

Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage

 ,
, 
Abstract
1. Introduction
2. Results
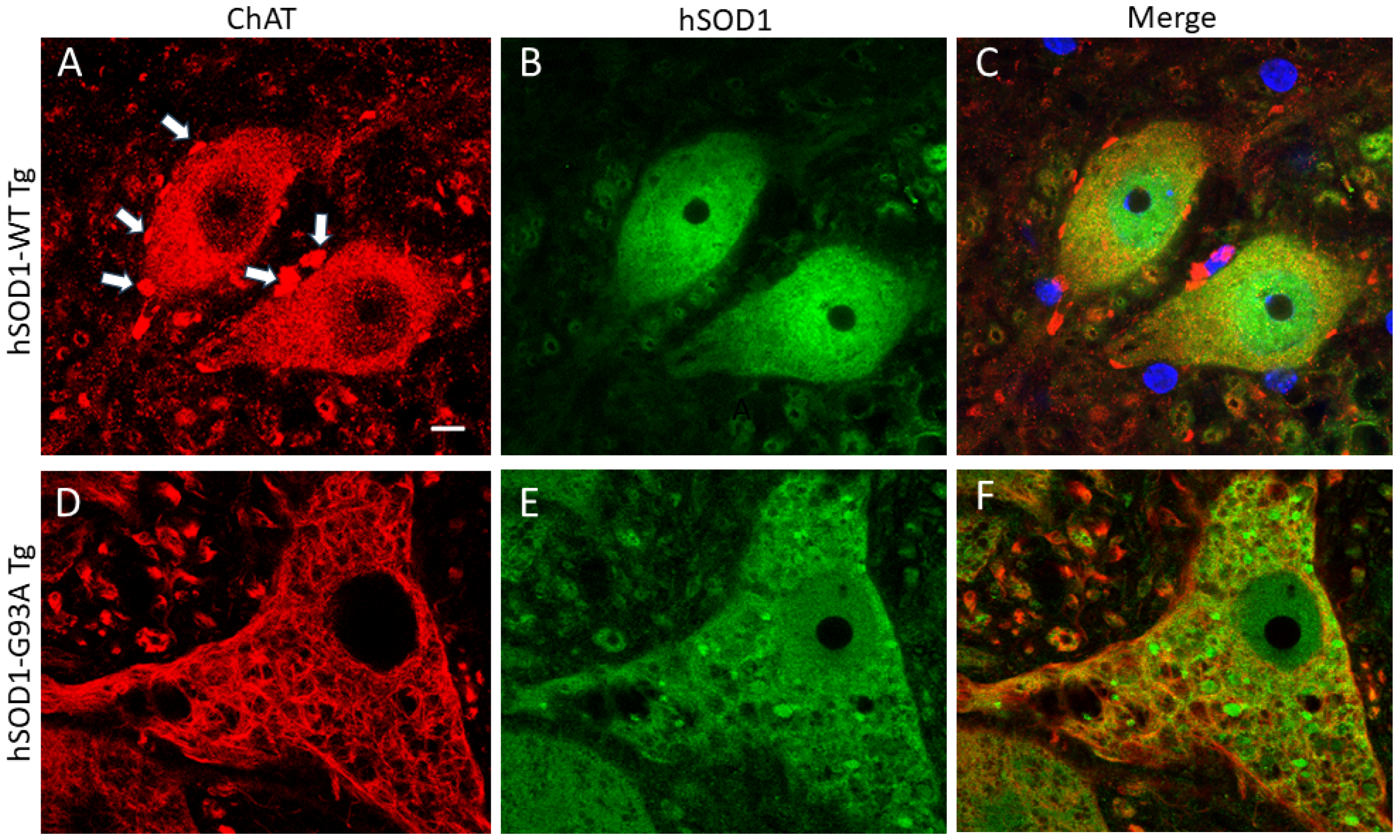
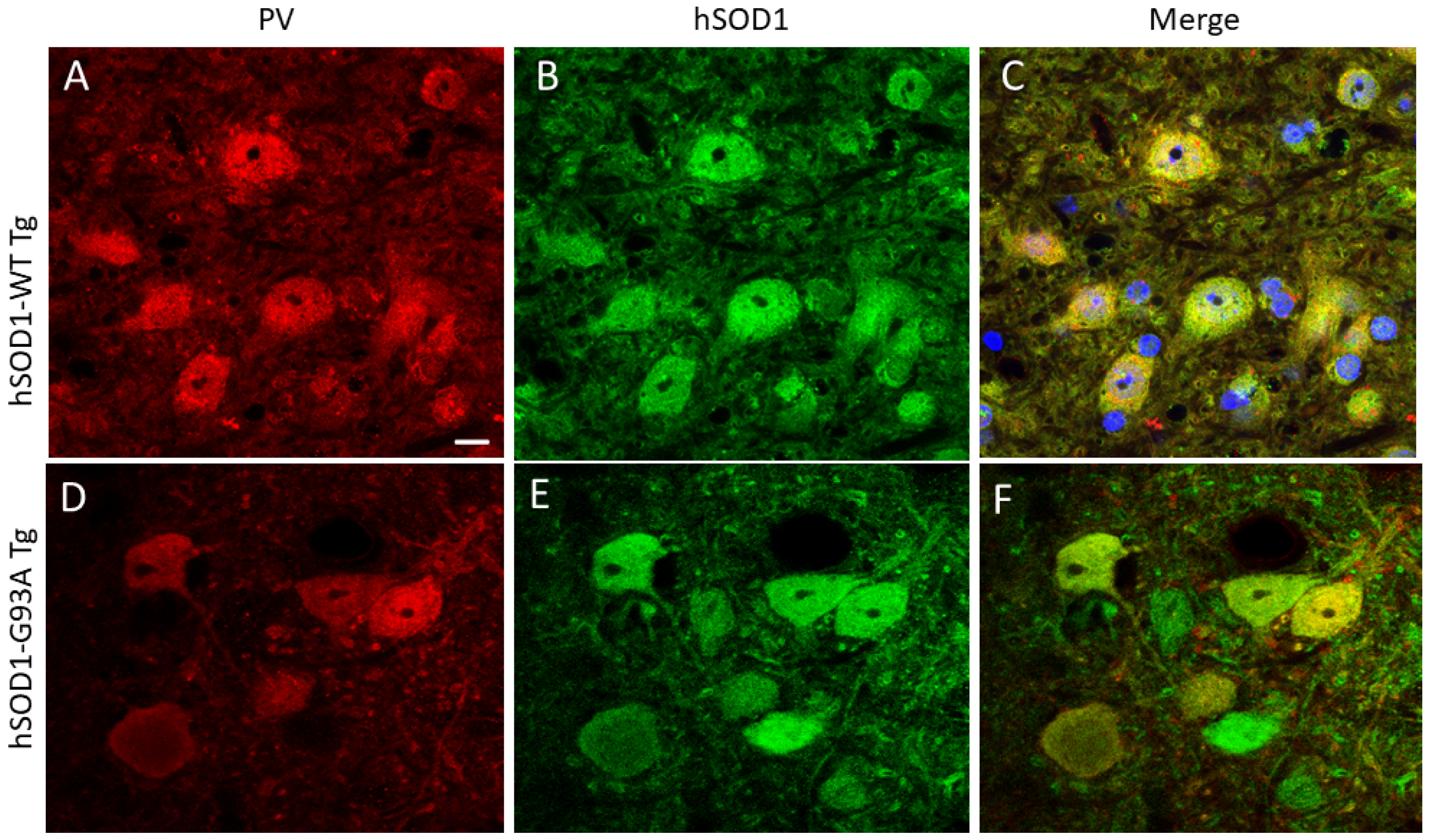
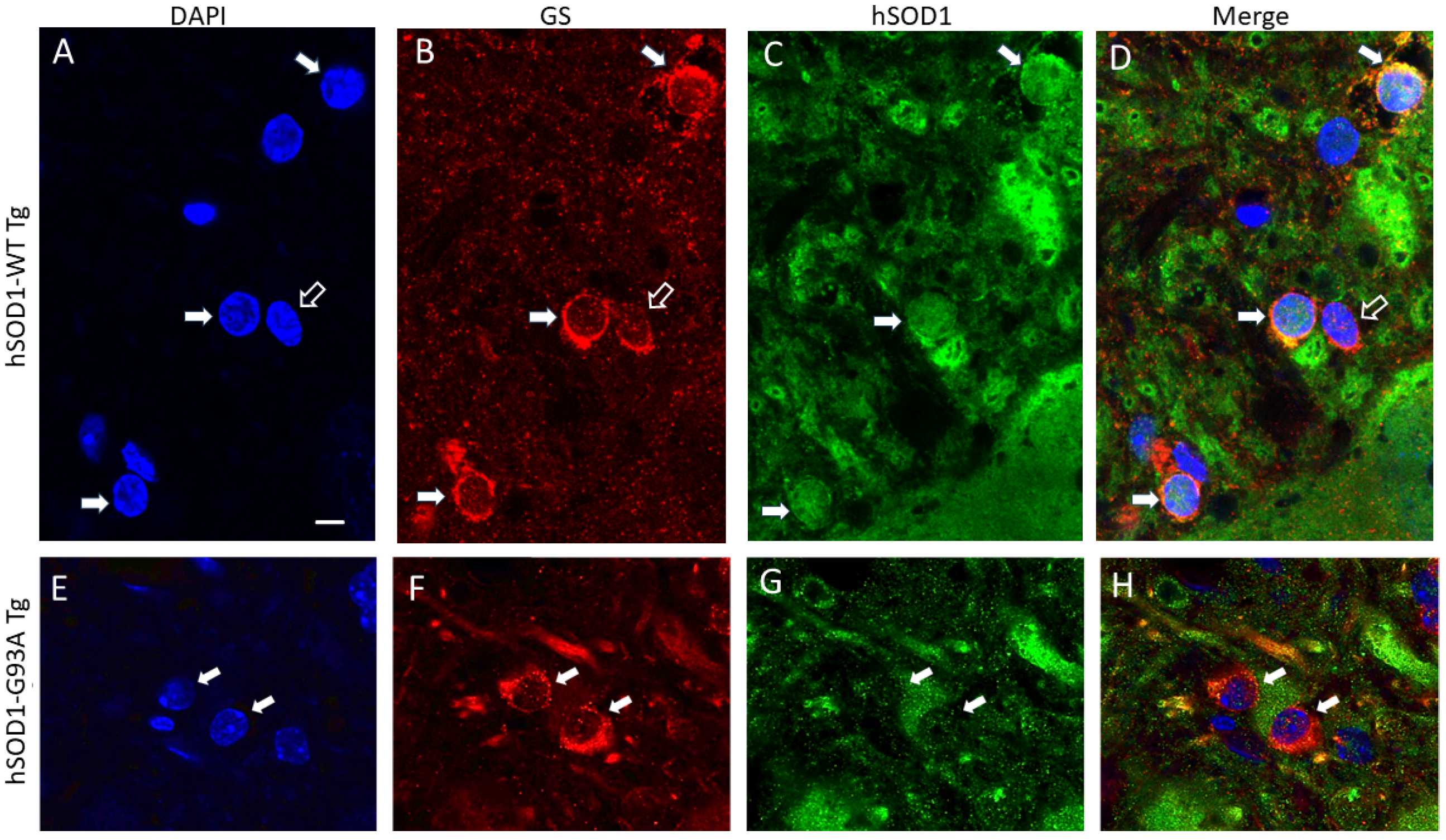
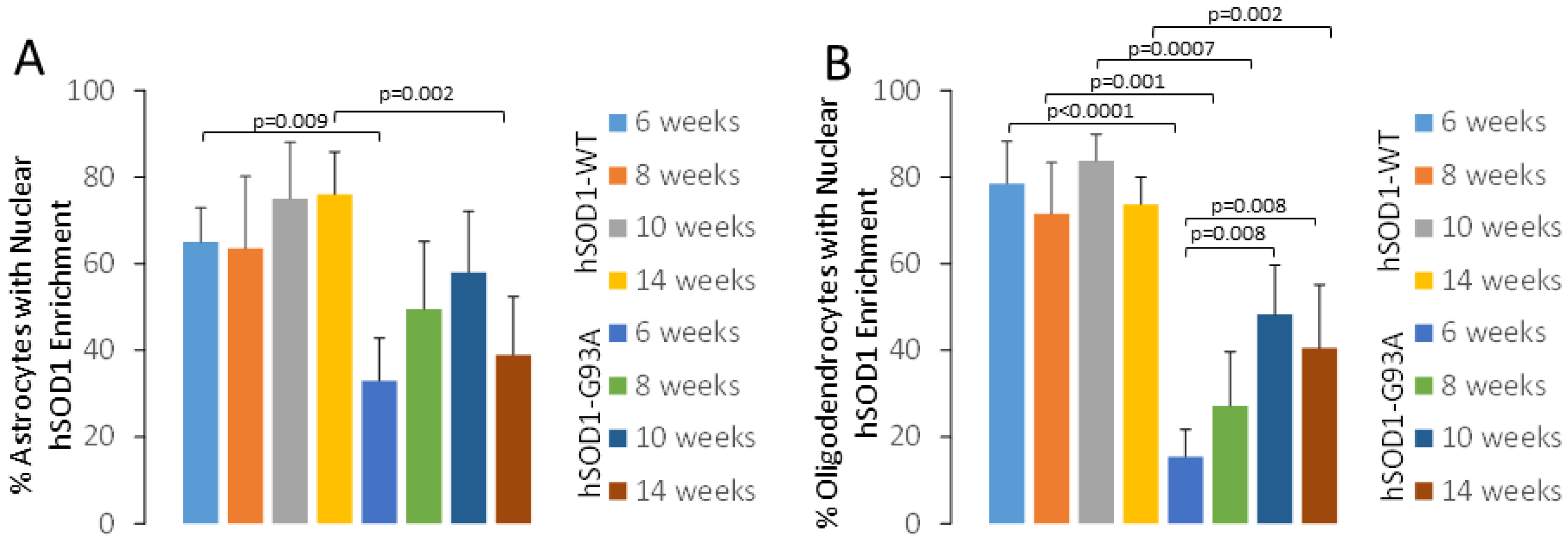
2.1. hSOD1 Nuclear Localization in Tg Mouse Spinal Cord Is Cell-Type Differential and Disease-Related
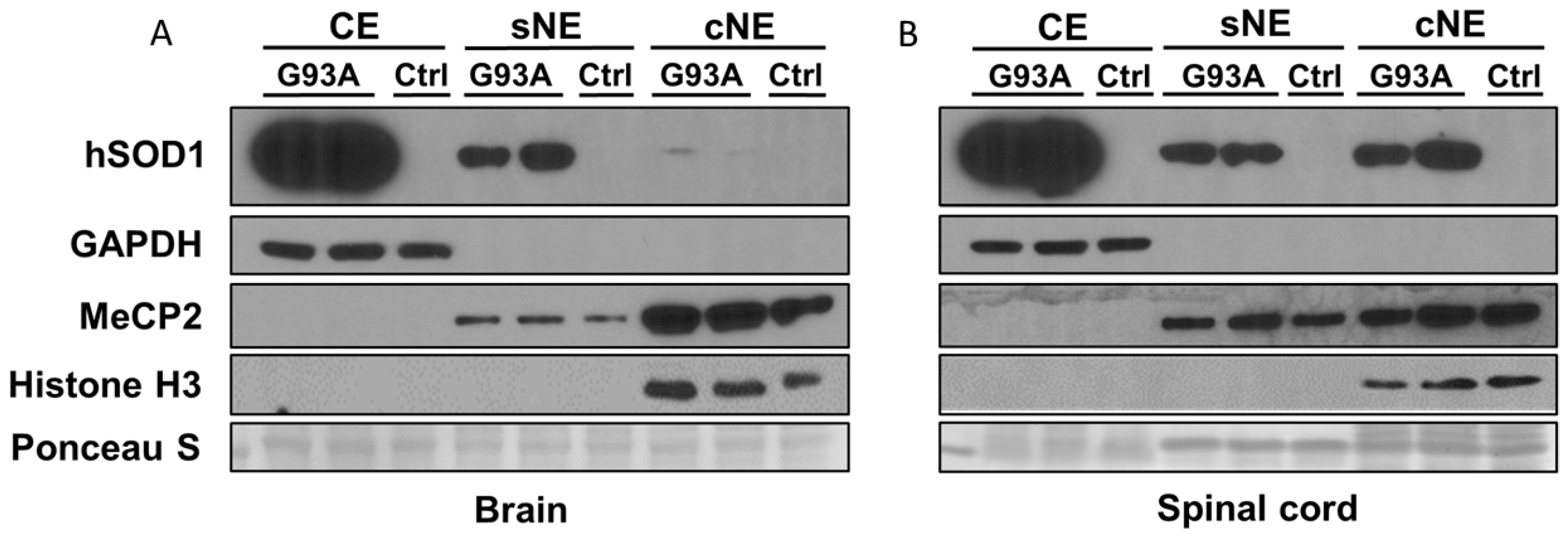
2.2. Subcellular Fractionation of Brain and Spinal Cord of hSOD1-G93A Mice Shows Differential Enrichment of hSOD1 in Nuclear Chromatin Extracts
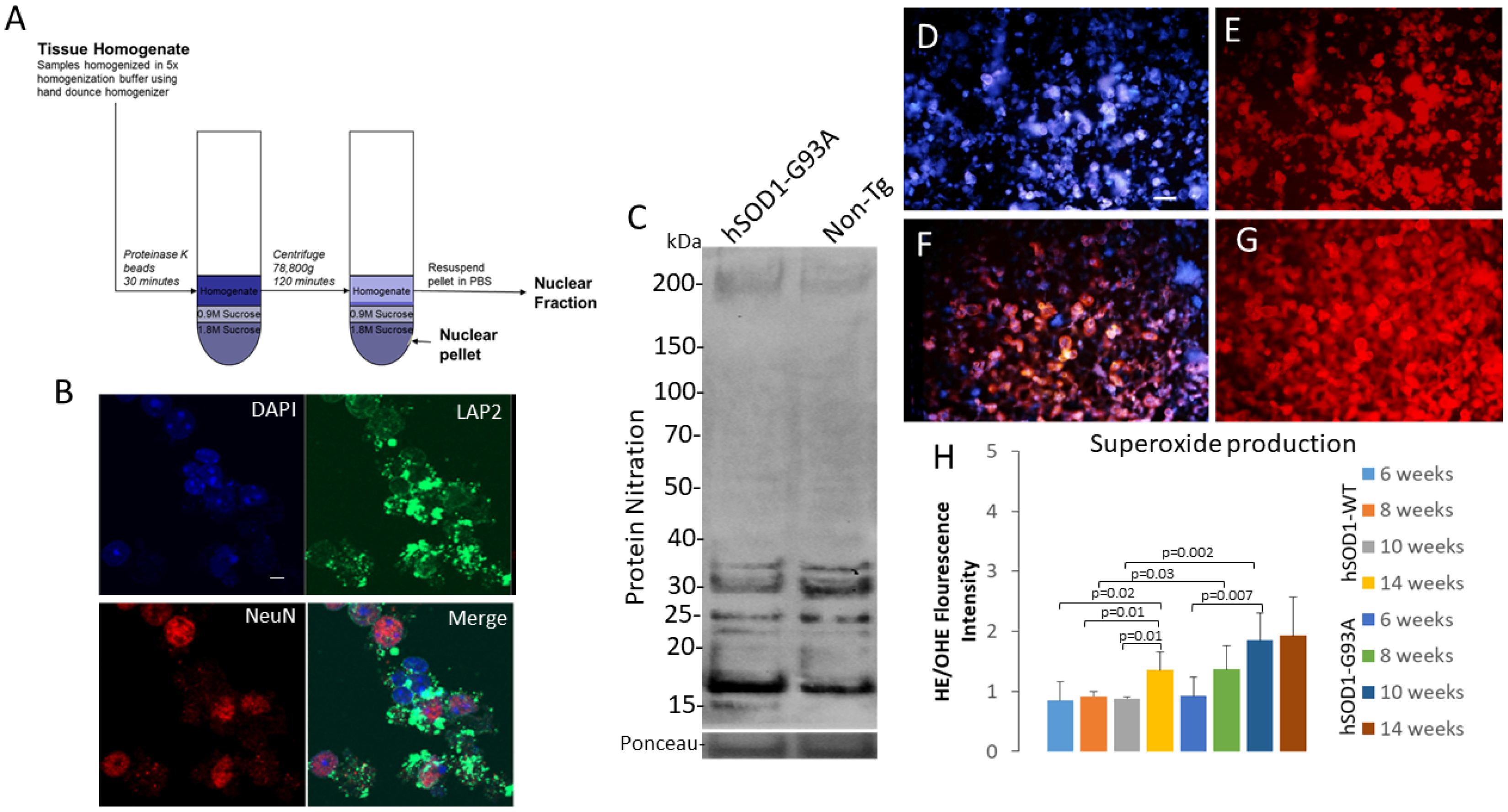
2.3. Isolation of an Intact Nuclear Fraction of Spinal Cord for Acute Live-Nucleus Analyses of Oxidative Stress and DNA Damage
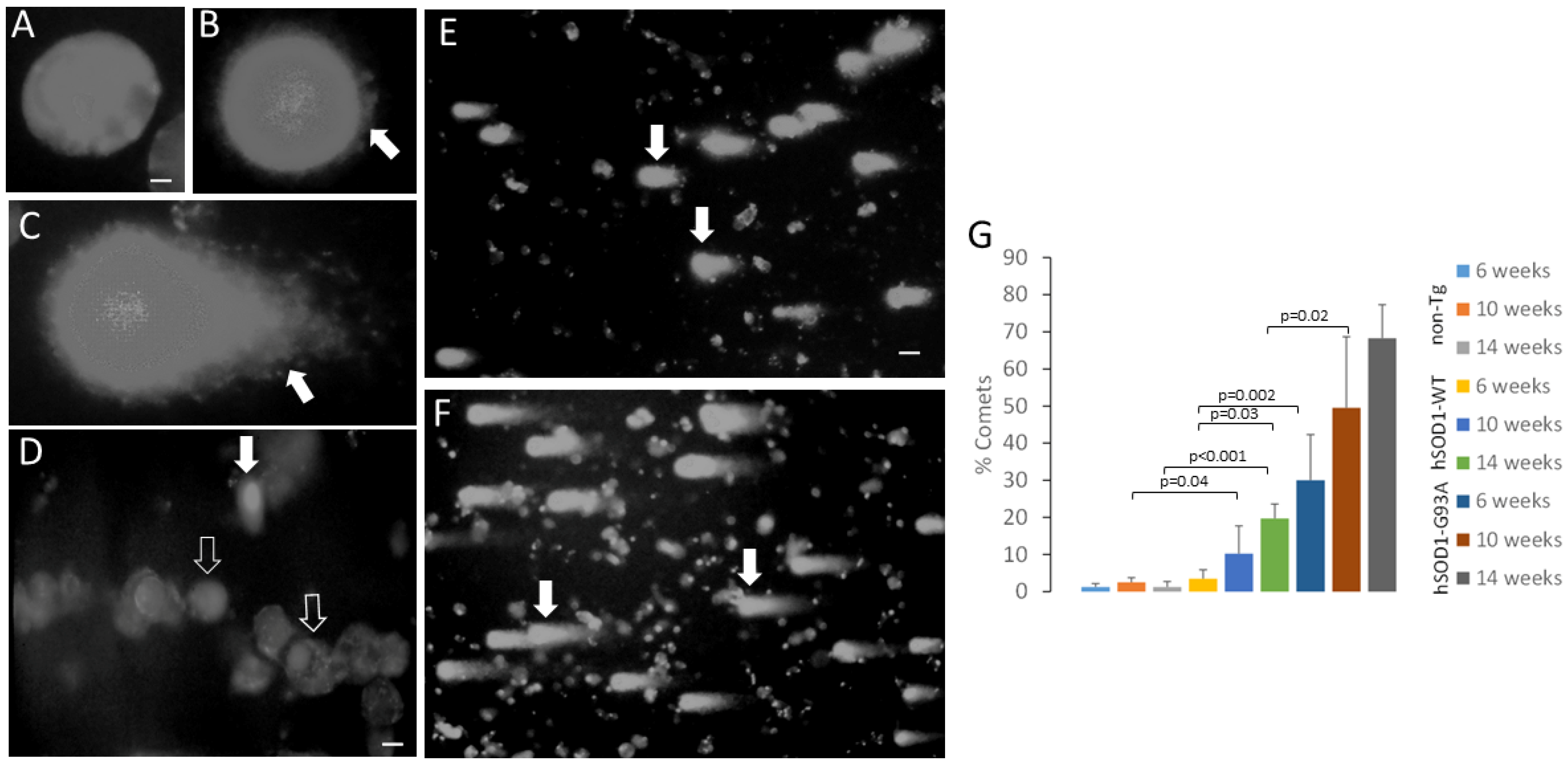
2.4. MNs from Spinal Cord of Tg Mice Have hSOD1 Variant-Related Accumulation of DNA Single-Strand Breaks
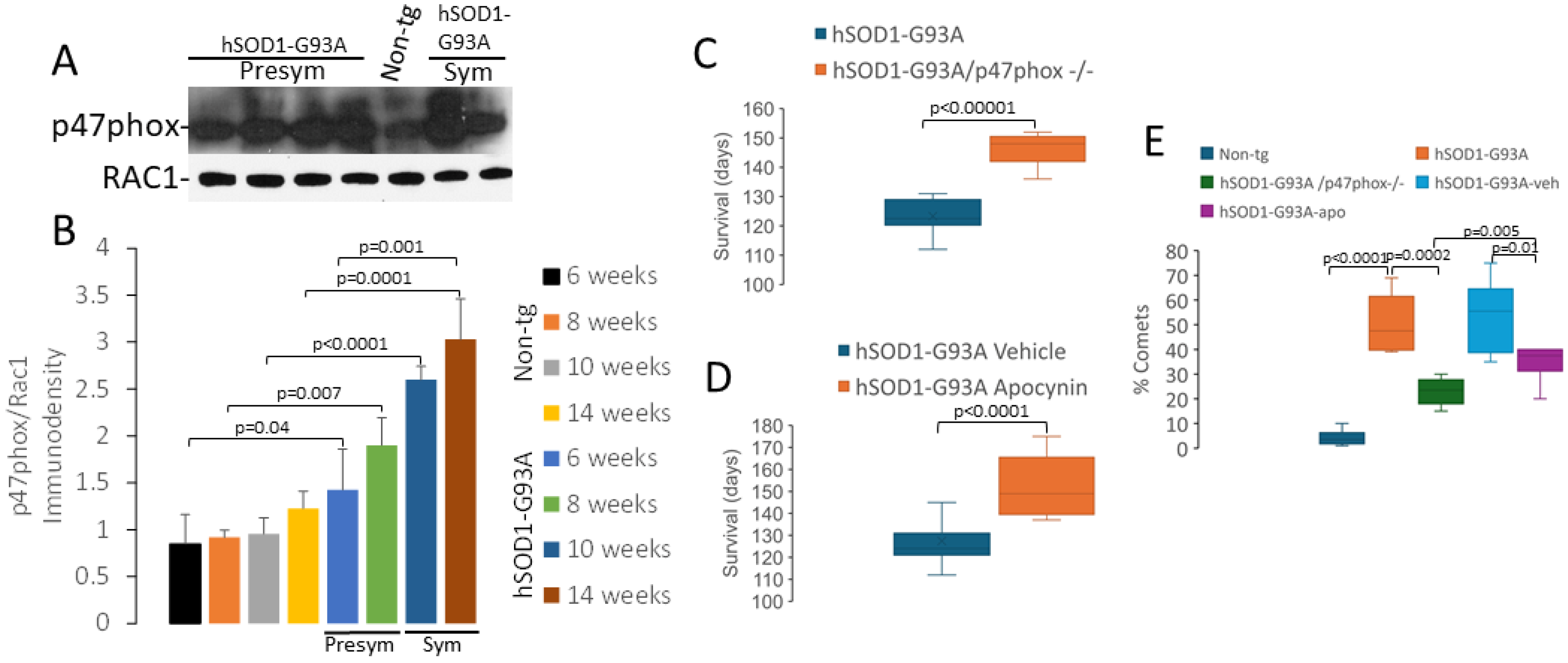
2.5. NADPH Oxidase Has a Role in Disease in hSOD1-G93A Mice
2.6. SOD1 Localizes the Nucleus in MNs and Glia in Human CNS: Nuclear Presence Increases in ALS Upper and Lower MNs
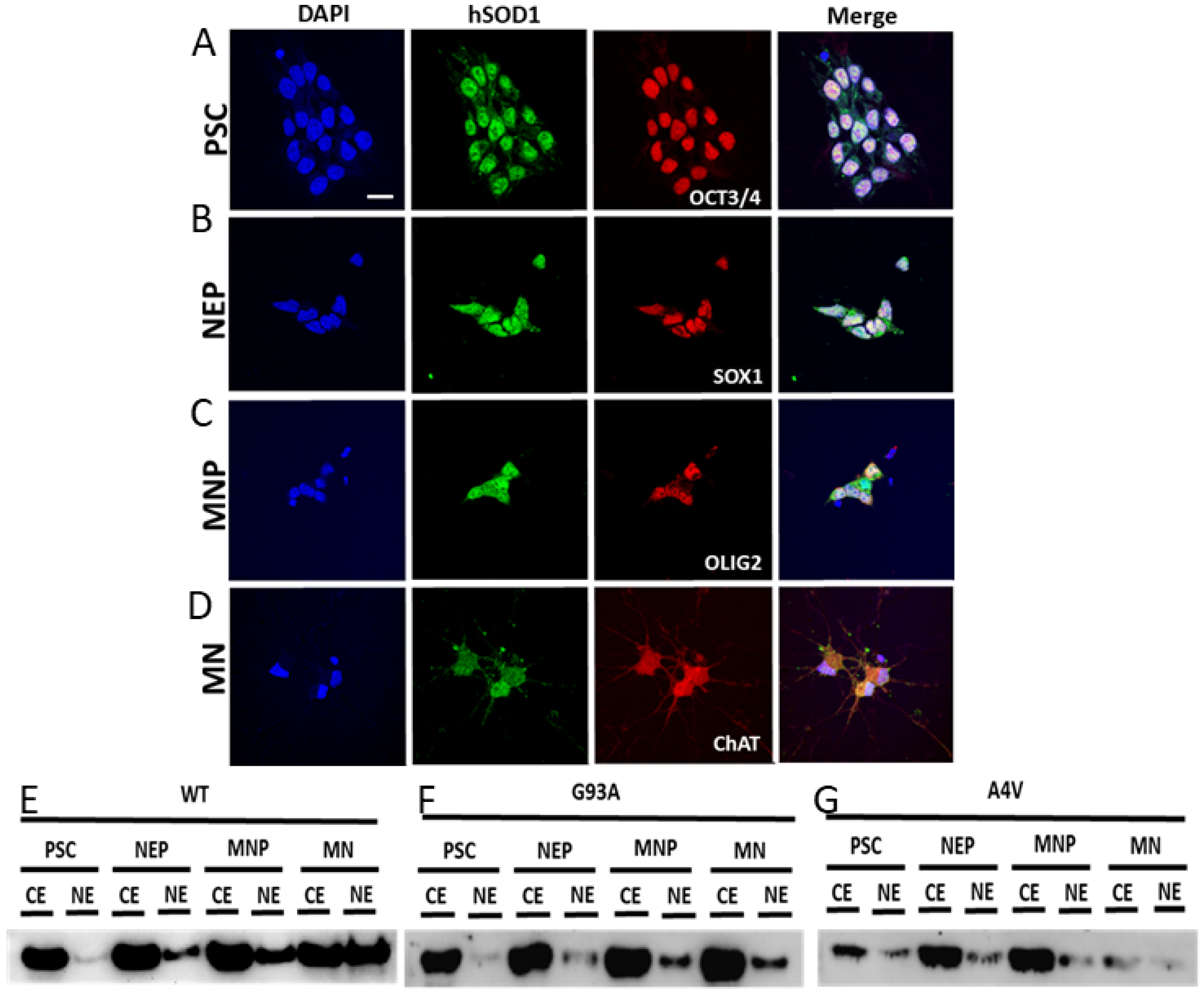
2.7. Nuclear SOD1 Is Present in Human iPS Cells at Different Stages of Directed Differentiation to MNs, but Familial Mutant SOD1 ALS MNs Have Aberrant Subcellular Compartmentation of SOD1
3. Discussion
3.1. hSOD1-G93A MNs with Nuclear SOD1 Lose Their Cholinergic Phenotype of the Nucleus
3.2. Spinal Cord Oligodendrocytes in hSOD1-G93A Mice Adopt a Nuclear hSOD1 Phenotype during Disease
3.3. Oxidative Stress within the Nucleus and DNA Damage Occur in MNs in hSOD1-G93A Mouse Spinal Cord
3.4. NADPH Oxidase Is Involved in the Disease Process in hSOD1-G93A Mice
3.5. SOD1 Localizes to the Nucleus of MNs in Human ALS Brains and Spinal Cords and in Patient iPS Cell-Derived MNs in Cell Culture
4. Materials and Methods
4.1. Tg Mice
4.2. Immunohistochemistry on Mouse Spinal Cord
4.3. Preparation of Spinal Cord Tissue for Isolation of MNs and Cell Nuclei
4.4. Preparation of MN-Enriched Cell Suspensions and MN Nuclei
4.5. Preparation of Brain and Spinal Cord Subcellular Fractions
4.6. Immunoblotting
4.7. Comet Assay
4.8. NCF1 Gene Knockout Experiment
4.9. Pharmacological Intervention Experiment with Apocynin
4.10. Human Tissues
4.11. Human iPS Cells and Directed Differentiation
