
Molecular Aspects in the Development of Type 2 Diabetes and Possible Preventive and Complementary Therapies
 ,
,
Abstract
:1. Introduction
1.1. History of Diabetes
1.2. Type 2 Diabetes Mellitus
2. Metabolic Syndrome (MetS)
2.1. Hypertension
2.2. Dyslipidemia
2.3. Abdominal Obesity
2.4. Insulin Action and Insulin Resistance
2.4.1. Normal Function of Insulin
2.4.2. Insulin Pathway
Mitogenic Pathway
Metabolic Pathway
- The glycogen synthase kinase 3 (GSK3) is one of the Akt substrates in the cytosol. This is responsible for the regulation of glycogen synthase (GS), and the eukaryotic initiation factor 2B (eIF2B), among others. Inactivation of GSK3 by Akt activates GS, which then induces glycogenesis [62], as well as the eIF2B, which then enhances the cell growth (Figure 1B) [63].
- Forkhead transcription factors (FOXOs) are another targets of Akt. FOXOs acting in glucose metabolism increase the gene expression of key enzymes of gluconeogenesis, such as glucose-6-phosphatase and phospho-enol-pyruvate carboxykinase (PEPCK) [64]. When FOXOs are inactivated through phosphorylation by Akt, they exit the nucleus and are degraded by proteasomes; thereby Akt inhibit gluconeogenesis (Figure 1B) [65]. FOXOs also control gene expression that regulates the cell cycle, apoptosis, oxidative stress resistance, differentiation, and muscle atrophy as well as energy homeostasis. Thus, this can induce apoptosis through activation of FasL and Bim, and can promote cell-cycle arrest and stress resistance by induction of catalase and manganese superoxide dismutase (MnSOD) for inactivation of reactive oxygen species (ROS) as well as by facilitating DNA repair [66].
- The GTPase-activating protein Akt substrate of 160 kDa (AS160), also known as TBC1D4, is also an Akt substrate [67]. It is attached to vesicles of glucose transporter 4 (GLUT4)—an insulin-dependent transporter in muscle and adipose tissues [68]—and inhibits the efflux, and translocation of these vesicles from the Golgi to the plasma membrane through inactivation of Rab protein, thus restraining GLUT4 exocytosis (Figure 1B) [69]. When AS160 activity is blocked by Akt via phosphorylation, AS160 detaches from the GLUT4 vesicles and increases their efflux and exocytosis (Figure 1B) [70]. However, insulin-stimulated phosphorylation of AS160 impaired the skeletal muscle in T2DM [23]. Similarly to AS160, the AS160 paralog TBC1D1, which is highly expressed in skeletal muscle, is also phosphorylated by insulin, as well as by exercise and AMP kinase (AMPK); moreover, AMPK is suggested as the most robust regulator of this signaling molecule [23,71,72]. GLUT4 exocytosis can be also stimulated by PDK1 through PKCλ phosphorylation. PKCλ then stimulates Rab4 activity, which initiates GLUT4 exocytosis [73]. Additionally, TC10 lipid raft microdomain is also associated with and promotes GLUT4 fusion to the plasma membrane lipid rafts [74].
- Akt activates mammalian target of rapamycin complex 1 (mTORC1) by inhibiting tuberous sclerosis complex 2 (TSC2), which forms heterodimer with tuberous sclerosis complex 1 (TSC1) (Figure 1B) [77]. TSC2 is a GTPase-activating protein (GAP) for Ras homologue enriched in brain (Rheb) protein. When inhibition of Rheb is blocked by inactivated TSC complex, mTORC1 become activated, and then throughout the activation of p70 ribosomal protein S6 kinase (S6K1) and inhibition of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), the protein synthesis is enhanced (Figure 1B) [78].
- Akt, mTOR, and PKC λ/ζ can activate sterol regulatory binding proteins (SREBPs) to enhance lipid synthesis and regulate cholesterol homeostasis (Figure 1B). SREBPs are localized in the ER membrane and bind to the sterol cleavage protein (SCA) and sterol regulatory element (SRE). At low sterol levels, this complex is transported to the Golgi, where the transcription factor is deliberated by proteases. After translocation to the nucleus, genes of lipid synthesis will be induced (Figure 1B) [81,82].
2.4.3. Insulin Resistance
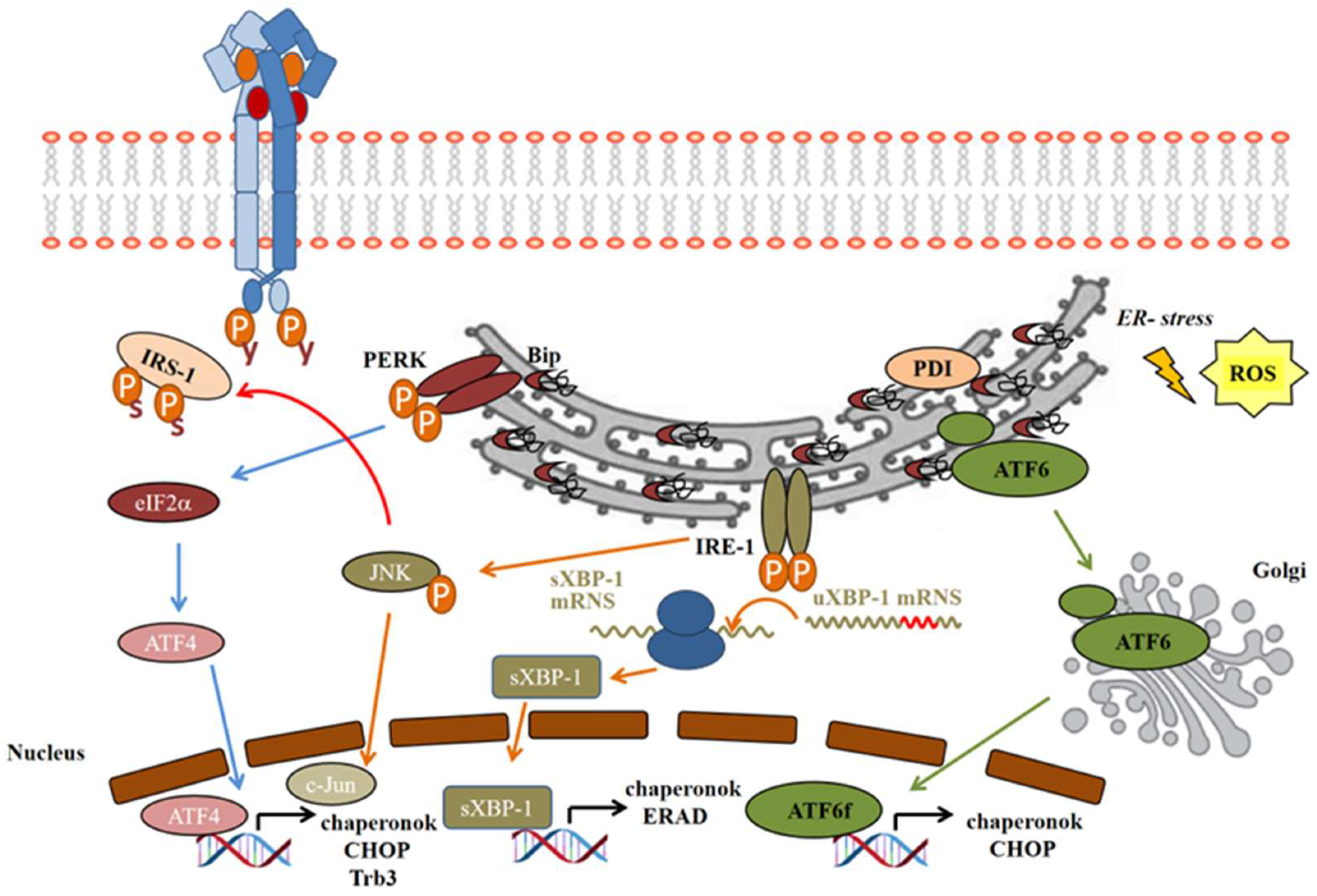
Endoplasmic Reticulum (ER) Stress
- On the IRE-1 pathway, activating phosphorylation of IRE-1α leads to the recruitment of tumor necrosis factor receptor-associated factor 2 (TRAF2), which then activates apoptosis signal-regulating kinase 1 (ASK1) and the stress-activated c-Jun N-terminal kinase (JNK) [111]. JNK then inactivates the insulin signaling by serine phosphorylation of IRS-1 (Figure 3), as well as inducing apoptosis, while inhibiting autophagy [103]. IRE-1α has endonuclease activity as well, which cleaves 26 base–pair sequences from the mRNA of X-box binding protein-1 (XBP1). This spliced variant sXBP-1 is facilitating the transcription of chaperons to enhance the folding capacity of ER. The misfolded proteins are removed from the ER, ubiquitinated, and transferred to proteasomal degradation; this process is called endoplasmic reticulum-associated degradation (ERAD). sXBP-1 also induces factors involved in ERAD to facilitate damaged protein clearance from ER lumen [109,112].
