
Prostate Cancer’s Silent Partners: Fibroblasts and Their Influence on Glutamine Metabolism Manipulation
 ,
, 

Abstract
1. Introduction
2. Results
2.1. Established Prostate Cancer Cell Lines Expressing mKATE2-NLS to Trace Cell Proliferation and Clonogenic Potential
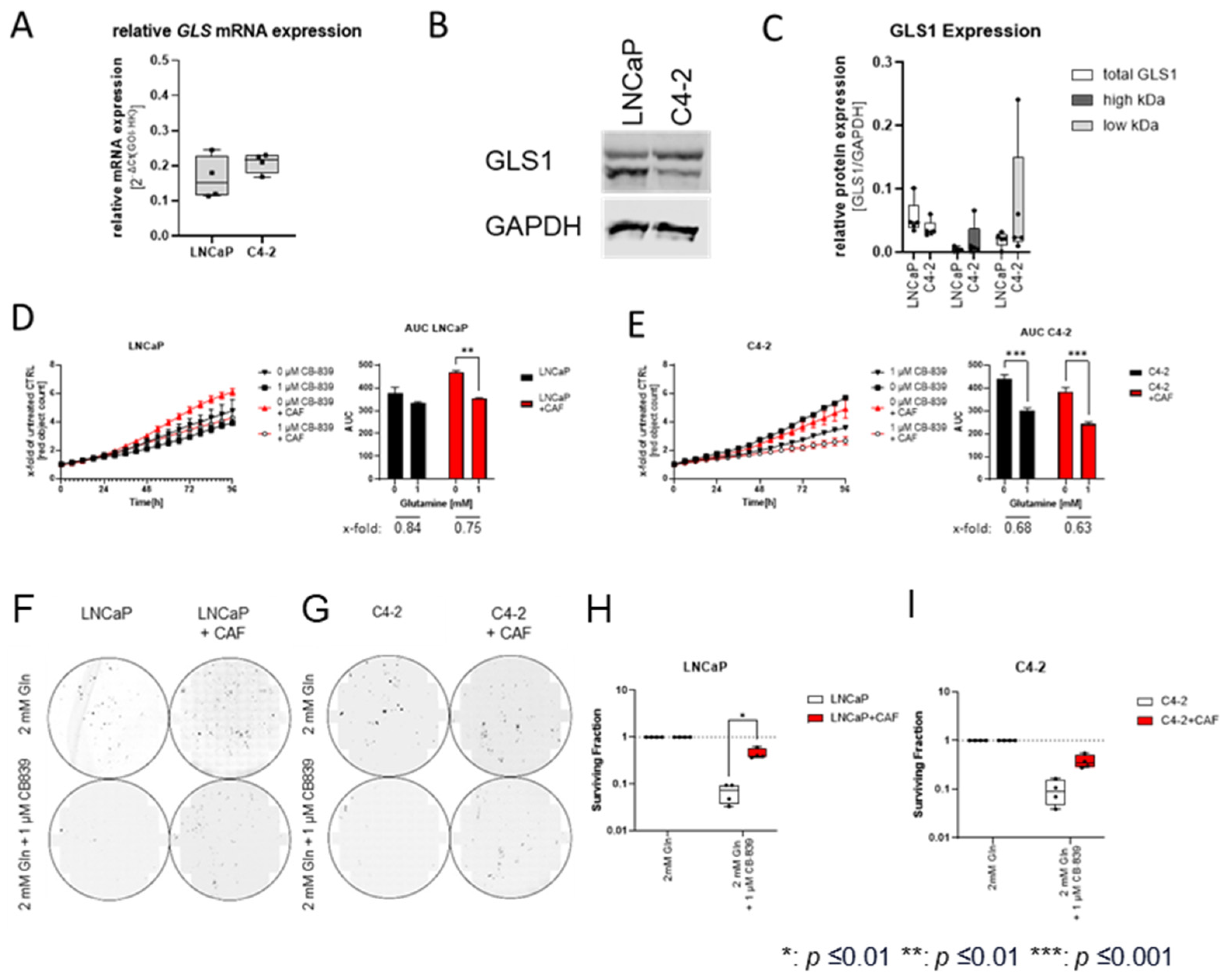
2.2. Cancer-Associated Fibroblasts Sensitise the Hormone-Sensitive LNCaPcell Line to Glutamine Deprivation
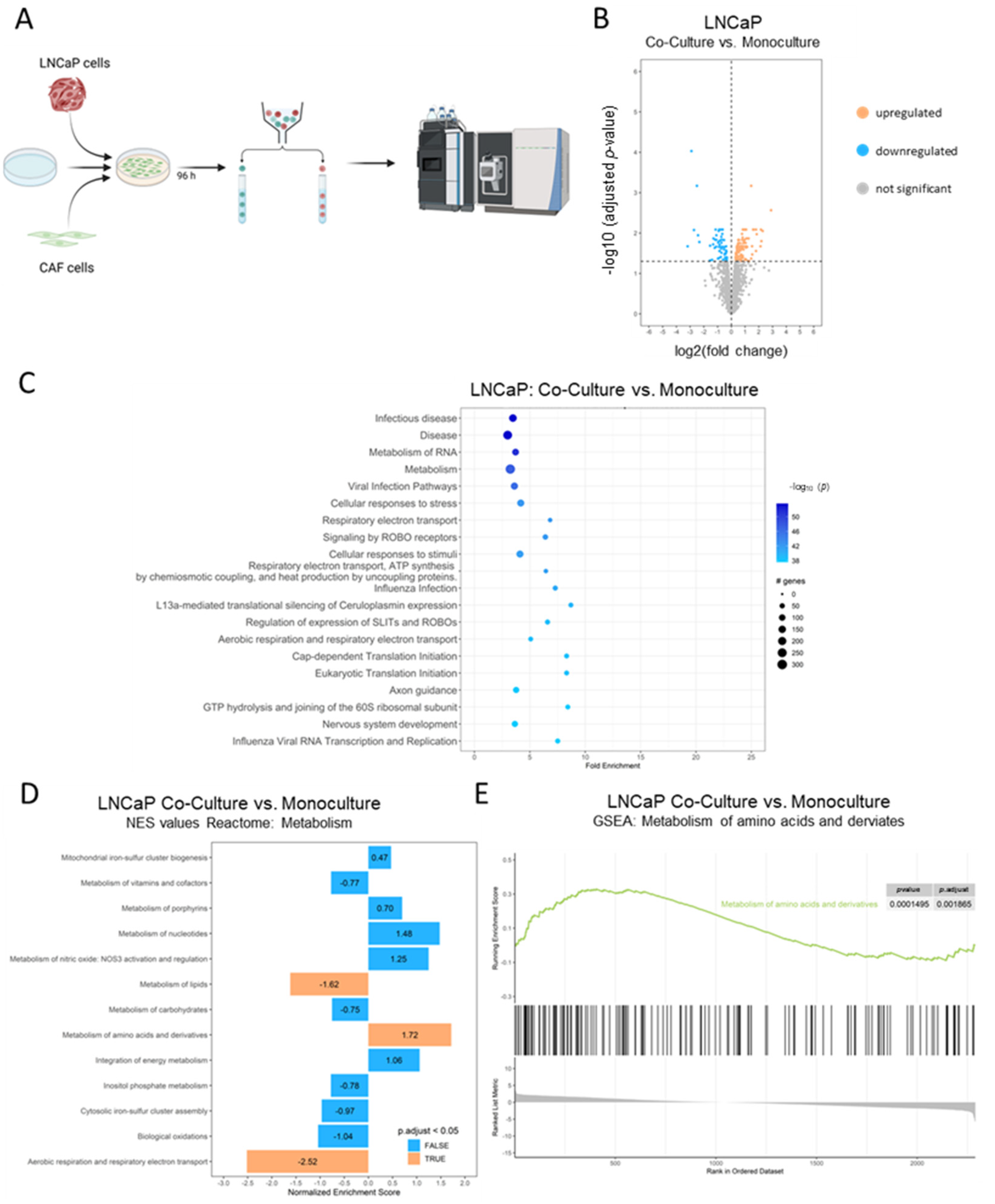
2.3. Cancer-Associated Fibroblasts Reprogram LNCaP Cell’s Metabolic Needs
2.4. Cancer-Associated Fibroblasts Reduce the Inhibitory Effect of CB839 on Clonogenic Potential
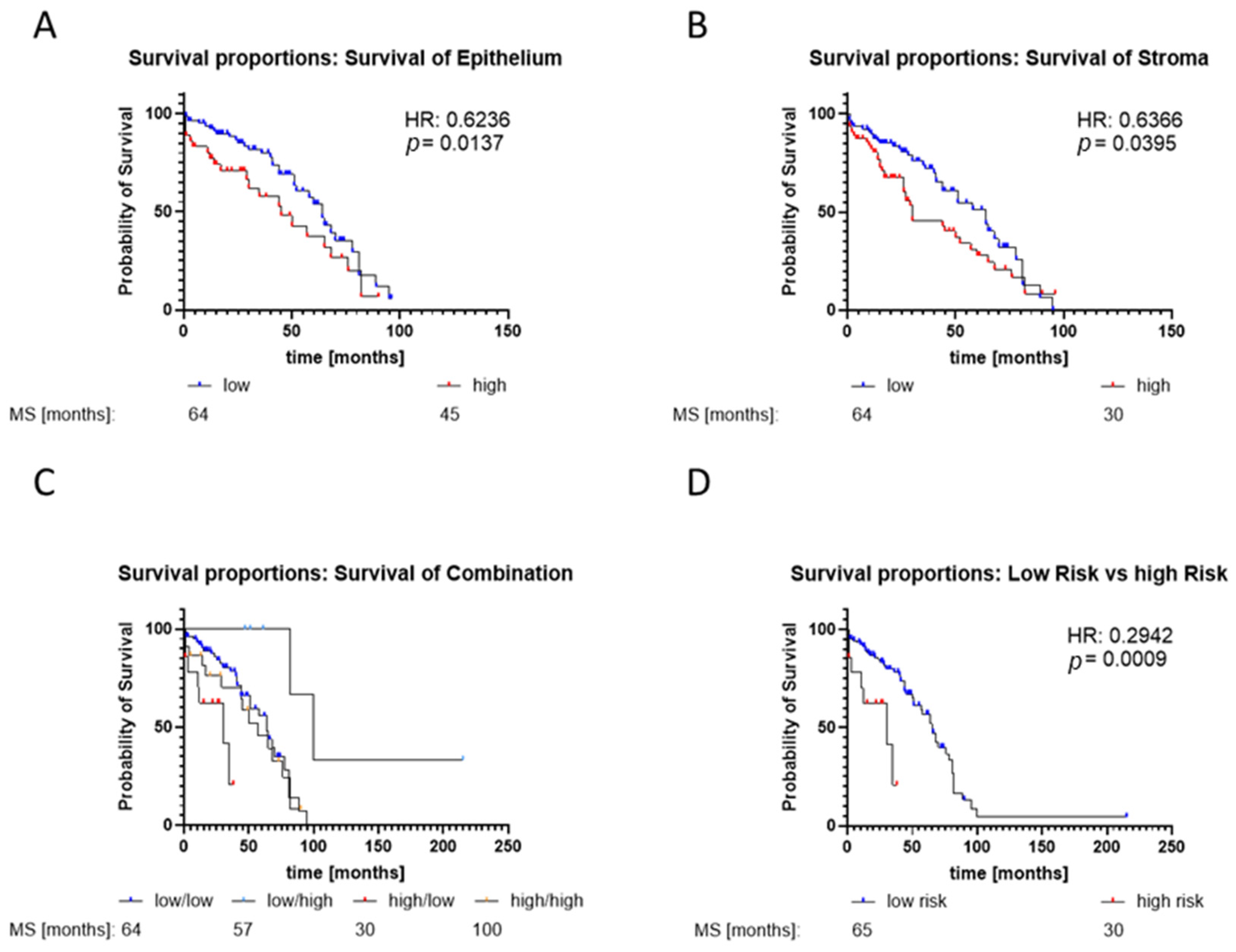
2.5. Low GLS1 Expression in Stroma and High Expression in Epithelial Cells Is a Marker for Bad Prognosis
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Lentivirus Production, eGFP-NLS and mKATE2-NLS Nuclei Labelling, and Image Cytometry
4.3. Gln Deprivation and CB-839 Treatment
4.4. Proliferation Assay with the IncuCyte® S3 Live-Cell Analysis System
4.5. Clonogenic Assay
4.6. Western Blot
4.7. Data Preparation, Imputation, Overrepresentation Analysis (ORA), and Gene Set Enrichment Analysis (GSEA) of the Mass Spectrometry Data
4.8. Patients and Study Design
4.9. Immunohistochemistry
4.10. qPCR
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bedeschi, M.; Marino, N.; Cavassi, E.; Piccinini, F.; Tesei, A. Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance. Cells 2023, 12, 802. [Google Scholar] [CrossRef]
- Yu, S.; Jiang, Y.; Wan, F.; Wu, J.; Gao, Z.; Liu, D. Immortalized Cancer-associated Fibroblasts Promote Prostate Cancer Carcinogenesis, Proliferation and Invasion. Anticancer Res. 2017, 37, 4311–4318. [Google Scholar] [PubMed]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef]
- Levesque, C.; Nelson, P.S. Cellular constituents of the prostate stroma: Key contributors to prostate cancer progression and therapy resistance. Cold Spring Harb. Perspect. Med. 2018, 8, a030510. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. Cancer-associated fibroblasts: Secretions, interactions, and therapy. J. Cell. Biochem. 2019, 120, 2791–2800. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Beier, A.K.; Ebersbach, C.; Siciliano, T.; Scholze, J.; Hofmann, J.; Hönscheid, P.; Baretton, G.B.; Woods, K.; Guezguez, B.; Dubrovska, A.; et al. Targeting the glutamine metabolism to suppress cell proliferation in mesenchymal docetaxel-resistant prostate cancer. Oncogene 2024, 43, 2038–2050. [Google Scholar] [CrossRef]