4.12. Quantitative and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 2015, 33, 855–876. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.S.; Guarnieri, M.; Xu, Z.S.; Wong, P.C.; Brown, R.H., Jr.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar] [CrossRef]
- Yim, M.B.; Kang, J.H.; Yim, H.S.; Kwak, H.S.; Chock, P.B.; Stadtman, E.R. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1996, 93, 5709–5714. [Google Scholar] [CrossRef]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Rouleau, G.A. Oxidized/misfolded superoxide dismutase-1: The cause of all amyotrophic lateral sclerosis? Ann. Neurol. 2007, 62, 553–559. [Google Scholar] [CrossRef]
- Rotunno, M.S.; Bosco, D.A. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 2013, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in amyotrophic lateral sclerosis: “Ambivalent” behavior connected to the disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar] [CrossRef]
- Bendotti, C.; Carrì, M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004, 10, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: Translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef]
- Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA methylation occur in spinal cord and skeletal muscle of human SOD1 mouse models of ALS and in human ALS: Targeting DNA methylation is therapeutic. Cells 2022, 11, 3448. [Google Scholar] [CrossRef]
- Raoul, C.; Estévez, A.G.; Nishimune, H.; Cleveland, D.W.; deLapeyrière, O.; Henderson, C.E.; Haase, G.; Pettmann, B. Motoneuron death triggered by a specific pathway downstream of Fas: Potentiation by ALS-linked SOD1 mutations. Neuron 2002, 35, 1067–1083. [Google Scholar] [CrossRef]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, M.M.; Barbeito, L.; Beckman, J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 1999, 286, 2498–2500. [Google Scholar] [CrossRef]
- Martin, L.J.; Kaiser, A.; Price, A.C. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J. Neurobiol. 1999, 40, 185–201. [Google Scholar] [CrossRef]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 2010, 214, 219–234. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2006, 65, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Beal, M.F.; Manfredi, G. The role of mitochondria in inherited neurodegenerative diseases. J. Neurochem. 2006, 97, 1659–1675. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Matveeva, L.A.; Belosludtsev, K.N. Mitochondrial dyshomeostasis as an early hallmark and a therapeutic target in amyotrophic lateral sclerosis. Intl. J. Mol. Sci. 2023, 24, 16833. [Google Scholar] [CrossRef]