- The PERK pathway: after PERK activation by autophosphorylation, it subsequently phosphorylates the eukaryotic translation initiation factor 2α (eIF2α), generally inhibiting the translation of the proteins. However, some proteins will be translated properly in this unusual translational condition, such as the activating transcription factor 4 (ATF4), a member of cAMP-responsive element-binding protein (CREB) family [113], which induces the transcription of ER chaperons and the C/EBP homologous protein (CHOP), or, by another name, growth-arrest and DNA-damage-inducible protein (GADD153) (Figure 3) [23].
- In normal conditions, ATF6 occurs in its monomer, dimer, and oligomer forms in the ER membrane. Upon ER stress, it is reduced and forms monomers, which translocate to the Golgi apparatus, where it is activated by the cleavage of serine protease site-1 protease (S1P) and metalloprotease site 2 protease (S2P) [114]. Only the reduced monomer form can reach the Golgi and be released as an activated cytosolic fragment (ATF6f), which then enters the nucleus and enhances transcription of ER chaperons and CHOP (Figure 3) [110].
Inducing IR by Ser/Thr Phosphorylation of the IRS-1
IR by Protein Tyrosine Phosphatase 1B (PTP1B)
IR by Lipid Phosphatases—SHIP and PTEN
Role of Phosphoprotein Phosphatases in IR
Role of Adapter Proteins in IR
3. Molecular Targets in Insulin Signaling/Resistance in Preventive, Complementary, or Drug Treatment Strategies
3.1. Preventive or Complementary Treatment Options
3.1.1. Diet and Supplementation

{kind=link}
{kind=link}
{kind=link}
| Diet/Supplementation | Physiological and Cellular Effect(s) on T2DM/IR | Cellular and Molecular Target(s) | Reference Number |
|---|---|---|---|
| phytochemicals | |||
| polyphenols | •decreased inflammation and mitogenic processes | •inactivated NFκB, MAPK and arachidonic acids pathways | [155] |
| resveratrol | •diminish IR, decreased FFA, lipotoxycity and inflammation | •activates SIRT1 and AMPK; these activate PPARα and inhibit PPARγ, SREBP1c and NFκB, decrease TNFα and IL-6, management of AGEs | [19,20,104] [156] |
| support gluconeogenesis/glucose uptake, ER stress- resistance, apoptosis | by activating FOXOs, increasing GLUT4, inhibiting mTORC1 | ||
| carotenoid | |||
| astaxanthin | •attenuates IR, glucose intolerance, enhance exercise tolerance/FFA metabolism, stimulates anti-inflammatory effects | •oxidative phosphorylation and biogenesis through AMPK, through its antioxidant properties | [157] |
| lycopene | •promote angiogenesis for utilization of glucose and energy metabolism improves IR and vascular aging | •reactivation of SIRT1 | [158] |
| phytosterols | •decrease the absorption of cholesterol, LDL cholesterol, promote insulin action through its receptors | •normalizing lipid metabolism increased GLUT4 translocation to plasma membrane | [159] |
| inhibit mitochondrial dysfunction and ROS production | inhibit TNFα induced IKKβ/NFκB and JNK signaling | ||
| phytosterols & saponins | •hypolipidemic and angioprotective effects | [159] | |
| micronutrients | |||
| vitamin D3 | •increases glucose uptake inhibit oxidative stress | •inducing insulin-independent SIRT1/AMPK/IRS1/GLUT4 signaling pathway | [160] [161] |
| optimal glucose and lipid homeostasis | adiponectin, AMPK | [20] | |
| selenium | •necessary for normal inulin action, prevent IR | •support glucagon-like peptid receptor expression in beta cells necessary in the phosphorylation of insulin receptor | [162] |
| Zn&Mg | •necessary for normal inulin action, prevent IR | •necessary for autophosphorylation of insulin receptor, activation of PI3K and Akt and appropriate GLUT4 translocation to plasma membrane | [163,164] |
| lipids | |||
| n-3 PUFAs | •increase β-oxidation of TGs and protein synthesis in skeletal muscle | •activates mTORC1 in skeletal muscle | [165] |
| decrease lipogenesis in liver | inhibit diacylglycerol and SREBP1c | ||
| decrease lipogenesis in adipose tissue | activate AMPK, β-oxidation, increase adiponectin level | ||
| other natural compounds | improve IR and support glucose utilization | [166] |
3.1.2. Physical Activity and Exercise
Systemic Effects of Physical Activity: Inflammation
Systemic Effects of Physical Activity: Hormone Secretion
Tissue-Specific Effects of Physical Activity and Exercise
3.2. Targets of Currently Used Drugs in Treatments of T2DM and IR
4. In Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4E-BP1 | eukaryotic translation initiation factor 4E-binding protein 1. |
| AGEs | glycation end-products. |
| AMPK | AMP-kinase. |
| AS160 (TBC1D4) | Akt substrate of 160 kDa. |
| ASK1 | apoptosis signal-regulating kinase 1. |
| ATF4 | activating transcription factor 4. |
| ATF6 | activating transcription factor-6. |
| ATF6f | activating transcription factor-6 cytosolic fragment. |
| Bad | BCL2-antagonist of cell death. |
| BiP | immunoglobulin heavy-chain-binding protein. |
| BMI | body mass index. |
| CCs and CXCs | chemokines. |
| CHOP | C/EBP homologous protein. |
| CREB | cAMP-responsive element-binding protein. |
| CSFs | colony-stimulating factors. |
| CVD | cardiovascular disease. |
| DHA | docosahexaenoic acid. |
| DPP-4 | dipeptidyl-peptidase 4. |
| eIF2B | eukaryotic initiation factor 2B. |
| eIF2α | eukaryotic translation initiation factor 2α. |
| EPA | eicosapentaenoic acid. |
| ER stress | endoplasmic reticulum stress. |
| ERAD | endoplasmic reticulum-associated degradation. |
| FFA | fatty acid. |
| FOXOs | forkhead transcription factors. |
| GADD153 | growth-arrest and DNA-damage-inducible protein. |
| GAP | GTPase-activating protein. |
| GCs | glucocorticoids. |
| GEF | guanine exchange factor. |
| GFs | growth factors. |
| GH | growth hormone. |
| GK | glucokinase. |
| GLP | glucagon-like peptide. |
| GLUT4 | glucose transporter 4. |
| GRB2 | growth factor receptor-bound protein 2. |
| GRP78 | 78 kDa glucose-regulated protein. |
| GS | glycogen synthase. |
| GSK3 | glycogen synthase kinase 3. |
| HDL-C | high-density lipoprotein cholesterol. |
| HOMA-IR | homeostasis model assessment of IR. |
| IFNs | interferons. |
| IGF-1 | insulin-like growth factor 1. |
| IGF-2 | insulin-like growth factor 2. |
| IKKβ | inhibitor of nuclear factor kappa-B kinase subunit beta. |
| IL-6 | interleukin-6. |
| IR | insulin resistance. |
| IRE1 | inositol requiring enzyme 1. |
| IRS | insulin receptor substrate. |
| JNK | c-Jun N-terminal kinase. |
| LDL-C | low-density lipoprotein cholesterol. |
| MAPK | mitogen activated protein kinase. |
| MCP-1 | monocyte chemoattractant protein-1. |
| MetS | metabolic syndrome. |
| MIP-1 | macrophage inflammatory protein 1. |
