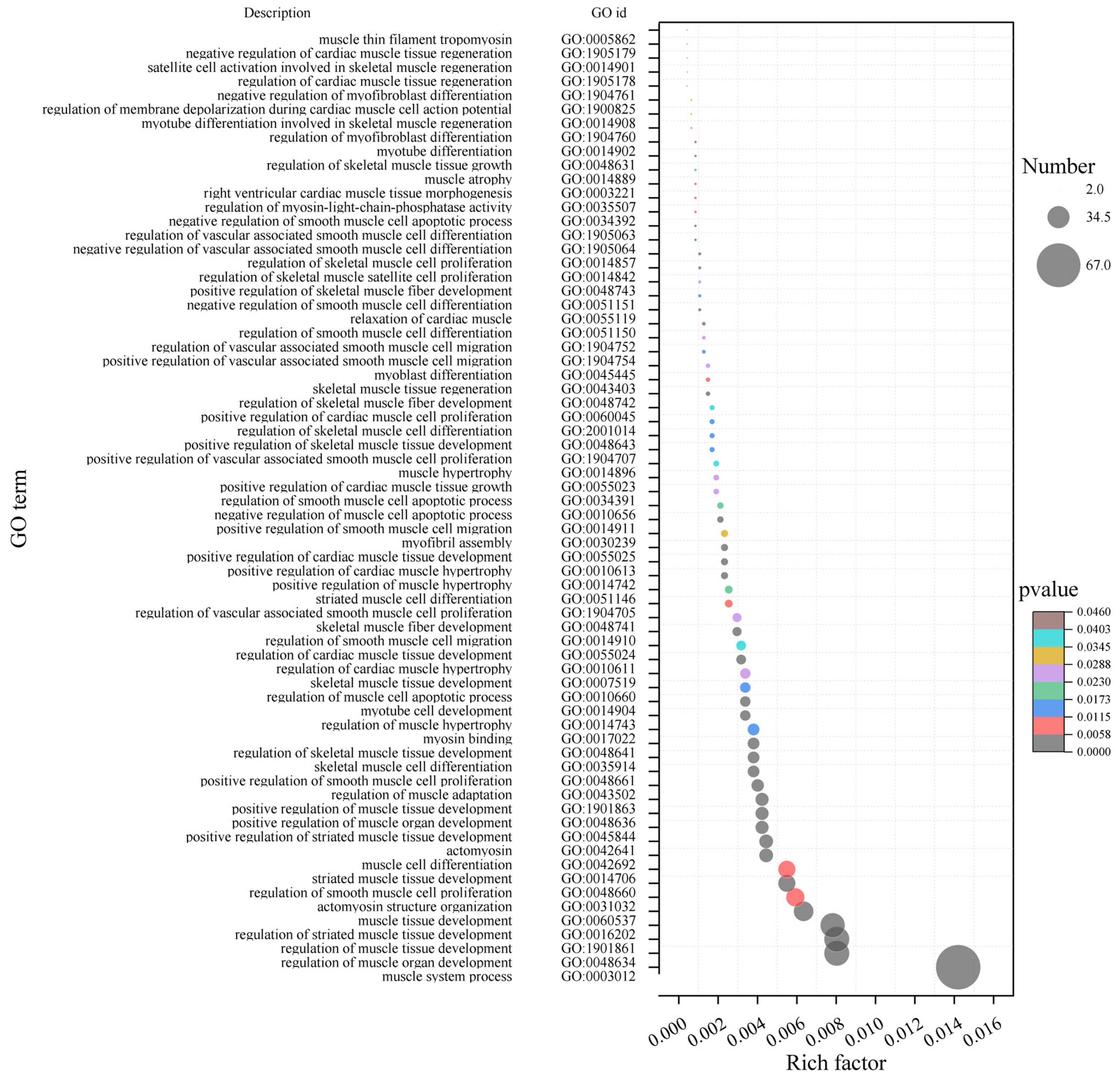
2.3.1. Functional Enrichment Analysis of Hub Genes of Related Modules in Muscle Development Stage
2.3.2. Functional Enrichment Analysis of Characteristic Genes of Muscle Fiber-Related Modules
2.3.3. Enrichment Analysis of Characteristic Genes of Fatty Acid-Related Modules in LD
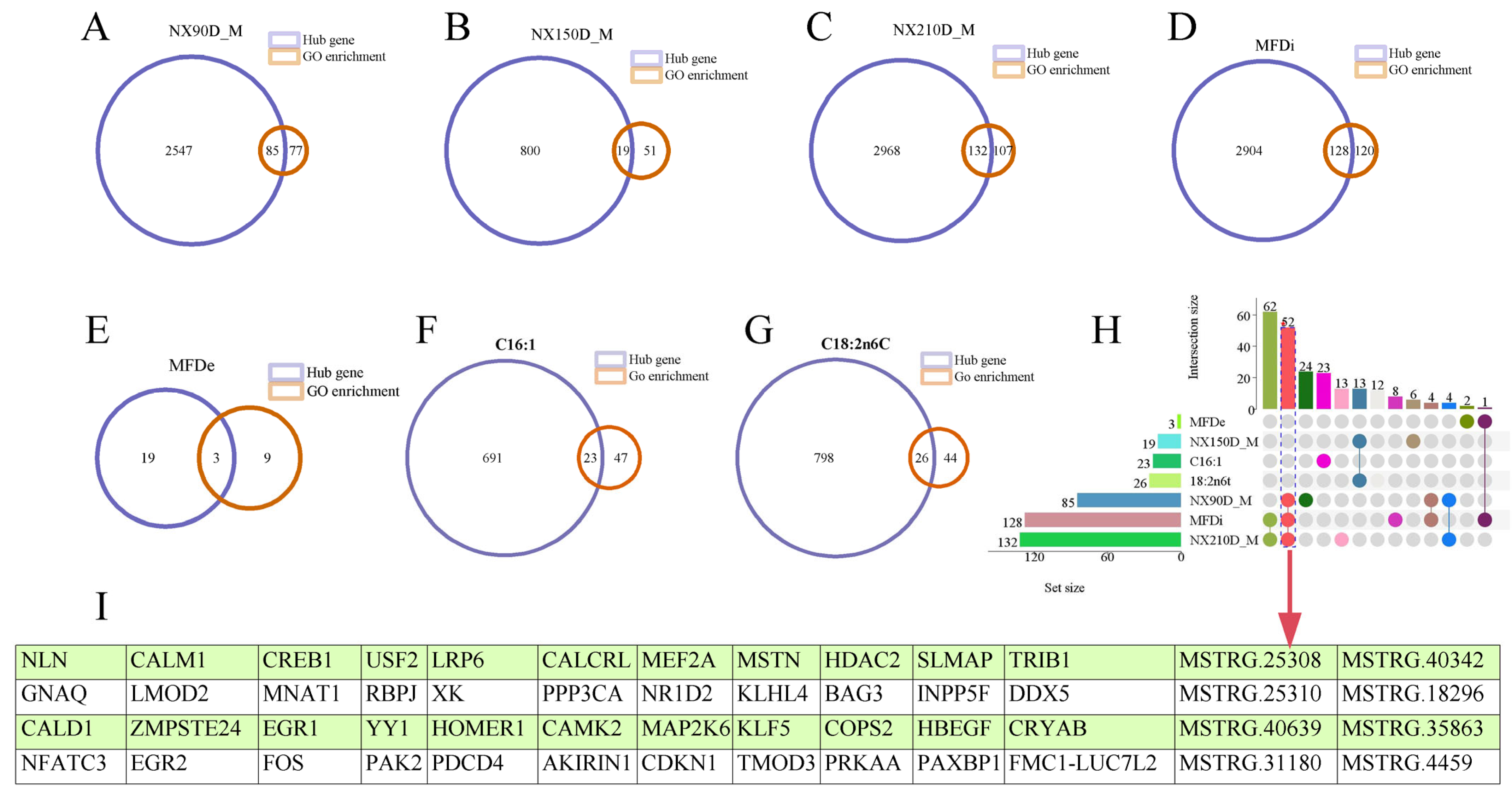
2.3.4. Overlapping Analysis of Characteristic Genes and Hub Genes of Muscle Phenotype
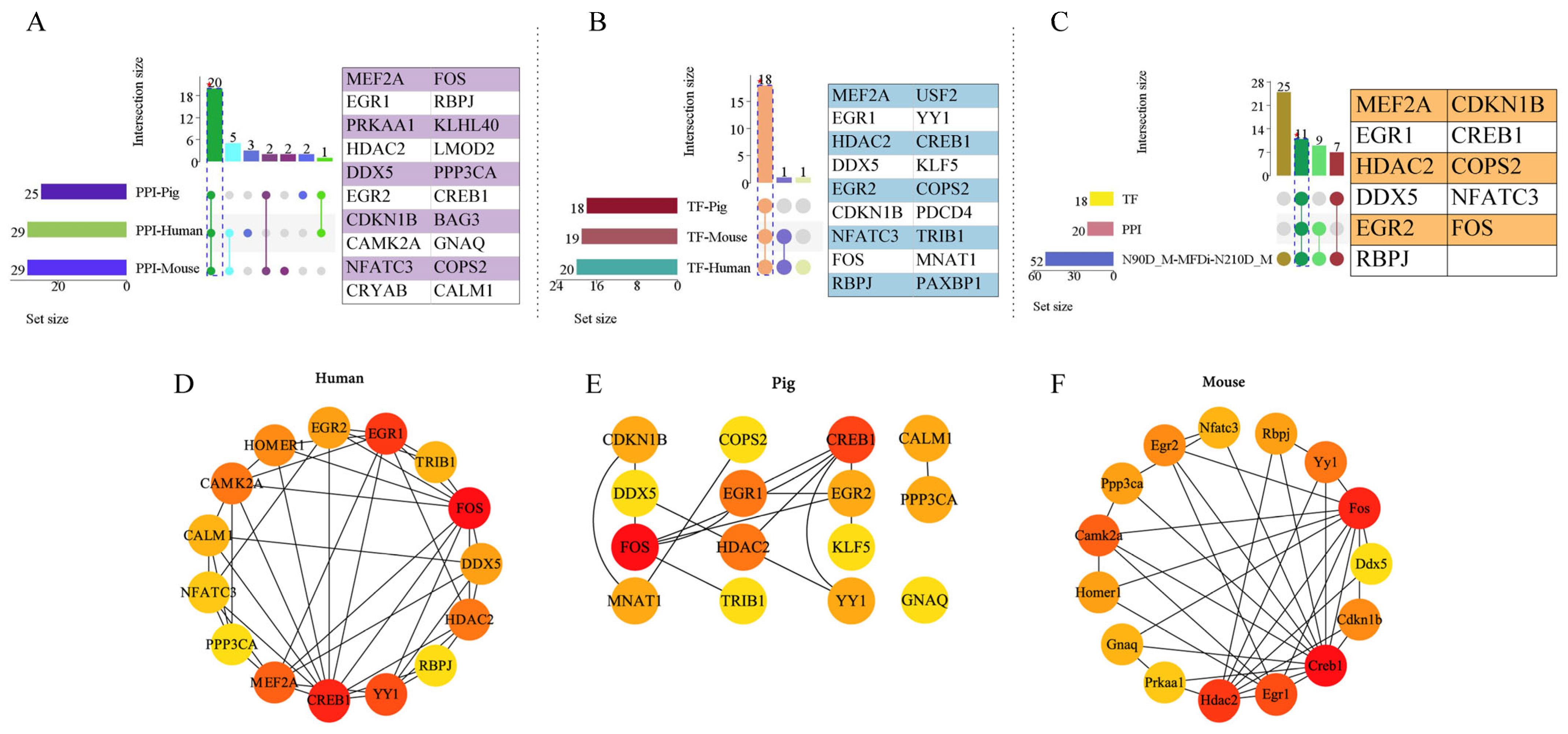
2.4. Protein–Protein Interaction (PPI) and Transcription Factor (TF) Analysis of Muscle Phenotype-Related Genes
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Sample Preparation
4.2. Phenotypic Detection
4.2.1. Detection of Type Proportion, Diameter and Density of Muscle Fibers
4.2.2. Detection of Fatty Acids and Amino Acids
4.3. RNA-Seq
4.4. Bioinformatics Analysis
4.4.1. Sequencing Data Sorting and Trimming
4.4.2. WGCNA Analysis
4.4.3. Enrichment Analysis of GO and KEGG
4.4.4. Prediction and Analysis of Transcription Factors
4.4.5. Protein–Protein Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, J.; Chen, F.; Lin, X.; Wang, Y.; He, J.; Zhao, Y. Effect of Excessive or Restrictive Energy on Growth Performance, Meat Quality, and Intramuscular Fat Deposition in Finishing Ningxiang Pigs. Animals 2021, 11, 27. [Google Scholar] [CrossRef]
- Xing, Y.; Wu, X.; Xie, C.; Xiao, D.; Zhang, B. Meat Quality and Fatty Acid Profiles of Chinese Ningxiang Pigs Following Supplementation with N-Carbamylglutamate. Animals 2020, 10, 88. [Google Scholar] [CrossRef]