- Baev, A.Y.; Vinokurov, A.Y.; Potapova, E.V.; Dunaev, A.V.; Angelova, P.R.; Abramov, A.Y. Mitochondrial permeability transition, cell death and neurodegeneration. Cells 2024, 13, 648. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Ito, H.; Hirano, A.; Fujita, K.; Wate, R.; Nakamura, M.; Kaneko, S.; Nakano, S.; Kusaka, H. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 1184–1192. [Google Scholar] [CrossRef]
- Zhang, J.; Ito, H.; Wate, R.; Ohnishi, S.; Nakano, S.; Kusaka, H. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006, 112, 673–680. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Seilhean, D.; Takahashi, J.; El Hachimi, K.H.; Fujigasaki, H.; Lebre, A.S.; Biancalana, V.; Dürr, A.; Salachas, F.; Hogenhuis, J.; de Thé, H.; et al. Amyotrophic lateral sclerosis with neuronal intranuclear protein inclusions. Acta Neuropathol. 2004, 108, 81–87. [Google Scholar] [CrossRef]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef]
- Ito, D.; Seki, M.; Tsunoda, Y.; Uchiyama, H.; Suzuki, N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 2011, 69, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. Endogenous oxidative DNA damage, aging, and cancer. Free Rad. Res. Comm. 1989, 7, 121–128. [Google Scholar] [CrossRef]
- Fitzmaurice, P.S.; Shaw, I.C.; Kleiner, H.E.; Miller, R.T.; Monks, T.J.; Lau, S.S.; Mitchell, J.D.; Lynch, P.G. Evidence for DNA damage in amyotrophic lateral sclerosis. Muscle Nerve 1996, 19, 797–798. [Google Scholar]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7 Pt B, 613–622. [Google Scholar] [CrossRef]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Comm. 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromol. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef]
- Kisby, G.E.; Milne, J.; Sweatt, C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport 1997, 8, 1337–1340. [Google Scholar] [CrossRef]
