
Proteomic Investigation of Immune Checkpoints and Some of Their Inhibitors

Abstract
:1. Introduction
2. Discussion
2.1. Predictive Biomarkers in Response to Immune Checkpoint Inhibitors
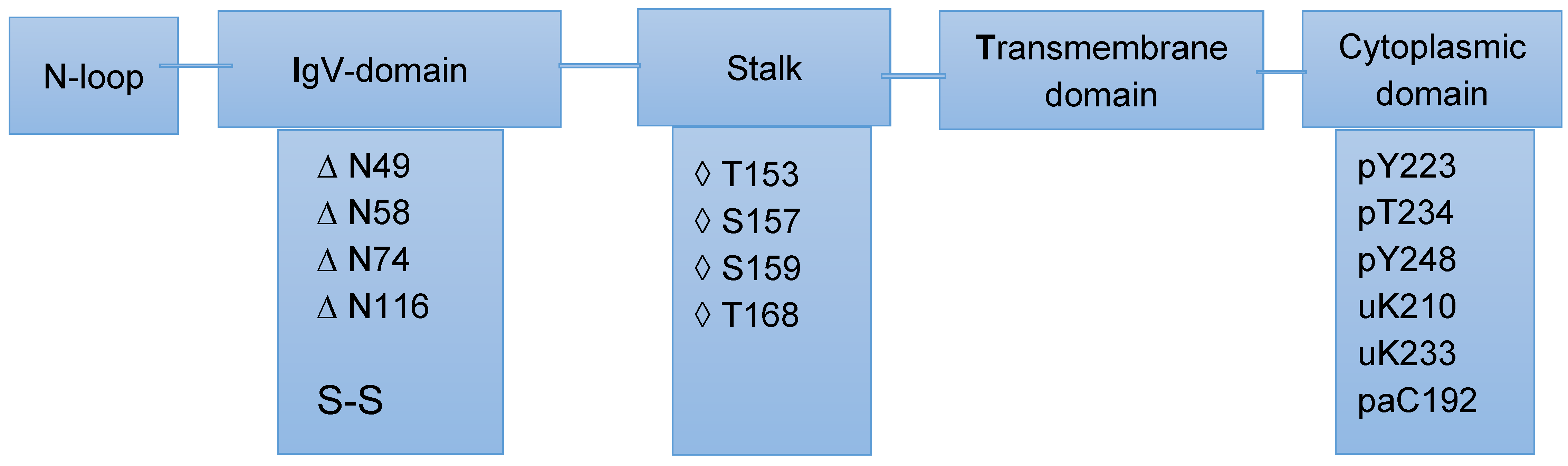
2.2. Post-Translational Modifications of PD-L1/PD-1 and Their Potential Role in Cancer Therapy
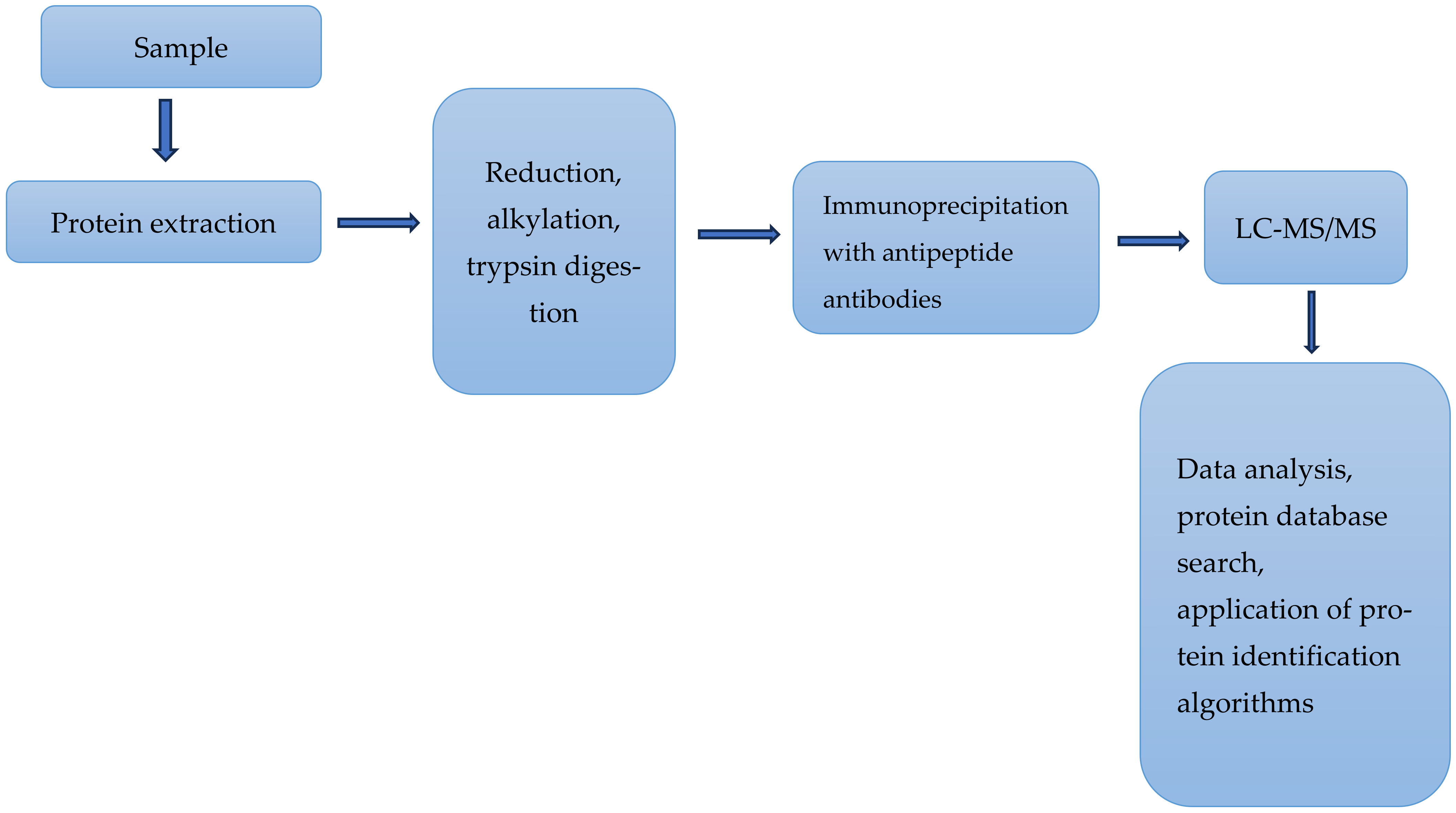
2.3. Mass Spectrometry-Based Analyses of Some Proteins Relevant to Immune Responses
2.4. Comments
2.4.1. Drug Resistance to Immune Therapies
2.4.2. The Promise of Bispecific Antibodies
2.4.3. MS-Based Investigation of Immune Checkpoints Is Still below Its Real Potential
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| bsAbs | Bispecific antibodies |
| CTCs | Circulating tumor cells. |
| CTLA-4 | Cytotoxic T-lymphocyte antigen-4 |
| EC | Electron capture |
| EGFR | Epidermal growth factor receptor. |
| EMA | European Medicines Agency |
| EpCAM | Epithelial cell adhesion molecule |
| ET | Electron transfer |
| Fc | Fragment crystallizable region (domain) |
| FDA | Food and Drug Administration |
| GSK3β | Glycogen synthase kinase 3β |
| HDXMS | Hydrogen/deuterium exchange mass spectrometry |
| HVEM | Herpes virus entry mediator |
| HEL2i | Helicase insertion domain |
| ICIs | Immune checkpoint inhibitors |
| ICTs | Immune checkpoint therapies |
| IgG | Immunoglobulin G |
| ITH | Intertumoral heterogeneity |
| LAG-3 | Lymphocyte activation gene-3 |
| LC-MS | Liquid chromatography–mass spectrometry |
| mIHC/IF MS/MS | Multiplex immunohistochemistry/immunofluorescence tandem mass spectrometry |
| MT | Single-molecule magnetic tweezers |
| NSCLC | Non-small cell lung cancer (NSCLC) |
| OS | Overall survival |
| PD-1 | Programmed cell death-1 |
| PD-L1 | Programmed cell death ligand-1 |
| PFS | Progression-free survival |
| PTMs | Post-translational modifications |
| RIG-I | Retinoic acid inducible gene-I (RIG-I) |
| SAP | Serum amyloid P component |
| SDS-PAGE | Sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
| TMB | Tumor mutational burden |
| UVPD | Ultraviolet photodissociation |
| WGCNA | Weighted correlation network analysis |
References
- Queirolo, P.; Boutros, A.; Tanda, E.; Spagnolo, F.; Quaglino, P. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: A model of cancer immunotherapy. Semin. Cancer Biol. 2019, 59, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 34, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.P.; Khavkine Binstock, S.S.; Thu, K.L. CD47: The Next Frontier in Immune Checkpoint Blockade for Non-Small Cell Lung Cancer. Cancers 2023, 15, 5229. [Google Scholar] [CrossRef]