| MnSOD | manganese superoxide dismutase |
| mTORC1 | mammalian target of rapamycin complex 1. |
| MyD88 | myeloid differentiation primary response gene 88. |
| NFκB | nuclear factor κB. |
| NO | nitrogen–monoxide. |
| OGTT | oral glucose tolerance test. |
| PDK-1 | phosphatidyl-inositol dependent kinase 1. |
| PEPCK | phospho-enol-pyruvate carboxykinase |
| PERK | PKR-like eukaryotic initiation factor 2α kinase |
| PGC-1α | proliferator-activated receptor gamma coactivator 1α |
| PHLPPs | PH domain and leucine rich repeat protein phosphatases |
| PI3K | phosphatidylinositol-3-kinase |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PIP3 | phosphatidylinositol 3,4,5-trisphosphate |
| PKB | protein kinase B. |
| PKC | protein kinase C. |
| PKCλ and PKCζ | PKC isomers. |
| PP2A | protein phosphatase 2A |
| PP2B | protein phosphatase 2B |
| PPARα and γ | peroxisome proliferator-activated receptor α and γ. |
| PTEN | phosphatase and tensin homolog deleted on chromosome ten |
| PTP1B | protein tyrosine phosphatase 1B |
| PUFA | polyunsaturated fatty acids |
| RAC1 | Ras-related C3 botulinum toxin substrate 1. |
| ROS | reactive oxygen species |
| S1P/S2P | site-1 protease/ site-2 protease |
| S6K1 | S6 kinase. |
| SCA | sterol cleavage protein |
| SELENOP/SELENOS | selenoprotein P/selenoprotein S. |
| Ser/Thr | serine/threonine |
| SGLT2 | sodium–glucose co-transporter-2 |
| SHIP1/SHIP2 | SH2 domain containing inositol 5’phosphatase 1 and 2. |
| SIRT1 | sirtuin 1 |
| SNP | single nucleotide polymorphism |
| SOCS1/3 | suppressor of cytokine signaling 1/3 |
| SOS | son of sevenless |
| SRE | sterol regulatory element |
| SREBP | sterol regulatory binding proteins |
| T1DM | Type 1 Diabetes Mellitus |
| T2DM | Type 2 Diabetes Mellitus |
| TBC1D1 | AS160 paralog. |
| TGs | triglycerides |
| TSC1/TSC2 | tuberous sclerosis complex 1 and 2 |
| UPR | unfolded protein response |
| VLDL | very-low-density lipoprotein |
| XBP1 | X-box binding protein-1 |
| Y | tyrosine |
References
- Ahmed, A.M. History of diabetes mellitus. Saudi Med. J. 2002, 23, 373–378. [Google Scholar] [PubMed]
- El-Seedi, H.R.; Yosri, N.; El-Aarag, B.; Mahmoud, S.H.; Zayed, A.; Du, M.; Saeed, A.; Musharraf, S.G.; El-Garawani, I.M.; Habib, M.R.; et al. Chemistry and the Potential Antiviral, Anticancer, and Anti-Inflammatory Activities of Cardiotonic Steroids Derived from Toads. Molecules 2022, 27, 6586. [Google Scholar] [CrossRef] [PubMed]
- Messina, G.; Alioto, A.; Parisi, M.C.; Mingrino, O.; Di Corrado, D.; Crescimanno, C.; Kuliś, S.; Nese Sahin, F.; Padua, E.; Canzone, A.; et al. Experimental study on physical exercise in diabetes: Pathophysiology and therapeutic effects. Eur. J. Transl. Myol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Rydén, L.; Lindsten, J. The history of the Nobel prize for the discovery of insulin. Diabetes Res. Clin. Pract. 2021, 175, 108819. [Google Scholar] [CrossRef] [PubMed]
- Karamanou, M.; Protogerou, A.; Tsoucalas, G.; Androutsos, G.; Poulakou-Rebelakou, E. Milestones in the history of diabetes mellitus: The main contributors. World J. Diabetes 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Paul Langerhans: A prilgrim “traveling” from functional histology to marine biology. Acta Med. Hist. Adriat. 2017, 15, 139–146. [Google Scholar] [CrossRef]
- Campbell, W.R. Paul Langerhans, 1847–1888. Can. Med. Assoc. J. 1958, 79, 855–856. [Google Scholar]
- Bliss, M. Rewriting medical history: Charles Best and the Banting and Best myth. J. Hist. Med. Allied Sci. 1993, 48, 253–274. [Google Scholar] [CrossRef]
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. 1), S67–S74. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, Z. Current views on type 2 diabetes. J. Endocrinol. 2010, 204, 1–11. [Google Scholar] [CrossRef]
- Ozougwu, O. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J. Physiol. Pathophysiol. 2013, 4, 46–57. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.S.; Wingard, D.L.; Kritz-Silverstein, D.; Barrett-Connor, E. Sensitivity and Specificity of Death Certificates for Diabetes. Diabetes Care 2008, 31, 279–284. [Google Scholar] [CrossRef]
- Fan, R.; Zhang, N.; Yang, L.; Ke, J.; Zhao, D.; Cui, Q. AI-based prediction for the risk of coronary heart disease among patients with type 2 diabetes mellitus. Sci. Rep. 2020, 10, 14457. [Google Scholar] [CrossRef]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449. [Google Scholar] [CrossRef]
- Holl, J.; Kowalewski, C.; Zimek, Z.; Fiedor, P.; Kaminski, A.; Oldak, T.; Moniuszko, M.; Eljaszewicz, A. Chronic Diabetic Wounds and Their Treatment with Skin Substitutes. Cells 2021, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, E.; Lim, S.; Lamptey, R.; Webb, D.R.; Davies, M.J. Type 2 diabetes. Lancet 2022, 400, 1803–1820. [Google Scholar] [CrossRef]
- Lemieux, I.; Despres, J.P. Metabolic Syndrome: Past, Present and Future. Nutrients 2020, 12, 3501. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.; Kiss, E.; Takacs, I. The Role of Epigenetic Regulator SIRT1 in Balancing the Homeostasis and Preventing the Formation of Specific “Soil” of Metabolic Disorders and Related Cancers. Front. Biosci. 2022, 27, 253. [Google Scholar] [CrossRef]
- Nemeth, Z.; Patonai, A.; Simon-Szabo, L.; Takacs, I. Interplay of Vitamin D and SIRT1 in Tissue-Specific Metabolism-Potential Roles in Prevention and Treatment of Non-Communicable Diseases Including Cancer. Int. J. Mol. Sci. 2023, 24, 6154. [Google Scholar] [CrossRef]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Ambroselli, D.; Masciulli, F.; Romano, E.; Catanzaro, G.; Besharat, Z.M.; Massari, M.C.; Ferretti, E.; Migliaccio, S.; Izzo, L.; Ritieni, A.; et al. New Advances in Metabolic Syndrome, from Prevention to Treatment: The Role of Diet and Food. Nutrients 2023, 15, 640. [Google Scholar] [CrossRef]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Kario, K.; Okura, A.; Hoshide, S.; Mogi, M. The WHO Global report 2023 on hypertension warning the emerging hypertension burden in globe and its treatment strategy. Hypertens. Res. 2024, 47, 1099–1102. [Google Scholar] [CrossRef]
- Messerli, F.H.; Williams, B.; Ritz, E. Essential hypertension. Lancet 2007, 370, 591–603. [Google Scholar] [CrossRef]
- Mao, Y.; Morrison, H.; MacWilliam, L.; White, M.; Davies, J.; Wigle, D. Risk factors for hypertension: Results from a cross sectional survey. J. Clin. Epidemiol. 1988, 41, 4. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Strain, W.D.; Paldanius, P.M. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 2018, 17, 57. [Google Scholar] [CrossRef]