- Zhu, B.; Gao, H.; Yang, F.; Li, Y.; Yang, Q.; Liao, Y.; Guo, H.; Xu, K.; Tang, Z.; Gao, N.; et al. Comparative Characterization of Volatile Compounds of Ningxiang Pig, Duroc and Their Crosses (Duroc × Ningxiang) by Using SPME-GC-MS. Foods 2023, 12, 1059. [Google Scholar] [CrossRef]
- Wang, Q.; Gao, H.; Fu, Y.; Chen, Y.; Song, G.; Jin, Z.; Zhang, Y.; Yin, J.; Yin, Y.; Xu, K. Comprehensive characterization of the differences in metabolites, lipids, and volatile flavor compounds between Ningxiang and Berkshire pigs using multi-omics techniques. Food Chem. 2024, 457, 139807. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Jin, Z.; Gao, H.; Fu, Y.; Ren, R.; Deng, X.; Chen, Y.; Hou, X.; Wang, Q.; Song, G.; Fan, N.; et al. Whole-Transcriptome Analysis Sheds Light on the Biological Contexts of Intramuscular Fat Deposition in Ningxiang Pigs. Genes 2024, 15, 642. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Y.-B.; Jiang, Q.; Yin, Y.-L.; Tan, B.-E.; Chen, J.-S. Analysis of transcriptome differences between subcutaneous and intramuscular adipose tissue of Ningxiang pigs. Yíchuán 2023, 45, 1147–1157. [Google Scholar] [CrossRef]
- Lan, Q.; Liufu, S.; Liu, X.; Ai, N.; Xu, X.; Li, X.; Yu, Z.; Yin, Y.; Liu, M.; Ma, H. Comprehensive analysis of transcriptomic and metabolomic profiles uncovered the age-induced dynamic development pattern of subcutaneous fat in Ningxiang pig. Gene 2023, 880, 147624. [Google Scholar] [CrossRef]
- Liufu, S.; Lan, Q.; Liu, X.; Chen, B.; Xu, X.; Ai, N.; Li, X.; Yu, Z.; Ma, H. Transcriptome Analysis Reveals the Age-Related Developmental Dynamics Pattern of the Longissimus Dorsi Muscle in Ningxiang Pigs. Genes 2023, 14, 1050. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Feng, S.; Xu, Y.; Dai, Z.; Yin, H.; Zhang, K.; Shen, Y. Integrative Analysis from Multicenter Studies Identifies a WGCNA-Derived Cancer-Associated Fibroblast Signature for Ovarian Cancer. Front. Immunol. 2022, 13, 951582. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, H.; Li, H.; Dou, W.; Wang, X. Weighted Gene Co-expression Network Analysis Identifies a Cancer-Associated Fibroblast Signature for Predicting Prognosis and Therapeutic Responses in Gastric Cancer. Front. Mol. Biosci. 2021, 8, 744677. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, H.; Wang, H.; He, X.; Wang, J.; Wei, W.; Liu, M.; Xu, J.; Liu, Y.; Jiang, R. Identification of Key Modules and Hub Genes Involved in Regulating the Color of Chicken Breast Meat Using WGCNA. Animals 2023, 13, 2356. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, P.; Fan, S.; Zhai, B.; Jin, W.; Li, D.; Li, H.; Sun, G.; Han, R.; Liu, X.; et al. Weighted gene co-expression network indicates that the DYNLL2 is an important regulator of chicken breast muscle development and is regulated by miR-148a-3p. BMC Genom. 2022, 23, 258. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Li, M.; Wang, S.; Chen, Q.; Lu, S. Identification of key sex-specific pathways and genes in the subcutaneous adipose tissue from pigs using WGCNA method. BMC Genom. Data 2022, 23, 35. [Google Scholar] [CrossRef]
- Osterhoff, M.A.; Frahnow, T.; Seltmann, A.C.; Mosig, A.S.; Neunübel, K.; Sales, S.; Sampaio, J.; Hornemann, S.; Kruse, M.; Pfeiffer, A. Identification of gene-networks associated with specific lipid metabolites by Weighted Gene Co-Expression Network Analysis (WGCNA). Exp. Clin. Endocrinol. Diabet. 