- Holder, A.M.; Dedeilia, A.; Sierra-Davidson, K.; Cohen, S.; Liu, D.; Parikh, A.; Boland, G.M. Defining clinically useful biomarkers of immune checkpoint inhibitors in solid tumours. Nat. Rev. Cancer 2024, 24, 498–512. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M.; Coupland, S.E.; Aittokallio, T.; Figueiredo, C.R. Resistance to immune checkpoint therapies by tumor-induced T-cell desertification and exclusion: Key mechanisms, prognostication and new therapeutic opportunities. Br. J. Cancer 2023, 129, 1212–1224. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Yates, J.R.; McCormack, A.L.; Schieltz, D.; Carmack, E.; Link, A. Direct analysis of protein mixtures by tandem mass spectrometry. J. Protein Chem. 1997, 16, 495–497. [Google Scholar] [CrossRef]
- May, J.C.; Morris, C.B.; McLean, J.A. Ion mobility collision cross section compendium. Anal. Chem. 2017, 89, 1032–1044. [Google Scholar] [CrossRef]
- Syka, J.E.P.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and Protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef]
- Zubarev, R.A.; Horn, D.M.; Fridriksson, E.K.; Kelleher, N.L.; Kruger, N.A.; Lewis, M.A.; Carpenter, B.K.; McLafferty, F.W. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000, 72, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Smyrnakis, A.; Levin, N.; Kosmopoulou, M.; Jha, A.; Fort, K.; Makarov, A.; Papanastasiou, D.; Mohammed, S. Characterization of an omnitrap-orbitrap platform equipped with Infrared multiphoton dissociation, ultraviolet photodissociation, and electron capture dissociation for the analysis of peptides and proteins. Anal. Chem. 2023, 95, 12039–12046. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Lin, S.; Karch, K.R.; Garcia, B.A. Bottom-up and middle-down proteomics have comparable accuracies in defining histone post-translational modification relative abundance and stoichiometry. Anal. Chem. 2015, 87, 3129–3133. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-generation sequencing technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Wenk, D.; Zuo, C.; Kislinger, T.; Sepiashvili, L. Recent developments in mass-spectrometry-based targeted proteomics of clinical cancer biomarkers. Clin. Proteom. 2024, 21, 6. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in Cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Eslami-S, Z.; Cortés-Hernández, L.E.; Sinoquet, L.; Gauthier, L.; Vautrot, V.; Cayrefourcq, L.; Avoscan, L.; Jacot, W.; Pouderoux, S.; Viala, M.; et al. Circulating tumor cells and PD-L1-positive small extracellular vesicles: The liquid biopsy combination for prognostic information in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2024, 130, 63–72. [Google Scholar] [CrossRef]