- Tropeano, A.I.; Boutouyrie, P.; Katsahian, S.; Laloux, B.; Laurent, S. Glucose level is a major determinant of carotid intima-media thickness in patients with hypertension and hyperglycemia. J. Hypertens. 2004, 22, 2153–2160. [Google Scholar] [CrossRef]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Mosca, S.; Araujo, G.; Costa, V.; Correia, J.; Bandeira, A.; Martins, E.; Mansilha, H.; Tavares, M.; Coelho, M.P. Dyslipidemia Diagnosis and Treatment: Risk Stratification in Children and Adolescents. J. Nutr. Metab. 2022, 2022, 4782344. [Google Scholar] [CrossRef]
- Nussbaumerova, B.; Rosolova, H. Obesity and Dyslipidemia. Curr. Atheroscler. Rep. 2023, 25, 947–955. [Google Scholar] [CrossRef]
- Wu, L.; Parhofer, K.G. Diabetic dyslipidemia. Metabolism 2014, 63, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. The metabolic syndrome: Time to get off the merry-go-round? J. Intern. Med. 2011, 269, 127–136. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Nutrition Physical Activity & Obesity (NAO); Office for Prevention & Control of NCDs (MOS). WHO European Regional Obesity Report 2022; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Townsend, N.; Scriven, A. (Eds.) Public Health Mini-Guides: Obesity, 1st ed.; Churchill Livingstone: London, UK, 2014; p. 144. [Google Scholar]
- World Health Organization. Physical status: The Use and Interpretation of Anthropometry, Report of a WHO Expert Committee; World Health Organization: Geneva, Switzerland, 1995. [Google Scholar]
- Pouliot, M.C.; Despres, J.P.; Lemieux, S.; Moorjani, S.; Bouchard, C.; Tremblay, A.; Nadeau, A.; Lupien, P.J. Waist circumference and abdominal sagittal diameter: Best simple anthropometric indexes of abdominal visceral adipose tissue accumulation and related cardiovascular risk in men and women. Am. J. Cardiol. 1994, 73, 460–468. [Google Scholar] [CrossRef]
- Lee, C.M.; Huxley, R.R.; Wildman, R.P.; Woodward, M. Indices of abdominal obesity are better discriminators of cardiovascular risk factors than BMI: A meta-analysis. J. Clin. Epidemiol. 2008, 61, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Richmond, P.; Avenell, A.; Lean, M.E. Waist circumference reduction and cardiovascular benefits during weight loss in women. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 127–134. [Google Scholar] [CrossRef]
- Riaz, H.; Khan, M.S.; Siddiqi, T.J.; Usman, M.S.; Shah, N.; Goyal, A.; Khan, S.S.; Mookadam, F.; Krasuski, R.A.; Ahmed, H. Association Between Obesity and Cardiovascular Outcomes: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. JAMA Netw. Open 2018, 1, e183788. [Google Scholar] [CrossRef] [PubMed]
- Le, T.K.C.; Dao, X.D.; Nguyen, D.V.; Luu, D.H.; Bui, T.M.H.; Le, T.H.; Nguyen, H.T.; Le, T.N.; Hosaka, T.; Nguyen, T.T.T. Insulin signaling and its application. Front. Endocrinol. 2023, 14, 1226655. [Google Scholar] [CrossRef] [PubMed]
- Jin Chan, S.; Steiner, D.F. Insulin Through the Ages: Phylogeny of a Growth Promoting and Metabolic Regulatory Hormone. Am. Zool. 2000, 40, 213–222. [Google Scholar] [CrossRef]
- Bedinger, D.H.; Adams, S.H. Metabolic, anabolic, and mitogenic insulin responses: A tissue-specific perspective for insulin receptor activators. Mol. Cell Endocrinol. 2015, 415, 143–156. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Lipson, K.E.; Kolhatkar, A.A.; Donner, D.B. Insulin stimulates proteolysis of the alpha-subunit, but not the beta-subunit, of its receptor at the cell surface in rat liver. Biochem. J. 1989, 261, 7. [Google Scholar] [CrossRef]
- Kasuga, M.; Zick, Y.; Blithe, D.L.; Crettaz, M.; Kahn, C.R. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 1982, 298, 667–669. [Google Scholar] [CrossRef]
- McDonald, N.Q.; Murray-Rust, J.; Blundell, T.L. The first structure of a receptor tyrosine kinase domain: A further step in understanding the molecular basis of insulin action. Structure 1995, 15, 6. [Google Scholar]
- Wilden, P.A.; Morrison, B.D.; Pessin, J.E. Relationship between insulin receptor subunit association and protein kinase activation: Insulin-dependent covalent and Mn/MgATP-dependent noncovalent association of alpha beta heterodimeric insulin receptors into an alpha 2 beta 2 heterotetrameric state. Biochemistry 1989, 28, 785–792. [Google Scholar] [CrossRef]
- Scapin, G.; Dandey, V.P.; Zhang, Z.; Prosise, W.; Hruza, A.; Kelly, T.; Mayhood, T.; Strickland, C.; Potter, C.S.; Carragher, B. Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature 2018, 556, 122–125. [Google Scholar] [CrossRef]
- Gutmann, T.; Schafer, I.B.; Poojari, C.; Brankatschk, B.; Vattulainen, I.; Strauss, M.; Coskun, U. Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain. J. Cell Biol. 2020, 219, e201907210. [Google Scholar] [CrossRef]
- Yunn, N.O.; Kim, J.; Ryu, S.H.; Cho, Y. A stepwise activation model for the insulin receptor. Exp. Mol. Med. 2023, 55, 2147–2161. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, H.; Lin, C.H.; Cong, L.N.; McGibbon, M.A.; Sciacchitano, S.; Lesniak, M.A.; Quon, M.J.; Taylor, S.I. Insulin receptor substrate-2 (IRS-2) can mediate the action of insulin to stimulate translocation of GLUT4 to the cell surface in rat adipose cells. J. Biol. Chem. 1997, 272, 29829–29833. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Matsuda, M.; Yamamoto, T.; Kanematsu, T.; Kikkawa, U.; Yagisawa, H.; Watanabe, Y.; Hirata, M. PTB domain of insulin receptor substrate-1 binds inositol compounds. Biochem. J. 1998, 334, 9. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 11. [Google Scholar] [CrossRef]
- Jeng, H.H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Di, L.J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef]
- Wang, X.; Janmaat, M.; Beugnet, A.; Paulin, F.E.M.; Proud, C.G. Evidence that the dephosphorylation of Ser(535) in the ∊-subunit of eukaryotic initiation factor (eIF) 2B is insufficient for the activation of eIF2B by insulin. Biochem. J. 2002, 367, 6. [Google Scholar] [CrossRef]
- Nakae, J.; Kitamura, T.; Silver, D.L.; Accili, D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Investig. 2001, 108, 1359–1367. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr. Biol. 2007, 17, R113–R114. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Koumanov, F.; Holman, G.D. GLUT4 On the move. Biochem. J. 2022, 479, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, P.; Koistinen, H.A.; Koivisto, V.A. Insulin-independent glucose transport regulates insulin sensitivity. FEBS Lett. 1998, 436, 3. [Google Scholar] [CrossRef]
- Miinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peranen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef]