- Beier, A.K.; Puhr, M.; Stope, M.B.; Thomas, C.; Erb, H.H.H. Metabolic changes during prostate cancer development and progression. J. Cancer Res. Clin. Oncol. 2023, 149, 2259–2270. [Google Scholar] [CrossRef]
- Mestre-Farrera, A.; Bruch-Oms, M.; Peña, R.; Rodríguez-Morató, J.; Alba-Castellón, L.; Comerma, L.; Quintela-Fandino, M.; Duñach, M.; Baulida, J.; Pozo Ó, J.; et al. Glutamine-Directed Migration of Cancer-Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res. 2021, 81, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Kunou, S.; Shimada, K.; Tsunoda, M.; Aoki, T.; Iriyama, C.; Tomita, A.; Nakamura, S.; Hayakawa, F.; Kiyoi, H. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 2019, 110, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Brunckhorst, O.; Darraugh, J.; Eberli, D.; De Meerleer, G.; De Santis, M.; Farolfi, A.; et al. EAU-EANM-ESTRO-ESUR-ISUP-SIOG Guidelines on Prostate Cancer-2024 Update. Part I: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2024, 71, 618–629. [Google Scholar] [CrossRef]
- Tilki, D.; Bergh, R.; Briers, E.; Van den Broeck, T.; Brunckhorst, O.; Darraugh, J.; Eberli, D.; Meerleer, G.; Santis, M.; Farolfi, A.; et al. EAU-EANM-ESTRO-ESUR-ISUP-SIOG Guidelines on Prostate Cancer. Part II—2024 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2024, 86, 164–182. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Dubrovska, A. Targeting glutamine metabolism and autophagy: The combination for prostate cancer radiosensitization. Autophagy 2021, 17, 3879–3881. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, O.; Balslev, E.; Høgdall, E. Targeting the Supply Lines of Cancer—A Possible Strategy for Combating the Disease? Anticancer Res. 2021, 41, 2737–2744. [Google Scholar] [CrossRef]
- Xu, L.; Yin, Y.; Li, Y.; Chen, X.; Chang, Y.; Zhang, H.; Liu, J.; Beasley, J.; McCaw, P.; Zhang, H.; et al. A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2012748118. [Google Scholar] [CrossRef] [PubMed]