- Aguirre, N.; Beal, M.F.; Matson, W.R.; Bogdanov, M.B. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Rad. Res. 2005, 39, 383–388. [Google Scholar] [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Gen. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.G.; Krasin, F. A new hypothesis of the etiology of amyotrophic lateral sclerosis. The DNA hypothesis. Arch. Neurol. 1982, 39, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Kisby, G.E.; Kabel, H.; Hugon, J.; Spencer, P. Damage and repair of nerve cell DNA in toxic stress. Drug Metabol. Rev. 1999, 31, 589–618. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Rad. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal cell death in nervous system development, disease, and injury. Intl. J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [CrossRef]
- Gertz, B.; Wong, M.; Martin, L.J. Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice. J. Neuropathol. Exp. Neurol. 2012, 71, 162–177. [Google Scholar] [CrossRef]
- Bordoni, M.; Pansarasa, O.; Dell’Orco, M.; Crippa, V.; Gagliardi, S.; Sproviero, D.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Tedeschi, G.; et al. Nuclear phospho-SOD1 protects DNA from oxidative stress damage in amyotrophic lateral sclerosis. J. Clin. Med. 2019, 8, 729. [Google Scholar] [CrossRef]
- Brasil, A.A.; Magalhães, R.S.S.; De Carvalho, M.D.C.; Paiva, I.; Gerhardt, E.; Pereira, M.D.; Outeiro, T.F.; Eleutherio, E.C.A. Implications of fALS mutations on Sod1 function and oligomerization in cell models. Mol. Neurobiol. 2018, 55, 5269–5281. [Google Scholar] [CrossRef]
- Kim, B.W.; Ryu, J.; Jeong, Y.E.; Kim, J.; Martin, L.J. Human motor neurons with SOD1-G93A mutation generated from CRISPR/Cas9 gene-edited iPSCs develop pathological features of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2020, 14, 604171. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Gen. 2010, 19, 2284–2302. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Skeletal muscle-restricted expression of human SOD1 in transgenic mice causes a fatal ALS-like syndrome. Front. Neurol. 2020, 11, 592851. [Google Scholar] [CrossRef]