- Treebak, J.T.; Taylor, E.B.; Witczak, C.A.; An, D.; Toyoda, T.; Koh, H.J.; Xie, J.; Feener, E.P.; Wojtaszewski, J.F.; Hirshman, M.F.; et al. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am. J. Physiol. Cell Physiol. 2010, 298, C377–C385. [Google Scholar] [CrossRef]
- Taylor, E.B.; An, D.; Kramer, H.F.; Yu, H.; Fujii, N.L.; Roeckl, K.S.; Bowles, N.; Hirshman, M.F.; Xie, J.; Feener, E.P.; et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J. Biol. Chem. 2008, 283, 9787–9796. [Google Scholar] [CrossRef]
- Imamura, T.; Huang, J.; Usui, I.; Satoh, H.; Bever, J.; Olefsky, J.M. Insulin-induced GLUT4 translocation involves protein kinase C-lambda-mediated functional coupling between Rab4 and the motor protein kinesin. Mol. Cell Biol. 2003, 23, 4892–4900. [Google Scholar] [CrossRef]
- Watson, R.T.; Shigematsu, S.; Chiang, S.H.; Mora, S.; Kanzaki, M.; Macara, I.G.; Saltiel, A.R.; Pessin, J.E. Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. J. Cell Biol. 2001, 154, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Corradetti, M.N.; Inoki, K.; Guan, K.L. TSC2: Filling the GAP in the mTOR signaling pathway. Trends Biochem. Sci. 2004, 29, 32–38. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Zhou, H.; Li, X.-M.; Meinkoth, J.; Pittman, R.N. Akt Regulates Cell Survival and Apoptosis at a Postmitochondrial Level. J. Cell Biol. 2000, 151, 11. [Google Scholar] [CrossRef]
- Allan, L.A.; Clarke, P.R. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J. 2009, 276, 6063–6073. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Yamamoto, T.; Watanabe, K.; Inoue, N.; Nakagawa, Y.; Ishigaki, N.; Matsuzaka, T.; Takeuchi, Y.; Kobayashi, K.; Yatoh, S.; Takahashi, A.; et al. Protein kinase Cbeta mediates hepatic induction of sterol-regulatory element binding protein-1c by insulin. J. Lipid Res. 2010, 51, 1859–1870. [Google Scholar] [CrossRef]
- Eyth, E.; Basit, H.; Swift, C. Glucose Tolerance Test. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Subramanian, V.; Bagger, J.I.; Harihar, V.; Holst, J.J.; Knop, F.K.; Villsboll, T. An extended minimal model of OGTT: Estimation of alpha- and beta-cell dysfunction, insulin resistance, and the incretin effect. Am. J. Physiol. Endocrinol. Metab. 2024, 326, E182–E205. [Google Scholar] [CrossRef]
- Tahapary, D.L.; Pratisthita, L.B.; Fitri, N.A.; Marcella, C.; Wafa, S.; Kurniawan, F.; Rizka, A.; Tarigan, T.J.E.; Harbuwono, D.S.; Purnamasari, D.; et al. Challenges in the diagnosis of insulin resistance: Focusing on the role of HOMA-IR and Tryglyceride/glucose index. Diabetes Metab. Syndr. 2022, 16, 102581. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ashford, M.L. AMPK: Regulating energy balance at the cellular and whole body levels. Physiology 2014, 29, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Vatier, C.; Antuna-Puente, B.; Fellahi, S.; Vigouroux, C.; Capeau, J.; Bastard, J.P.; Pour le Groupe de Travail RIHN Adipokines. The adiponectin to leptin ratio, a still unrecognized biomarker of insulin resistance and cardiometabolic risk. Ann. Biol. Clin. 2020, 78, 265–268. [Google Scholar] [CrossRef]
- Castela, I.; Morais, J.; Barreiros-Mota, I.; Silvestre, M.P.; Marques, C.; Rodrigues, C.; Ismael, S.; Araujo, J.R.; Angelo-Dias, M.; Martins, C.; et al. Decreased adiponectin/leptin ratio relates to insulin resistance in adults with obesity. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E115–E119. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef]
- Wu, H.; Deng, X.; Shi, Y.; Su, Y.; Wei, J.; Duan, H. PGC-1alpha, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol. 2016, 229, R99–R115. [Google Scholar] [CrossRef] [PubMed]
- Simon-Szabo, L.; Kokas, M.; Mandl, J.; Keri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS ONE 2014, 9, e97868. [Google Scholar] [CrossRef]
- Zambo, V.; Simon-Szabo, L.; Szelenyi, P.; Kereszturi, E.; Banhegyi, G.; Csala, M. Lipotoxicity in the liver. World J. Hepatol. 2013, 5, 550–557. [Google Scholar] [CrossRef]
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.A.; Montagnani, M.; Quon, M.J. Molecular and physiologic actions of insulin related to production of nitric oxide in vascular endothelium. Curr. Diab Rep. 2003, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Stepniakowski, K.T.; Goodfriend, T.L.; Egan, B.M. Fatty acids enhance vascular alpha-adrenergic sensitivity. Hypertension 1995, 25, 774–778. [Google Scholar] [CrossRef]
- Erdos, B.; Miller, A.W.; Busija, D.W. Alterations in KATP and KCa channel function in cerebral arteries of insulin-resistant rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2472–H2477. [Google Scholar] [CrossRef]
- Yang, R.; Trevillyan, J.M. c-Jun N-terminal kinase pathways in diabetes. Int. J. Biochem. Cell Biol. 2008, 40, 2702–2706. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Eo, H.; Valentine, R.J. Saturated Fatty Acid-Induced Endoplasmic Reticulum Stress and Insulin Resistance Are Prevented by Imoxin in C2C12 Myotubes. Front. Physiol. 2022, 13, 842819. [Google Scholar] [CrossRef]
- Huang, J.S.; Guo, B.B.; Wang, G.H.; Zeng, L.M.; Hu, Y.H.; Wang, T.; Wang, H.Y. DGAT1 inhibitors protect pancreatic beta-cells from palmitic acid-induced apoptosis. Acta Pharmacol. Sin. 2021, 42, 264–271. [Google Scholar] [CrossRef]
- Simon-Szabo, L.; Kokas, M.; Greff, Z.; Boros, S.; Banhegyi, P.; Zsakai, L.; Szantai-Kis, C.; Vantus, T.; Mandl, J.; Banhegyi, G.; et al. Novel compounds reducing IRS-1 serine phosphorylation for treatment of diabetes. Bioorg Med. Chem. Lett. 2016, 26, 424–428. [Google Scholar] [CrossRef]
- Ruderman, N.; Prentki, M. AMP kinase and malonyl-CoA: Targets for therapy of the metabolic syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Pervaiz, S. Apoptosis in the pathophysiology of diabetes mellitus. Int. J. Biochem. Cell Biol. 2007, 39, 497–504. [Google Scholar] [CrossRef]
- Mandl, J.; Meszaros, T.; Banhegyi, G.; Hunyady, L.; Csala, M. Endoplasmic reticulum: Nutrient sensor in physiology and pathology. Trends Endocrinol. Metab. 2009, 20, 194–201. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Ochiai, K.; Kondo, S.; Tsumagari, K.; Murakami, T.; Cavener, D.R.; Imaizumi, K. Endoplasmic reticulum stress response mediated by the PERK-eIF2α-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem. 2011, 286, 4809–4818. [Google Scholar] [CrossRef]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Pederson, T.M.; Kramer, D.L.; Rondinone, C.M. Serine/threonine phosphorylation of IRS-1 triggers its degradation: Possible regulation by tyrosine phosphorylation. Diabetes 2001, 50, 7. [Google Scholar] [CrossRef]