- Erb, H.H.H.; Polishchuk, N.; Stasyk, O.; Kahya, U.; Weigel, M.M.; Dubrovska, A. Glutamine Metabolism and Prostate Cancer. Cancers 2024, 16, 2871. [Google Scholar] [CrossRef]
- Krebs, H.A. Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem. J. 1935, 29, 1951–1969. [Google Scholar] [CrossRef]
- Myint, Z.W.; Sun, R.C.; Hensley, P.J.; James, A.C.; Wang, P.; Strup, S.E.; McDonald, R.J.; Yan, D.; St. Clair, W.H.; Allison, D.B. Evaluation of Glutaminase Expression in Prostate Adenocarcinoma and Correlation with Clinicopathologic Parameters. Cancers 2021, 13, 2157. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Giacobbe, A.; Bongiorno-Borbone, L.; Bernassola, F.; Terrinoni, A.; Markert, E.K.; Levine, A.J.; Feng, Z.; Agostini, M.; Zolla, L.; Agrò, A.F.; et al. p63 regulates glutaminase 2 expression. Cell Cycle 2013, 12, 1395–1405. [Google Scholar] [CrossRef]
- Velletri, T.; Romeo, F.; Tucci, P.; Peschiaroli, A.; Annicchiarico-Petruzzelli, M.; Niklison-Chirou, M.V.; Amelio, I.; Knight, R.A.; Mak, T.W.; Melino, G.; et al. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell Cycle 2013, 12, 3564–3573. [Google Scholar] [CrossRef] [PubMed]
- Campos-Sandoval, J.A.; López de la Oliva, A.R.; Lobo, C.; Segura, J.A.; Matés, J.M.; Alonso, F.J.; Márquez, J. Expression of functional human glutaminase in baculovirus system: Affinity purification, kinetic and molecular characterization. Int. J. Biochem. Cell Biol. 2007, 39, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Simeone, S.; Wu, R.; Grady, J.; Mandoiu, I.; Srivastava, P.K. Area under the curve as a tool to measure kinetics of tumor growth in experimental animals. J. Immunol. Methods 2012, 382, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.J.; Lee, R.J.; Jain, R.K.; Davis, N.B.; Appleman, L.J.; Goodman, O.B.; et al. CANTATA: Primary analysis of a global, randomized, placebo (Pbo)-controlled, double-blind trial of telaglenastat (CB-839) + cabozantinib versus Pbo + cabozantinib in advanced/metastatic renal cell carcinoma (mRCC) patients (pts) who progressed on immune checkpoint inhibitor (ICI) or anti-angiogenic therapies. J. Clin. Oncol. 2021, 39, 4501. [Google Scholar] [CrossRef]
- Zarling, L.; Emamekhoo, H.; Bhutani, G.; Ziemlewicz, T.; Matkowskyj, K.A.; Kyriakopoulos, C.E. Polycystic Liver Disease in a Patient With Metastatic Renal Cell Carcinoma: A Case Report. Anticancer Res. 2020, 40, 1527–1534. [Google Scholar] [CrossRef]