- Liu, Z.; Martin, L.J. Isolation of mature spinal motor neurons and single-cell analysis using the comet assay of early low-level DNA damage induced in vitro and in vivo. J. Histochem. Cytochem. 2001, 49, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Martin, L.J. Motor neurons rapidly accumulate DNA single-strand breaks after in vitro exposure to nitric oxide and peroxynitrite and in vivo axotomy. J. Comp. Neurol. 2001, 432, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Dis. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.P. Mitochondria in amyotrophic lateral sclerosis: A trigger and a target. Neurodegen. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef]
- Sorce, S.; Stocker, R.; Seredenina, T.; Holmdahl, R.; Aguzzi, A.; Chio, A.; Depaulis, A.; Heitz, F.; Olofsson, P.; Olsson, T.; et al. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Rad. Biol. Med. 2017, 112, 387–396. [Google Scholar] [CrossRef]
- Moloney, J.N.; Jayavelu, A.K.; Stanicka, J.; Roche, S.L.; O’Brien, R.L.; Scholl, S.; Böhmer, F.D.; Cotter, T.G. Nuclear membrane-localised NOX4D generates pro-survival ROS in FLT3-ITD-expressing AML. Oncotarget 2017, 8, 105440–105457. [Google Scholar] [CrossRef]
- Trumbull, K.A.; McAllister, D.; Gandelman, M.M.; Fung, W.Y.; Lew, T.; Brennan, L.; Lopez, N.; Morré, J.; Kalyanaraman, B.; Beckman, J.S. Diapocynin and apocynin administration fails to significantly extend survival in G93A SOD1 ALS mice. Neurobiol. Dis. 2012, 45, 137–144. [Google Scholar] [CrossRef]
- Dale, H. Pharmacology and nerve-endings. Proc. Royal Soc. Med. 1935, 28, 319–332. [Google Scholar] [CrossRef]
- Resendes, M.C.; Dobransky, T.; Ferguson, S.S.; Rylett, R.J. Nuclear localization of the 82-kDa form of human choline acetyltransferase. J. Biol. Chem. 1999, 274, 19417–19421. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.K.; Ishak, M.; Dobransky, T.; Haroutunian, V.; Davis, K.L.; Rylett, R.J. 82-kDa choline acetyltransferase is in nuclei of cholinergic neurons in human CNS and altered in aging and Alzheimer disease. Neurobiol. Aging 2007, 28, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Winick-Ng, W.; Caetano, F.A.; Winick-Ng, J.; Morey, T.M.; Heit, B.; Rylett, R.J. 82-kDa choline acetyltransferase and SATB1 localize to β-amyloid induced matrix attachment regions. Sci. Rep. 2016, 6, 23914. [Google Scholar] [CrossRef] [PubMed]