- Kiyotani, K.; Toyoshima, Y.; Nakamura, Y. Personalized immunotherapy in cancer precision medicine. Cancer Biol. Med. 2021, 18, 955–965. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Aggarwal, V.; Workman, C.J.; Vignali, D.A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.-L.; Wang, Y.-T.; Fu, W.-J.; Nan Lu, N.; Liu, Z.-S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, G.; Zhang, Q.; Zhao, S.; Li, X.; Cao, W.; Luo, H.; Zhou, C. In depth proteomic analysis identifies key gene signatures predicting therapeutic efficacy of anti-PD-1/PD-L1 monotherapy in non-small cell lung cancer. Transl. Lung Cancer Res. 2024, 13, 34–45. [Google Scholar] [CrossRef]
- Tan, Q.; Gao, R.; Zhang, X.; Yang, J.; Xing, P.; Yang, S.; Wang, D.; Wang, G.; Wang, S.; Yao, J.; et al. Longitudinal plasma proteomic analysis identifies biomarkers and combinational targets for anti-PD1-resistant cancer patients. Cancer Immunol. Immunother. 2024, 73, 47. [Google Scholar] [CrossRef]
- Administration FaD. 2023. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 20 August 2024).
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA approval summary: Pembrolizumab for the treatment of microsatellit instability-high solid tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA approval summary: Pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef] [PubMed]
- Peled, M.; Tocheva, A.S.; Sandigursky, S.; Mor, A. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Biol. Sci. 2017, 115, E468–E477. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Li, H.; van der Merwe, P.A.; Sivakumar, S. Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer 2022, 126, 1663–1675. [Google Scholar] [CrossRef]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, Q.; Wang, Y.N.; Jin, Y.; He, M.M.; Liu, Z.X.; Xu, R.H. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019, 5, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Gjoerup, O.; Brown, C.A.; Ross, J.S.; Huang, R.S.P.; Schrock, A.; Creeden, J.; Fabrizio, D.; Tolba, K. Identification and utilization of biomarkers to predict response to immune checkpoint inhibitors. AAPS J. 2020, 22, 132. [Google Scholar] [CrossRef] [PubMed]
- Arshiya, M.; Suneel, D.K. Biomarkers for response to anti–PD-1/anti–PD-L1 immune checkpoint Inhibitors: A Large meta-analysis. Oncology 2023, 37, 210–219. [Google Scholar]