- Lam, E.T.; Su, L.-J.; Salzmann-Sullivan, M.; Nordeen, S.K.; Flaig, T.W. Preclinical evaluation of teleglenastat (CB-839) in prostate cancer. J. Clin. Oncol. 2023, 41, 378. [Google Scholar] [CrossRef]
- Hennequin, C.; Hannoun-Lévi, J.M.; Rozet, F. Management of local relapse after prostate cancer radiotherapy: Surgery or radiotherapy? Cancer Radiother. 2017, 21, 433–436. [Google Scholar] [CrossRef]
- Renard-Penna, R.; Zhang-Yin, J.; Montagne, S.; Aupin, L.; Bruguière, E.; Labidi, M.; Latorzeff, I.; Hennequin, C. Targeting Local Recurrence After Surgery With MRI Imaging for Prostate Cancer in the Setting of Salvage Radiation Therapy. Front. Oncol. 2022, 12, 775387. [Google Scholar] [CrossRef]
- Suardi, N.; Porter, C.R.; Reuther, A.M.; Walz, J.; Kodama, K.; Gibbons, R.P.; Correa, R.; Montorsi, F.; Graefen, M.; Huland, H.; et al. A nomogram predicting long-term biochemical recurrence after radical prostatectomy. Cancer 2008, 112, 1254–1263. [Google Scholar] [CrossRef]
- Chun, F.K.H.; Graefen, M.; Zacharias, M.; Haese, A.; Steuber, T.; Schlomm, T.; Walz, J.; Karakiewicz, P.I.; Huland, H. Anatomic radical retropubic prostatectomy—Long-term recurrence-free survival rates for localized prostate cancer. World J. Urol. 2006, 24, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, M.; Akre, O.; Bock, D.; Carlsson, S.V.; Carlsson, S.; Hugosson, J.; Lantz, A.; Steineck, G.; Stranne, J.; Tyritzis, S.; et al. Risk of Recurrent Disease 6 Years After Open or Robotic-assisted Radical Prostatectomy in the Prospective Controlled Trial LAPPRO. Eur. Urol. Open Sci. 2020, 20, 54–61. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Matos, A.; Carvalho, M.; Bicho, M.; Ribeiro, R. Arginine and Arginases Modulate Metabolism, Tumor Microenvironment and Prostate Cancer Progression. Nutrients 2021, 13, 4503. [Google Scholar] [CrossRef] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Elia, I.; Haigis, M.C. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef]
- Pederzoli, F.; Raffo, M.; Pakula, H.; Ravera, F.; Nuzzo, P.V.; Loda, M. “Stromal cells in prostate cancer pathobiology: Friends or foes?”. Br. J. Cancer 2023, 128, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.-Y.; Wu, J.-Q.; He, Z.-H.; He, M.-F.; Sun, H.-B. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-β signaling pathway. Life Sci. 2019, 235, 116791. [Google Scholar] [CrossRef]