- Potashnik, R.; Bloch-Damti, A.; Bashan, N.; Rudich, A. IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia 2003, 46, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Lair, B.; Laurens, C.; Van Den Bosch, B.; Moro, C. Novel Insights and Mechanisms of Lipotoxicity-Driven Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6358. [Google Scholar] [CrossRef]
- Jung, T.W.; Lee, M.W.; Lee, Y.J.; Kim, S.M. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem. Biophys. Res. Commun. 2012, 417, 147–152. [Google Scholar] [CrossRef]
- Kim, D.S.; Jeong, S.K.; Kim, H.R.; Kim, D.S.; Chae, S.W.; Chae, H.J. Metformin regulates palmitate-induced apoptosis and ER stress response in HepG2 liver cells. Immunopharmacol. Immunotoxicol. 2010, 32, 251–257. [Google Scholar] [CrossRef]
- Unger, E.K.; Piper, M.L.; Olofsson, L.E.; Xu, A.W. Functional role of c-Jun-N-terminal kinase in feeding regulation. Endocrinology 2010, 151, 671–682. [Google Scholar] [CrossRef]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.H.; Corbould, A.; Dunaif, A.; White, M.F. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 9. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 2003, 27 (Suppl. 3), S53–S55. [Google Scholar] [CrossRef]
- Roche, H.M. Fatty acids and the metabolic syndrome. Proc. Nutr. Soc. 2005, 64, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef] [PubMed]
- Kanety, H.; Feinstein, R.; Papa, M.Z.; Hemi, R.; Karasik, A. Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J. Biol. Chem. 1995, 270, 23780–23784. [Google Scholar] [CrossRef] [PubMed]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006, 3, 393–402. [Google Scholar] [CrossRef]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Torocsik, B.; Szeberenyi, J. Anisomycin uses multiple mechanisms to stimulate mitogen-activated protein kinases and gene expression and to inhibit neuronal differentiation in PC12 phaeochromocytoma cells. Eur. J. Neurosci. 2000, 12, 527–532. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, H.; Zhao, Z.; Huang, M.; Wang, S.; Zhan, J. Status of research on natural protein tyrosine phosphatase 1B inhibitors as potential antidiabetic agents: Update. Biomed. Pharmacother. 2023, 157, 113990. [Google Scholar] [CrossRef]
- Krishnan, N.; Bonham, C.A.; Rus, I.A.; Shrestha, O.K.; Gauss, C.M.; Haque, A.; Tocilj, A.; Joshua-Tor, L.; Tonks, N.K. Harnessing insulin- and leptin-induced oxidation of PTP1B for therapeutic development. Nat. Commun. 2018, 9, 283. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Gu, F.; Nguyen, D.T.; Stuible, M.; Dube, N.; Tremblay, M.L.; Chevet, E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 49689–49693. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, P.A.; Delile, E.; Coquerel, D.; Brunel, J.M.; Renet, S.; Tamion, F.; Richard, V. Protein tyrosine phosphatase 1B regulates endothelial endoplasmic reticulum stress; role in endothelial dysfunction. Vasc. Pharmacol. 2018, 109, 36–44. [Google Scholar] [CrossRef]
- Prestwich, G.D. Phosphoinositide signaling; from affinity probes to pharmaceutical targets. Chem. Biol. 2004, 11, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Clement, S.; Krause, U.; Desmedt, F.; Tanti, J.F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 2001, 409, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, S.; Soeda, Y.; Ishihara, H.; Oya, T.; Sasahara, M.; Yaguchi, S.; Oshita, R.; Wada, T.; Tsuneki, H.; Sasaoka, T. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology 2008, 149, 642–650. [Google Scholar] [CrossRef]
- Li, Y.Z.; Di Cristofano, A.; Woo, M. Metabolic Role of PTEN in Insulin Signaling and Resistance. Cold Spring Harb. Perspect. Med. 2020, 10, a036137. [Google Scholar] [CrossRef]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Ni, Y.G.; Wang, N.; Cao, D.J.; Sachan, N.; Morris, D.J.; Gerard, R.D.; Kuro, O.M.; Rothermel, B.A.; Hill, J.A. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc. Natl. Acad. Sci. USA 2007, 104, 20517–20522. [Google Scholar] [CrossRef]
- Chen, Z. Adapter proteins regulate insulin resistance and lipid metabolism in obesity. Sci. Bull. 2016, 61, 8. [Google Scholar] [CrossRef]
- Ord, T.; Ord, D.; Koivomagi, M.; Juhkam, K.; Ord, T. Human TRB3 is upregulated in stressed cells by the induction of translationally efficient mRNA containing a truncated 5′-UTR. Gene 2009, 444, 24–32. [Google Scholar] [CrossRef]
- Fang, D.; Liu, Y.C. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol. 2001, 2, 870–875. [Google Scholar] [CrossRef]
- Nakao, R.; Hirasaka, K.; Goto, J.; Ishidoh, K.; Yamada, C.; Ohno, A.; Okumura, Y.; Nonaka, I.; Yasutomo, K.; Baldwin, K.M.; et al. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol. Cell Biol. 2009, 29, 4798–4811. [Google Scholar] [CrossRef]
- Mahmassani, Z.S.; Reidy, P.T.; McKenzie, A.I.; Petrocelli, J.J.; Matthews, O.; de Hart, N.M.; Ferrara, P.J.; O’Connell, R.M.; Funai, K.; Drummond, M.J. Absence of MyD88 from Skeletal Muscle Protects Female Mice from Inactivity-Induced Adiposity and Insulin Resistance. Obesity 2020, 28, 772–782. [Google Scholar] [CrossRef]
- Thomas, M.S.; Calle, M.; Fernandez, M.L. Healthy plant-based diets improve dyslipidemias, insulin resistance, and inflammation in metabolic syndrome. A narrative review. Adv. Nutr. 2023, 14, 44–54. [Google Scholar] [CrossRef]
- Tuso, P.J.; Ismail, M.H.; Ha, B.P.; Bartolotto, C. Nutritional update for physicians: Plant-based diets. Perm. J. 2013, 17, 61–66. [Google Scholar] [CrossRef]
- Ito, H.; Ishida, H.; Takeuchi, Y.; Antoku, S.; Abe, M.; Mifune, M.; Togane, M. Long-term effect of metformin on blood glucose control in non-obese patients with type 2 diabetes mellitus. Nutr. Metab. 2010, 7, 83. [Google Scholar] [CrossRef]
- Sigal, R.J.; Kenny, G.P.; Boule, N.G.; Wells, G.A.; Prud’homme, D.; Fortier, M.; Reid, R.D.; Tulloch, H.; Coyle, D.; Phillips, P.; et al. Effects of aerobic training, resistance training, or both on glycemic control in type 2 diabetes: A randomized trial. Ann. Intern. Med. 2007, 147, 357–369. [Google Scholar] [CrossRef]
- Kumar, A.; P, N.; Kumar, M.; Jose, A.; Tomer, V.; Oz, E.; Proestos, C.; Zeng, M.; Elobeid, T.; K, S.; et al. Major Phytochemicals: Recent Advances in Health Benefits and Extraction Method. Molecules 2023, 28, 887. [Google Scholar] [CrossRef]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The Immunomodulatory and Anti-Inflammatory Role of Polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef]
- Pannucci, E.; Spagnuolo, L.; De Gara, L.; Santi, L.; Dugo, L. Phenolic Compounds as Preventive and Therapeutic Agents in Diabetes-Related Oxidative Stress, Inflammation, Advanced Glycation End-Products Production and Insulin Sensitivity. Discov. Med. 2023, 35, 715–732. [Google Scholar] [CrossRef]