- Stieber, A.; Gonatas, J.O.; Gonatas, N.K. Aggregates of mutant protein appear progressively in dendrites, in periaxonal processes of oligodendrocytes, and in neuronal and astrocytic perikarya of mice expressing the SOD1(G93A) mutation of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 2000, 177, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free radicals, lipid peroxidation, and cell damage. Lancet 1984, 2, 1095. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Kanski, J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Devel. 2001, 122, 945–962. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Chiu, A.Y.; Zhai, P.; Dal Canto, M.C.; Peters, T.M.; Kwon, Y.W.; Prattis, S.M.; Gurney, M.E. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 1995, 6, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Figlewicz, D.A.; Bohn, M.C. Selective loss of alpha motoneurons innervating the medial gastrocnemius muscle in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 1998, 150, 329–336. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271 Pt 1, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Carson, M.; Smith, C.D.; Koppenol, W.H. ALS, SOD and peroxynitrite. Nature 1993, 364, 584. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef]
- Martin, L.J.; Chen, K.; Liu, Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J. Neurosci. 2005, 25, 6449–6459. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Enforced DNA repair enzymes rescue neurons from apoptosis induced by target deprivation and axotomy in mouse models of neurodegeneration. Mech. Ageing Devel. 2017, 161 Pt A, 149–162. [Google Scholar] [CrossRef]
- Shadfar, S.; Parakh, S.; Jamali, M.S.; Atkin, J.D. Redox dysregulation as a driver for DNA damage and its relationship to neurodegenerative diseases. Transl. Neurodegr. 2023, 12, 18. [Google Scholar] [CrossRef]
- Wu, D.C.; Ré, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. he inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef]
- Soubannier, V.; Chaineau, M.; Gursu, L.; Lépine, S.; Kalaydjian, D.; Sirois, J.; Haghi, G.; Rouleau, G.; Durcan, T.M.; Stifani, S. Early nuclear phenotypes and reactive transformation in human iPSC-derived astrocytes from ALS patients with SOD1 mutations. Glia 2024. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef]