- Tagat, A.; Ayoubian, H.; Zaccagnino, A.; Stöckle, M.; Linxweiler, J.; Junker, K. Abstract B039: Influence of tumor-associated fibroblasts and their exosomes in the development and progression of prostate cancer. Cancer Res. 2023, 83, B039. [Google Scholar] [CrossRef]
- Sasaki, T.; Yoshikawa, Y.; Kageyama, T.; Sugino, Y.; Kato, M.; Masui, S.; Nishikawa, K.; Inoue, T. Prostate fibroblasts enhance androgen receptor splice variant 7 expression in prostate cancer cells. Prostate 2023, 83, 364–375. [Google Scholar] [CrossRef]
- Da, J.; Lu, M.; Wang, Z. Estrogen receptor alpha (ERα)-associated fibroblasts promote cell growth in prostate cancer. Cell Biochem. Biophys. 2015, 73, 793–798. [Google Scholar] [CrossRef]
- Levenberg, S.; Yarden, A.; Kam, Z.; Geiger, B. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene 1999, 18, 869–876. [Google Scholar] [CrossRef]
- Pavel, M.; Renna, M.; Park, S.J.; Menzies, F.M.; Ricketts, T.; Füllgrabe, J.; Ashkenazi, A.; Frake, R.A.; Lombarte, A.C.; Bento, C.F.; et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat. Commun. 2018, 9, 2961. [Google Scholar] [CrossRef] [PubMed]
- Leontieva, O.V.; Demidenko, Z.N.; Blagosklonny, M.V. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc. Natl. Acad. Sci. USA 2014, 111, 8832–8837. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Deuster, O.; Rinn, B.; Schulz, M.; Kautz, A.; Dodel, R.; Meyer, B.; Al-Abed, Y.; Balakrishnan, K.; Reese, J.P.; et al. Restoration of contact inhibition in human glioblastoma cell lines after MIF knockdown. BMC Cancer 2009, 9, 464. [Google Scholar] [CrossRef]
- Blom, S.; Erickson, A.; Östman, A.; Rannikko, A.; Mirtti, T.; Kallioniemi, O.; Pellinen, T. Fibroblast as a critical stromal cell type determining prognosis in prostate cancer. Prostate 2019, 79, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.J.; Ma, S. The Role of Cancer-Associated Fibroblast as a Dynamic Player in Mediating Cancer Stemness in the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 727640. [Google Scholar] [CrossRef]