- Nishida, Y.; Nawaz, A.; Kado, T.; Takikawa, A.; Igarashi, Y.; Onogi, Y.; Wada, T.; Sasaoka, T.; Yamamoto, S.; Sasahara, M.; et al. Astaxanthin stimulates mitochondrial biogenesis in insulin resistant muscle via activation of AMPK pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 241–258. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Zeng, X.; Cheng, Y.; Tang, L.; Hong, D.; Yang, X. Lycopene ameliorates insulin resistance and increases muscle capillary density in aging via activation of SIRT1. J. Nutr. Biochem. 2022, 99, 108862. [Google Scholar] [CrossRef]
- Normen, L.; Dutta, P.; Lia, A.; Andersson, H. Soy sterol esters and beta-sitostanol ester as inhibitors of cholesterol absorption in human small bowel. Am. J. Clin. Nutr. 2000, 71, 908–913. [Google Scholar] [CrossRef]
- Manna, P.; Achari, A.E.; Jain, S.K. 1,25(OH)2-vitamin D3 upregulates glucose uptake mediated by SIRT1/IRS1/GLUT4 signaling cascade in C2C12 myotubes. Mol. Cell. Biochem. 2018, 444, 103–108. [Google Scholar] [CrossRef]
- Manna, P.; Achari, A.E.; Jain, S.K. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch. Biochem. Biophys. 2017, 615, 22–34. [Google Scholar] [CrossRef]
- Morais, J.B.S.; Cruz, K.J.C.; de Oliveira, A.R.S.; Cardoso, B.E.P.; da Silva Dias, T.M.; de Sousa Melo, S.R.; Dos Santos, L.R.; Severo, J.S.; de Freitas, S.T.; Henriques, G.S.; et al. Association Between Parameters of Cortisol Metabolism, Biomarkers of Minerals (Zinc, Selenium, and Magnesium), and Insulin Resistance and Oxidative Stress in Women with Obesity. Biol. Trace Elem. Res. 2023, 201, 5677–5691. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Pigera, S.; Galappatthy, P.; Katulanda, P.; Constantine, G.R. Zinc and diabetes mellitus: Understanding molecular mechanisms and clinical implications. Daru 2015, 23, 44. [Google Scholar] [CrossRef]
- Barbagallo, M.; Dominguez, L.J. Magnesium metabolism in type 2 diabetes mellitus, metabolic syndrome and insulin resistance. Arch. Biochem. Biophys. 2007, 458, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Albracht-Schulte, K.; Kalupahana, N.S.; Ramalingam, L.; Wang, S.; Rahman, S.M.; Robert-McComb, J.; Moustaid-Moussa, N. Omega-3 fatty acids in obesity and metabolic syndrome: A mechanistic update. J. Nutr. Biochem. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Fei, Z.; Xu, Y.; Zhang, G.; Liu, Y.; Li, H.; Chen, L. Natural products with potential hypoglycemic activity in T2DM: 2019–2023. Phytochemistry 2024, 223, 114130. [Google Scholar] [CrossRef] [PubMed]
- Di Pino, A.; Currenti, W.; Urbano, F.; Mantegna, C.; Purrazzo, G.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Low advanced glycation end product diet improves the lipid and inflammatory profiles of prediabetic subjects. J. Clin. Lipidol. 2016, 10, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Nawaz, A.; Hecht, K.; Tobe, K. Astaxanthin as a Novel Mitochondrial Regulator: A New Aspect of Carotenoids, beyond Antioxidants. Nutrients 2021, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Jayaraman, S.; Eladl, M.A.; El-Sherbiny, M.; Abdelrahman, M.A.E.; Veeraraghavan, V.P.; Vengadassalapathy, S.; Umapathy, V.R.; Jaffer Hussain, S.F.; Krishnamoorthy, K.; et al. A Comprehensive Review on Therapeutic Perspectives of Phytosterols in Insulin Resistance: A Mechanistic Approach. Molecules 2022, 27, 1595. [Google Scholar] [CrossRef]
- Valitova, J.N.; Sulkarnayeva, A.G.; Minibayeva, F.V. Plant Sterols: Diversity, Biosynthesis, and Physiological Functions. Biochemistry 2016, 81, 819–834. [Google Scholar] [CrossRef]
- Misawa, E.; Tanaka, M.; Nomaguchi, K.; Yamada, M.; Toida, T.; Takase, M.; Iwatsuki, K.; Kawada, T. Administration of phytosterols isolated from Aloe vera gel reduce visceral fat mass and improve hyperglycemia in Zucker diabetic fatty (ZDF) rats. Obes. Res. Clin. Pract. 2008, 2, I–II. [Google Scholar] [CrossRef]
- Munoz Garcia, M.; Perez Menendez-Conde, C.; Bermejo Vicedo, T. [Advances in the knowledge of the use of micronutrients in artificial nutrition]. Nutr. Hosp. 2011, 26, 37–47. [Google Scholar]
- Berger, M.M.; Shenkin, A.; Schweinlin, A.; Amrein, K.; Augsburger, M.; Biesalski, H.K.; Bischoff, S.C.; Casaer, M.P.; Gundogan, K.; Lepp, H.L.; et al. ESPEN micronutrient guideline. Clin. Nutr. 2022, 41, 1357–1424. [Google Scholar] [CrossRef]
- Ideraabdullah, F.Y.; Zeisel, S.H. Dietary Modulation of the Epigenome. Physiol. Rev. 2018, 98, 667–695. [Google Scholar] [CrossRef]
- Charoenngam, N.; Holick, M.F. Immunologic Effects of Vitamin D on Human Health and Disease. Nutrients 2020, 12, 2097. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats. Nutrients 2017, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; O’Keefe, J.H. Magnesium and Vitamin D Deficiency as a Potential Cause of Immune Dysfunction, Cytokine Storm and Disseminated Intravascular Coagulation in covid-19 patients. Mo. Med. 2021, 118, 68–73. [Google Scholar]
- Steinbrenner, H.; Duntas, L.H.; Rayman, M.P. The role of selenium in type-2 diabetes mellitus and its metabolic comorbidities. Redox Biol. 2022, 50, 102236. [Google Scholar] [CrossRef]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Schoenmakers, E.; Agostini, M.; Mitchell, C.; Schoenmakers, N.; Papp, L.; Rajanayagam, O.; Padidela, R.; Ceron-Gutierrez, L.; Doffinger, R.; Prevosto, C.; et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Investig. 2010, 120, 4220–4235. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.Y.; Soma Roy, M. Dietary copper and selenium are associated with insulin resistance in overweight and obese Malaysian adults. Nutr. Res. 2021, 93, 38–47. [Google Scholar] [CrossRef]
- Carpentier, Y.A.; Portois, L.; Malaisse, W.J. n-3 fatty acids and the metabolic syndrome. Am. J. Clin. Nutr. 2006, 83, 1499S–1504S. [Google Scholar] [CrossRef]
- Pedersen, B.K. Anti-inflammatory effects of exercise: Role in diabetes and cardiovascular disease. Eur. J. Clin. Investig. 2017, 47, 600–611. [Google Scholar] [CrossRef]
- Petersen, A.M.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E.; Team, C.P. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef]
- Malkowska, P.; Sawczuk, M. Cytokines as Biomarkers for Evaluating Physical Exercise in Trained and Non-Trained Individuals: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 11156. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.E.; et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Quail, D.F.; Zhang, X.; White, R.M.; Jones, L.W. Exercise-dependent regulation of the tumour microenvironment. Nat. Rev. Cancer 2017, 17, 620–632. [Google Scholar] [CrossRef]