- Barbosa, L.F.; Cerqueira, F.M.; Macedo, A.F.; Garcia, C.C.; Angeli, J.P.; Schumacher, R.I.; Sogayar, M.C.; Augusto, O.; Carrì, M.T.; Di Mascio, P.; et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta 2010, 1802, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Liguori, F.; Alberti, F.; Amadio, S.; Angelini, D.F.; Pilesi, E.; Vitale, G.; Tesoriere, G.; Borsellino, G.; Vernì, F.; Volonté, C. Pan-neuronal expression of human mutant SOD1 in Drosophila impairs survival and motor performance, induces early neuroinflammation and chromosome aberrations. Biochim. Biophys. Acta 2024, 1870, 167192. [Google Scholar] [CrossRef]
- Tanigami, H.; Rebel, A.; Martin, L.J.; Chen, T.Y.; Brusilow, S.W.; Traystman, R.J.; Koehler, R.C. Effect of glutamine synthetase inhibition on astrocyte swelling and altered astroglial protein expression during hyperammonemia in rats. Neuroscience 2005, 131, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Liu, D.; Jiang, D.; Kulikowicz, E.; Tekes, A.; Liu, P.; Qin, Q.; Koehler, R.C.; Aggarwal, M.; Zhang, J.; et al. Fractional anisotropy from diffusion tensor imaging correlates with acute astrocyte and myelin swelling in neonatal swine models of excitotoxic and hypoxic-ischemic brain injury. J. Comp. Neurol. 2021, 529, 2750–2770. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Case Number | Age (Years)/Sex | Postmortem Delay (Hours) | Cause of Death |
|---|---|---|---|---|
| Controls (Neurological Disease Free) | 487 | 73/male | 22 | Pancreatic cancer |
| 515 | 62/male | 21 | Aortic aneurysm | |
| 712 | 44/female | 20 | Pneumonia | |
| 719 | 66/male | 10 | Myocardial infarction | |
| 961 | 59/female | 6 | Myocardial infarction | |
| 993 | 66/male | 12 | Prostatic carcinoma | |
| 1277 | 80/female | 8 | Lymphoma | |
| 1344 | 53/male | 12 | Metastatic carcinoma | |
| 1348 | 44/male | 18 | Lymphoma | |
| 1361 | 49/female | 15 | Thromboembolic disease | |
| 1517 | 71/female | 16 | Heart disease | |
| 1591 | 94/male | 16 | Pneumonia | |
| 1603 | 89/male | 16 | Pulmonary embolism | |
| 1613 | 74/male | 4 | Myocardial infarction | |
| 1683 | 91/female | 8 | Cardiomyopathy | |
| ALS | 345 | 59/female | 3 | Respiratory arrest |
| 414 | 65/male | 4 | Respiratory arrest | |
| 433 | 71/male | 17 | Respiratory arrest | |
| 447 | 69/female | 15 | Respiratory arrest | |
| 492 | 68/female | 18 | Respiratory arrest | |
| 834F | 46/male | 3 | Respiratory arrest | |
| 875 | 70/female | 24 | Respiratory arrest | |
| 950F | 38/male | 22 | Respiratory arrest | |