- Samoszuk, M.; Tan, J.; Chorn, G. Clonogenic growth of human breast cancer cells co-cultured in direct contact with serum-activated fibroblasts. Breast Cancer Res. 2005, 7, R274. [Google Scholar] [CrossRef]
- Eder, T.; Weber, A.; Neuwirt, H.; Grünbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-Associated Fibroblasts Modify the Response of Prostate Cancer Cells to Androgen and Anti-Androgens in Three-Dimensional Spheroid Culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef]
- Eigentler, A.; Handle, F.; Schanung, S.; Degen, A.; Hackl, H.; Erb, H.H.H.; Fotakis, G.; Hoefer, J.; Ploner, C.; Jöhrer, K.; et al. Glucocorticoid treatment influences prostate cancer cell growth and the tumor microenvironment via altered glucocorticoid receptor signaling in prostate fibroblasts. Oncogene 2024, 43, 235–247. [Google Scholar] [CrossRef]
- Heidegger, I.; Frantzi, M.; Salcher, S.; Tymoszuk, P.; Martowicz, A.; Gomez-Gomez, E.; Blanca, A.; Lendinez Cano, G.; Latosinska, A.; Mischak, H.; et al. Prediction of Clinically Significant Prostate Cancer by a Specific Collagen-related Transcriptome, Proteome, and Urinome Signature. Eur. Urol. Oncol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Erb, H.H.; Langlechner, R.V.; Moser, P.L.; Handle, F.; Casneuf, T.; Verstraeten, K.; Schlick, B.; Schäfer, G.; Hall, B.; Sasser, K.; et al. IL6 sensitizes prostate cancer to the antiproliferative effect of IFNα2 through IRF9. Endocr. Relat. Cancer 2013, 20, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 42. [Google Scholar] [CrossRef]
- Mauer, J.; Denson, J.L.; Brüning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Copeland, C.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2021, 1311, 17–38. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef]
- Lee, C.H.; Motzer, R.; Emamekhoo, H.; Matrana, M.; Percent, I.; Hsieh, J.J.; Hussain, A.; Vaishampayan, U.; Liu, S.; McCune, S.; et al. Telaglenastat plus Everolimus in Advanced Renal Cell Carcinoma: A Randomized, Double-Blinded, Placebo-Controlled, Phase II ENTRATA Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 3248–3255. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.-Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef]