- Zouhal, H.; Jayavel, A.; Parasuraman, K.; Hayes, L.D.; Tourny, C.; Rhibi, F.; Laher, I.; Abderrahman, A.B.; Hackney, A.C. Effects of Exercise Training on Anabolic and Catabolic Hormones with Advanced Age: A Systematic Review. Sports Med. 2022, 52, 1353–1368. [Google Scholar] [CrossRef]
- Elias, C.F.; Aschkenasi, C.; Lee, C.; Kelly, J.; Ahima, R.S.; Bjorbaek, C.; Flier, J.S.; Saper, C.B.; Elmquist, J.K. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 1999, 23, 775–786. [Google Scholar] [CrossRef]
- Mastorakos, G.; Pavlatou, M.; Diamanti-Kandarakis, E.; Chrousos, G.P. Exercise and the stress system. Hormones 2005, 4, 73–89. [Google Scholar] [PubMed]
- Rasmussen, M.H. Obesity, growth hormone and weight loss. Mol. Cell Endocrinol. 2010, 316, 147–153. [Google Scholar] [CrossRef]
- Sylow, L.; Moller, L.L.V.; Kleinert, M.; D’Hulst, G.; De Groote, E.; Schjerling, P.; Steinberg, G.R.; Jensen, T.E.; Richter, E.A. Rac1 and AMPK Account for the Majority of Muscle Glucose Uptake Stimulated by Ex Vivo Contraction but Not In Vivo Exercise. Diabetes 2017, 66, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, C.; Ke, C.; Huang, C.; Pan, B.; Wan, C. The activation of AMPK/PGC-1alpha/GLUT4 signaling pathway through early exercise improves mitochondrial function and mitigates ischemic brain damage. Neuroreport 2024, 35, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Duchen, M.R. PPARgamma and PGC-1alpha as therapeutic targets in Parkinson’s. Neurochem. Res. 2015, 40, 308–316. [Google Scholar] [CrossRef]
- Kennedy, J.W.; Hirshman, M.F.; Gervino, E.V.; Ocel, J.V.; Forse, R.A.; Hoenig, S.J.; Aronson, D.; Goodyear, L.J.; Horton, E.S. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes 1999, 48, 1192–1197. [Google Scholar] [CrossRef]
- Kristiansen, S.; Hargreaves, M.; Richter, E.A. Exercise-induced increase in glucose transport, GLUT-4, and VAMP-2 in plasma membrane from human muscle. Am. J. Physiol. 1996, 270, E197–E201. [Google Scholar] [CrossRef]
- Douen, A.G.; Ramlal, T.; Rastogi, S.; Bilan, P.J.; Cartee, G.D.; Vranic, M.; Holloszy, J.O.; Klip, A. Exercise induces recruitment of the “insulin-responsive glucose transporter”. Evidence for distinct intracellular insulin- and exercise-recruitable transporter pools in skeletal muscle. J. Biol. Chem. 1990, 265, 13427–13430. [Google Scholar] [CrossRef]
- Ploug, T.; Wojtaszewski, J.; Kristiansen, S.; Hespel, P.; Galbo, H.; Richter, E.A. Glucose transport and transporters in muscle giant vesicles: Differential effects of insulin and contractions. Am. J. Physiol. 1993, 264, E270–E278. [Google Scholar] [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Anirudhan, A.; Ahmad, S.F.; Emran, T.B.; Angulo-Bejarano, P.I.; Sharma, A.; Ahmed, S. Comparative Efficacy of Metformin and Glimepiride in Modulating Pharmacological Network to Increase BDNF Levels and Benefit Type 2 Diabetes-Related Cognitive Impairment. Biomedicines 2023, 11, 2939. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Dokmak, G.; Bader, M.; Karaman, R. A Comprehensive Review on Weight Loss Associated with Anti-Diabetic Medications. Life 2023, 13, 1012. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Yang, N.; He, L.Y.; Liu, P.; Li, Z.Y.; Yang, Y.C.; Ping, F.; Xu, L.L.; Li, W.; Zhang, H.B.; Li, Y.X. Dipeptidyl peptidase-4 inhibitors and the risk of infection: A systematic review and meta-analysis of cardiovascular outcome trials. World J. Diabetes 2024, 15, 1011–1020. [Google Scholar] [CrossRef]
- Haddad, D.; Dsouza, V.S.; Al-Mulla, F.; Al Madhoun, A. New-Generation Glucokinase Activators: Potential Game-Changers in Type 2 Diabetes Treatment. Int. J. Mol. Sci. 2024, 25, 571. [Google Scholar] [CrossRef]
- Shrestha, S.C.; Gupta, S. Imeglimin: The New Kid on the Block. Curr. Diab Rep. 2024, 24, 13–18. [Google Scholar] [CrossRef]
- Hassan, H.A.; Nageeb, M.M.; Mohammed, H.O.; Samy, W.; Fawzy, A.; Afifi, R.; Abbas, N.A.T. Dapagliflozin dampens liver fibrosis induced by common bile duct ligation in rats associated with the augmentation of the hepatic Sirt1/AMPK/PGC1alpha/FoxO1 axis. Toxicol. Appl. Pharmacol. 2024, 489, 116991. [Google Scholar] [CrossRef]
- Suaifan, G.; Alkhawaja, B.; Shehadeh, M.B.; Sharmaa, M.; Hor Kuan, C.; Okechukwu, P.N. Glucosamine substituted sulfonylureas: IRS-PI3K-PKC-AKT-GLUT4 insulin signalling pathway intriguing agent. RSC Med. Chem. 2024, 15, 695–703. [Google Scholar] [CrossRef] [PubMed]


| Drugs | Physiological and Cellular Effect(s) on T2DM/IR | Cellular and Molecular Target(s) | Reference Number |
|---|---|---|---|
| Biguanides Metformin | Improved IR, decreased inflammation | AMPK and AMPK-independent mechanisms | [104,207,208,209] |
| DPP-4 inhibitors | Increased insulin secretion | β-cells in pancreas | [208,210] |
| GLP-1 receptor agonists Liraglutid Semaglutid | Improved insulin production and weight loss | β-cells in pancreas | [191,208] |
| Glucocorticoids | Peripheral IR, insulin secretion | PGC-1α | [92] |
| Glucokinase activators | Glucose-6-phosphate production, subsequent insulin release | Glucose | [211] |
| α-glucosidases inhibitors | Inhibit breakdown of carbohydrates in small intestine | Alpha-glucosidase enzymes in small intestine | [208] |
| Glimins Imeglimin | Increased mitochondrial function, increased insulin secretion, reduced hepatic gluconeogenesis, increased glucose uptake in muscle | Mitochondria, ROS β-cells in pancreas AMPK | [212] |
| SGLT2 inhibitor Dapagliflozin | Decreased oxidative stress and inflammation, increased insulin sensitivity | SIRT1/AMPK/PGC1α/FOXO1 axis | [208,213] |
| Sulfonylureas Glimepiride Glycosylated sul fonylurea | Improved insulin production Improved insulin sensitivity | β-cells in pancreas IRS–PI3K–PKC–AKT–GLUT4 | [207] [214] |
| Thiazolidinediones | Improved insulin resistance, decreased inflammation | AMPK | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon-Szabó, L.; Lizák, B.; Sturm, G.; Somogyi, A.; Takács, I.; Németh, Z. Molecular Aspects in the Development of Type 2 Diabetes and Possible Preventive and Complementary Therapies. Int. J. Mol. Sci. 2024, 25, 9113. https://doi.org/10.3390/ijms25169113
Simon-Szabó L, Lizák B, Sturm G, Somogyi A, Takács I, Németh Z. Molecular Aspects in the Development of Type 2 Diabetes and Possible Preventive and Complementary Therapies. International Journal of Molecular Sciences. 2024; 25(16):9113. https://doi.org/10.3390/ijms25169113
Chicago/Turabian StyleSimon-Szabó, Laura, Beáta Lizák, Gábor Sturm, Anikó Somogyi, István Takács, and Zsuzsanna Németh. 2024. "Molecular Aspects in the Development of Type 2 Diabetes and Possible Preventive and Complementary Therapies" International Journal of Molecular Sciences 25, no. 16: 9113. https://doi.org/10.3390/ijms25169113