- Nie, X.; Xia, L.; Gao, F.; Liu, L.; Yang, Y.; Chen, Y.; Duan, H.; Yao, Y.; Chen, Z.; Lu, S.; et al. Serum Metabolite Biomarkers Predictive of Response to PD-1 Blockade Therapy in non-small cell lung cancer. Front. Mol. Biosci. 2021, 8, 678753. [Google Scholar] [CrossRef]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E.; Baruch, E.N.; Tripple, V.; Arama-Chayoth, M.; Greenberg, E.; et al. Proteomics of melanoma response to immunotherapy reveals mitochondrial dependence. Cell 2019, 179, 236–250.e18. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Wang, Q.; Chen, P.; Zhao, L.; Gao, Y. Proteomic profiling and biomarker discovery for predicting the response to PD-1 inhibitor immunotherapy in gastric cancer patients. Front. Pharmacol. 2024, 15, 1349459. [Google Scholar] [CrossRef]
- Babačić, H.; Lehtiö, J.; de Coaña, Y.P.; Pernemalm, M.; Eriksson, H. In-depth plasma proteomics reveals increase in circulating PD-1 during anti-PD-1 immunotherapy in patients with metastatic cutaneous melanoma. J. Immunother. Cancer 2020, 8, e000204. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Klein, C.; Brinkmann, U.; Reichert, J.M.; Kontermann, R.E. The present and future of bispecific antibodies for cancer therapy. Nat. Rev. Drug Discov. 2024, 23, 301–319. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. Bispecific antibodies. Science 2021, 372, 916–917. [Google Scholar] [CrossRef]
- Mnatsakanyan, R.; Shema, G.; Basik, M.; Batist, G.; Borchers, C.H.; Sickmann, A.; Zahedi, R.P. Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev. Proteom. 2018, 6, 515–535. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.M.; Doig, A.J. Properties and identification of human protein drug targets. Bioinformatics 2009, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Comamala, G.; Krogh, C.C.; Nielsen, V.S.; Kutter, J.P.; Voglmeir, J.; Rand, K.D. Hydrogen/deuterium exchange mass spectrometry with Integrated electrochemical reduction and microchip-enabled deglycosylation for epitope mapping of heavily glycosylated and disulfide-bonded proteins. Anal. Chem. 2021, 93, 16330–16340. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 5, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, H.; Chai, Y.; Song, H.; Tong, Z.; Wang, Q.; Qi, J.; Wong, G. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 2017, 8, 14369. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Tit-oon, P.; Wonglangka, A.; Boonkanta, K.; Ruchirawat, M.; Fuangthong, M.; Sasisekharan, R.; Khongmanee, A. Intact mass analysis reveals the novel O-linked glycosylation on the stalk region of PD-1 protein. Sci. Rep. 2023, 13, 9631. [Google Scholar] [CrossRef]