- Xie, B.-X.; Zhang, H.; Yu, L.; Wang, J.; Pang, B.; Wu, R.-Q.; Qian, X.-L.; Li, S.-H.; Shi, Q.-G.; Wang, L.-L.; et al. The radiation response of androgen-refractory prostate cancer cell line C4-2 derived from androgen-sensitive cell line LNCaP. Asian J. Androl. 2010, 12, 405–414. [Google Scholar] [CrossRef]
- Siciliano, T.; Simons, I.H.; Beier, A.-M.K.; Ebersbach, C.; Aksoy, C.; Seed, R.I.; Stope, M.B.; Thomas, C.; Erb, H.H.H. A Systematic Comparison of Antiandrogens Identifies Androgen Receptor Protein Stability as an Indicator for Treatment Response. Life 2021, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, F.; Geisslinger, F.; Gabriel, A.; von Schwarzenberg, K.; Vollmar, A.M.; Bartel, K. A metabolic shift toward glycolysis enables cancer cells to maintain survival upon concomitant glutamine deprivation and V-ATPase inhibition. Front. Nutr. 2023, 10, 1124678. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Wojtkowiak, J.; Neilson, A.; Gillies, R.J. Metabolic Profiling of healthy and cancerous tissues in 2D and 3D. Sci. Rep. 2017, 7, 15285. [Google Scholar] [CrossRef]
- Thalmann, G.N.; Sikes, R.A.; Wu, T.T.; Degeorges, A.; Chang, S.M.; Ozen, M.; Pathak, S.; Chung, L.W. LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate 2000, 44, 91–103. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Seed, R.I.; Taurozzi, A.J.; Wilcock, D.J.; Nappo, G.; Erb, H.H.H.; Read, M.L.; Gurney, M.; Archer, L.K.; Ito, S.; Rumsby, M.G.; et al. The putative tumour suppressor protein Latexin is secreted by prostate luminal cells and is downregulated in malignancy. Sci. Rep. 2019, 9, 5120. [Google Scholar] [CrossRef]
- KÜSTER, J.H.S.; ERB, H.H.H.; AHREND, H.; ABAZID, A.; STOPE, M.B. Modulation of the Prostate Cancer Resistance Factor Hsp27 by the Chemotherapeutic Drugs Abiraterone, Cabazitaxel, Docetaxel and Enzalutamide. Anticancer Res. 2024, 44, 2815–2821. [Google Scholar] [CrossRef]
- Müller, H.; Lesur, A.; Dittmar, G.; Gentzel, M.; Kettner, K. Proteomic consequences of TDA1 deficiency in Saccharomyces cerevisiae: Protein kinase Tda1 is essential for Hxk1 and Hxk2 serine 15 phosphorylation. Sci. Rep. 2022, 12, 18084. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; François, R.; Henry, L.; Müller, K.; Vaughan, D. dplyr: A Grammar of Data Manipulation. R package version 1.1.4. 2023. Available online: https://dplyr.tidyverse.org (accessed on 16 August 2024).