2014, 122, 98. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, X.; Ai, N.; Wang, K.; Zhang, P.; Li, X.; LiuFu, S.; Liu, X.; Jiang, J.; Gu, J.; et al. Integrated analysis of circRNA, lncRNA, miRNA and mRNA to reveal the ceRNA regulatory network of postnatal skeletal muscle development in Ningxiang pig. Front. Cell. Dev. Biol. 2023, 11, 1185823. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, pdb-top084970. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhao, Z.; Cai, Q.; Zhang, Y.; Zhang, P.; Shi, S.; Xie, H.; Peng, X.; Yin, W.; Tao, Y.; et al. miRNA-based biomarkers, therapies, and resistance in Cancer. Int. J. Biol. Sci. 2020, 16, 2628–2647. [Google Scholar] [CrossRef]
- Ketkar, S.; Burrage, L.C.; Lee, B. RNA Sequencing as a Diagnostic Tool. JAMA-J. Am. Med. Assoc. 2023, 329, 85–86. [Google Scholar] [CrossRef]
- Chen, G.; Qian, H.; Chen, J.; Wang, J.; Guan, J.; Chi, Z. Whole transcriptome sequencing identifies key circRNAs, lncRNAs, and miRNAs regulating neurogenesis in developing mouse retina. BMC Genom. 2021, 22, 779. [Google Scholar] [CrossRef]
- Kuksin, M.; Morel, D.; Aglave, M.; Danlos, F.; Marabelle, A.; Zinovyev, A.; Gautheret, D.; Verlingue, L. Applications of single-cell and bulk RNA sequencing in onco-immunology. Eur. J. Cancer 2021, 149, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Streeper, R.S.; Farese, R.V.; Yamamoto, K.R. Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc. Natl. Acad. Sci. USA 2006, 103, 15675–15680. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S.; Schneider, J.W.; Nadal-Ginard, B.; Mahdavi, V. Activation of the myogenic lineage by MEF2A, a factor that induces and cooperates with MyoD. Science 1994, 266, 1236–1240. [Google Scholar] [CrossRef]