- Iwai, Y.; Terawaki, S.; Ikegawa, M.; Okazaki, T.; Honjo, T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med. 2003, 198, 39–50. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member led to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.A.; Tsai, E.Y.; Liu, S.H.; Hsu Hung, S.D.; Chang, S.J.; Chao, C.H.; Lai, Y.J.; Yamaguchi, H.; Li, C.W. Post-translational Modification of PD-1: Potential Targets for Cancer Immunotherapy. Cancer Res. 2024, 84, 800–807. [Google Scholar]
- Larsen, M.R.; Højrup, P.; Roepstorff, P. Characterization of Gel-separated Glycoproteins Using Two-step Proteolytic Digestion Combined with Sequential Microcolumns and Mass Spectrometry. Mol. Cell. Proteom. 2005, 4, 107–119. [Google Scholar] [CrossRef]
- Darula, Z.; Medzihradszky, K.F. Glycan side reaction may compromise ETD-based glycopeptide identification. J. Am. Soc. Mass Spectrom. 2014, 25, 977–987. [Google Scholar] [CrossRef]
- Xi, X.; Zhao, W. Anti-Tumor Potential of Post-Translational Modifications of PD-1. Curr. Issues Mol. Biol. 2024, 46, 2119–2132. [Google Scholar] [CrossRef]
- Illiano, A.; Pinto, G.; Melchiorre, C.; Carpentieri, A.; Faraco, V.; Amoresano, A. Protein Glycosylation Investigated by Mass Spectrometry: An Overview. Cells 2020, 9, 1986. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, N.; Zaia, J. Effective use of mass spectrometry for glycan and glycopeptide structural analysis. Anal. Chem. 2012, 84, 3040–3048. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhu, J.; Xu, J.; Gu, B.; Zhao, Q.; Luo, C.; Gao, Z.; Chin, Y.E.; Cheng, X. Anti-tumor potential of PD-L1/PD-1 post-translational modifications. Immunology 2022, 167, 471–481. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Brown, C.; Sekhavati, F.; Cardenes, R.; Windmueller, C.; Dacosta, K.; Rodriguez-Canales, J.; Steele, K.E. CTLA-4 Immunohistochemistry and quantitative image analysis for Profiling of human cancers. J. Histochem. Cytochem. 2019, 67, 901–918. [Google Scholar] [CrossRef]
- Miller, R.E.; Fayen, J.D.; Mohammad, S.F.; Stein, K.; Kadereit, S.; Woods, K.D.; Sramkoski, R.M.; Jacobberger, J.W.; Templeton, D.; Shurin, S.B.; et al. Reduced CTLA-4 protein and messenger RNA expression in umbilical cord blood T lymphocytes. Exp. Hematol. 2002, 30, 738–744. [Google Scholar] [CrossRef]
- Wei, D.; Horton, K.L.; Chen, J.; Dong, L.; Chen, S.; Abdul-Hadi, K.; Zhang, T.T.; Casson, C.N.; Shaw, M.; Shiraishi, T.; et al. Development of a highly sensitive hybrid LC/MS assay for the quantitative measurement of CTLA-4 in human T Cells. Molecules 2023, 28, 3311. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, M.; Spodzieja, M.; Kuncewicz, K.; Rodziewicz-Motowidło, S.; Orlikowska, M. CD160 protein as a new therapeutic target in a battle against autoimmune, infectious and lifestyle diseases. Analysis of the structure, interactions and functions. Eur. J. Med. Chem. 2021, 224, 113694. [Google Scholar] [CrossRef]