- Team, R. A language and environment for statistical computing. Computing 2006, 1, 3–24. [Google Scholar] [CrossRef]
- Okoye, K.; Hosseini, S. Introduction to R Programming and RStudio Integrated Development Environment (IDE); Springer: Berlin/Heidelberg, Germany, 2024; pp. 3–24. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Jin, L.; Bi, Y.; Hu, C.; Qu, J.; Shen, S.; Wang, X.; Tian, Y. A comparative study of evaluating missing value imputation methods in label-free proteomics. Sci. Rep. 2021, 11, 1760. [Google Scholar] [CrossRef] [PubMed]
- Stekhoven, D.J.; Buhlmann, P. MissForest--non-parametric missing value imputation for mixed-type data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis by H. WICKHAM. Biometrics 2011, 67, 678–679. [Google Scholar] [CrossRef]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021, 060012. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Sommer, U.; Siciliano, T.; Ebersbach, C.; Beier, A.K.; Stope, M.B.; Jöhrens, K.; Baretton, G.B.; Borkowetz, A.; Thomas, C.; Erb, H.H.H. Impact of Androgen Receptor Activity on Prostate-Specific Membrane Antigen Expression in Prostate Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1046. [Google Scholar] [CrossRef] [PubMed]
- Ebersbach, C.; Beier, A.-M.K.; Hönscheid, P.; Sperling, C.; Jöhrens, K.; Baretton, G.B.; Thomas, C.; Sommer, U.; Borkowetz, A.; Erb, H.H.H. Influence of Systemic Therapy on the Expression and Activity of Selected STAT Proteins in Prostate Cancer Tissue. Life 2022, 12, 240. [Google Scholar] [CrossRef] [PubMed]
- Erb, H.H.H.; Oster, M.A.; Gelbrich, N.; Cammann, C.; Thomas, C.; Mustea, A.; Stope, M.B. Enzalutamide-induced Proteolytic Degradation of the Androgen Receptor in Prostate Cancer Cells Is Mediated Only to a Limited Extent by the Proteasome System. Anticancer Res. 2021, 41, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Erb, H.H.; Bodenbender, J.; Handle, F.; Diehl, T.; Donix, L.; Tsaur, I.; Gleave, M.; Haferkamp, A.; Huber, J.; Fuessel, S. Assessment of STAT5 as a potential therapy target in enzalutamide-resistant prostate cancer. PLoS ONE 2020, 15, e0237248. [Google Scholar] [CrossRef]
- Voronov, D.A. Calculating the intrinsic growth rate: Comparison of definition and model. Zh. Obs. Biol. 2005, 66, 425–430. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All | HSPC | CRPC | |
|---|---|---|---|
| Patient Number | 108 | 26 | 82 |
| Median age at primary diagnosis, years | 71 | 72 | 71 |
| Median PSA at primary diagnosis, ng/mL (Interquartile range IQR) | 17 | 6.3 | 32.5 |
| (6.8; 73.0) | (2.7; 10.2) | (8.9; 101.6) | |
| Neuroendocrine differentiation at primary diagnosis, % | 1 | 0 | 1 |
| Presence of bone metastases at primary diagnosis, % | 25 | 3.8 | 32 |
| Presence of lymph node metastases at primary diagnosis, % | 14 | 12 | 11.7 |
| Presence of organ metastases at primary diagnosis, % | 1 | 1 | 1 |
| Median overall survival since the start of primary therapy, months | 59 | 47 | 81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hönscheid, P.V.; Baretton, G.B.; Puhr, M.; Siciliano, T.; Israel, J.S.; Stope, M.B.; Ebersbach, C.; Beier, A.-M.K.; Thomas, C.; Erb, H.H.H. Prostate Cancer’s Silent Partners: Fibroblasts and Their Influence on Glutamine Metabolism Manipulation. Int. J. Mol. Sci. 2024, 25, 9275. https://doi.org/10.3390/ijms25179275
Hönscheid PV, Baretton GB, Puhr M, Siciliano T, Israel JS, Stope MB, Ebersbach C, Beier A-MK, Thomas C, Erb HHH. Prostate Cancer’s Silent Partners: Fibroblasts and Their Influence on Glutamine Metabolism Manipulation. International Journal of Molecular Sciences. 2024; 25(17):9275. https://doi.org/10.3390/ijms25179275
Chicago/Turabian StyleHönscheid, Pia V., Gustavo B. Baretton, Martin Puhr, Tiziana Siciliano, Justus S. Israel, Matthias B. Stope, Celina Ebersbach, Alicia-Marie K. Beier, Christian Thomas, and Holger H. H. Erb. 2024. "Prostate Cancer’s Silent Partners: Fibroblasts and Their Influence on Glutamine Metabolism Manipulation" International Journal of Molecular Sciences 25, no. 17: 9275. https://doi.org/10.3390/ijms25179275
APA StyleHönscheid, P. V., Baretton, G. B., Puhr, M., Siciliano, T., Israel, J. S., Stope, M. B., Ebersbach, C., Beier, A.-M. K., Thomas, C., & Erb, H. H. H. (2024). Prostate Cancer’s Silent Partners: Fibroblasts and Their Influence on Glutamine Metabolism Manipulation. International Journal of Molecular Sciences, 25(17), 9275. https://doi.org/10.3390/ijms25179275