- Garg, A.; O’Rourke, J.; Long, C.; Doering, J.; Ravenscroft, G.; Bezprozvannaya, S.; Nelson, B.R.; Beetz, N.; Li, L.; Chen, S.; et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Investig. 2014, 124, 3529–3539. [Google Scholar] [CrossRef]
- Hayashi, S.; Manabe, I.; Suzuki, Y.; Relaix, F.; Oishi, Y. Klf5 regulates muscle differentiation by directly targeting muscle-specific genes in cooperation with MyoD in mice. eLife 2016, 5, e17462. [Google Scholar] [CrossRef]
- Salanova, M.; Volpe, P.; Blottner, D. Homer protein family regulation in skeletal muscle and neuromuscular adaptation. IUBMB Life 2013, 65, 769–776. [Google Scholar] [CrossRef]
- Miska, E.A. Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res. 2001, 29, 3439–3447. [Google Scholar] [CrossRef]
- Zhu, C.; Piao, Z.; Jin, L. HDAC5 inhibition attenuates ventricular remodeling and cardiac dysfunction. Orphanet J. Rare Dis. 2023, 18, 266. [Google Scholar] [CrossRef]
- Sakaida, M.; Sukeno, M.; Imoto, Y.; Tsuchiya, S.; Sugimoto, Y.; Okuno, Y.; Segi-Nishida, E. Electroconvulsive seizure-induced changes in gene expression in the mouse hypothalamic paraventricular nucleus. J. Psychopharmacol. 2013, 27, 1058–1069. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, C.; Yu, Y.; Xing, Y.; Xiao, D.; Zhang, B. Comparison of fatty acid profile of three adipose tissues in Ningxiang pigs. Anim. Nutr. 2018, 4, 256–259. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Ma, H.; Jiang, J.; He, J.; Liu, H.; Han, L.; Gong, Y.; Li, B.; Yu, Z.; Tang, S.; Zhang, Y.; et al. Long-read assembly of the Chinese indigenous Ningxiang pig genome and identification of genetic variations in fat metabolism among different breeds. Mol. Ecol. Resour. 2022, 22, 1508–1520. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000 Res. 2020, 9, 304. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome, P.D.P.S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Ai, N.; Xu, X.; Zhang, P.; Jin, Z.; Li, X.; Ma, H. Exploring the Molecular Mechanism of Skeletal Muscle Development in Ningxiang Pig by Weighted Gene Co-Expression Network Analysis. Int. J. Mol. Sci. 2024, 25, 9089. https://doi.org/10.3390/ijms25169089
Yu Z, Ai N, Xu X, Zhang P, Jin Z, Li X, Ma H. Exploring the Molecular Mechanism of Skeletal Muscle Development in Ningxiang Pig by Weighted Gene Co-Expression Network Analysis. International Journal of Molecular Sciences. 2024; 25(16):9089. https://doi.org/10.3390/ijms25169089
Chicago/Turabian StyleYu, Zonggang, Nini Ai, Xueli Xu, Peiwen Zhang, Zhao Jin, Xintong Li, and Haiming Ma. 2024. "Exploring the Molecular Mechanism of Skeletal Muscle Development in Ningxiang Pig by Weighted Gene Co-Expression Network Analysis" International Journal of Molecular Sciences 25, no. 16: 9089. https://doi.org/10.3390/ijms25169089
APA StyleYu, Z., Ai, N., Xu, X., Zhang, P., Jin, Z., Li, X., & Ma, H. (2024). Exploring the Molecular Mechanism of Skeletal Muscle Development in Ningxiang Pig by Weighted Gene Co-Expression Network Analysis. International Journal of Molecular Sciences, 25(16), 9089. https://doi.org/10.3390/ijms25169089