| 1014 | 72/male | 5 | Respiratory arrest | |
| 1088 | 66/male | 7 | Respiratory arrest | |
| 1108 | 64/female | 8 | Respiratory arrest | |
| 1151 | 57/female | 14 | Respiratory arrest | |
| 1161 | 47/male | 6 | Pneumonia | |
| 1169 | 67/female | 15 | Respiratory arrest | |
| 1176F | 27/male | 6 | Respiratory arrest | |
| 1359 | 61/female | 14 | Respiratory arrest | |
| 1365 | 59/male | 7 | Respiratory arrest | |
| 1386 | 69/female | 15 | Pneumonia | |
| 1413 | 79/male | 10 | Respiratory arrest | |
| 1452 | 65/female | 6 | Respiratory arrest | |
| 1453 | 60/male | 22 | Pneumonia | |
| 1485 | 61/female | 5 | Pneumonia | |
| 1589 | 55/male | 10 | Pneumonia | |
| 1614 | 69/male | 18 | Respiratory arrest | |
| 1620 | 63/male | 10 | Respiratory arrest | |
| 1623 | 71/female | 59 | Cardiopulmonary arrest | |
| 1629 | 55/female | 12 | Pneumonia | |
| 1635 | 68/female | 5 | Respiratory arrest | |
| 1668 | 76/male | 7 | Respiratory arrest | |
| 1693F | 42/male | 6 | Respiratory arrest | |
| 1713 | 54/female | 14 | Respiratory arrest | |
| 1742 | 55/male | 5 | Pneumonia | |
| 1755 | 72/female | 5 | Respiratory arrest | |
| 1789F-SOD1A4V | 55/male | 14 | Respiratory arrest |
| iPSC Lines (Clone) 1 | Gene | Mutation | Age of Donor | Gender |
|---|---|---|---|---|
| C3-1 | Control | N/A | 40 | F |
| C3-1 | SOD1 | G93A | 40 | F |
| GO013 | SOD1 | A4V | 63 | F |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, L.J.; Koh, S.J.; Price, A.; Park, D.; Kim, B.W. Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage. Int. J. Mol. Sci. 2024, 25, 9106. https://doi.org/10.3390/ijms25169106
Martin LJ, Koh SJ, Price A, Park D, Kim BW. Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage. International Journal of Molecular Sciences. 2024; 25(16):9106. https://doi.org/10.3390/ijms25169106
Chicago/Turabian StyleMartin, Lee J., Shannon J. Koh, Antionette Price, Dongseok Park, and Byung Woo Kim. 2024. "Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage" International Journal of Molecular Sciences 25, no. 16: 9106. https://doi.org/10.3390/ijms25169106
APA StyleMartin, L. J., Koh, S. J., Price, A., Park, D., & Kim, B. W. (2024). Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage. International Journal of Molecular Sciences, 25(16), 9106. https://doi.org/10.3390/ijms25169106