- Liu, W.; Garrett, S.C.; Fedorov, E.V.; Ramagopal, U.A.; Garforth, S.J.; Bonanno, J.B.; Almo, S.C. Structural Basis of CD160: HVEM. Recognit. Struct. 2019, 27, 1286–1295. [Google Scholar] [CrossRef]
- Liu, W.; Chou, T.F.; Garrett-Thomson, S.C.; Seo, G.Y.; Fedorov, E.; Ramagopal, U.A.; Bonanno, J.B.; Wang, Q.; Kim, K.; Garforth, S.J.; et al. HVEM structures and mutants reveal distinct functions of binding to LIGHT and BTLA/CD160. J. Exp. Med. 2021, 218, e20211112. [Google Scholar] [CrossRef] [PubMed]
- Kuncewicz, K.; Spodzieja, M.; Sieradzan, A.; Karczyńska, A.; Dąbrowska, K.; Dadlez, M.; Speiser, D.E.; Laurent Derre, L.; Rodziewicz-Motowidło, S. A structural model of the immune checkpoint CD160–HVEM complex derived from HDX-mass spectrometry and molecular modeling. Oncotarget 2019, 10, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Liu, K.; Xiong, H. Role of BTLA in Immunity and Immune disorders. Front. Immunol. 2021, 12, 654960. [Google Scholar] [CrossRef]
- Andrzejczak, A.; Karabon, L. BTLA biology in cancer: From bench discoveries to clinical potentials. Biomark. Res. 2024, 12, 8. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, C.; Chang, M.R.; Devarkar, S.C.; Schweibenz, B.; Crynen, G.C.; Garcia-Ordonez, R.D.; Pascal, B.D.; Novick, S.J.; Patel, S.S.; et al. HDX-MS reveals dysregulated checkpoints that compromise discrimination against self RNA during RIG-I mediated autoimmunity. Nat. Commun. 2018, 9, 5366. [Google Scholar] [CrossRef]
- Lei, Y.; Fei, P.; Song, B.; Shi, W.; Luo, C.; Luo, D.; Li, D.; Chen, W.; Zheng, J. A loosened gating mechanism of RIG-I leads to autoimmune disorders. Nucleic Acids Res. 2022, 50, 5850–5863. [Google Scholar]
- Kato, H.; Fujita, T. RIG-I-like receptors and autoimmune diseases. Curr. Opin. Immunol. 2015, 37, 40–45. [Google Scholar] [CrossRef]
- Tapia-Rojo, R.; Mora, M.; Garcia-Manyes, S. Single-molecule magnetic tweezers to probe the equilibrium dynamics of individual proteins at physiologically relevant forces and timescales. Nat. Protoc. 2024, 19, 1779–1806. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Modok, S.; Mellor, H.R.; Callaghan, R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr. Opin. Pharmacol. 2006, 6, 350–354. [Google Scholar] [CrossRef]
- Agostini, M.; Traldi, P.; Hamdan, M. Mass Spectrometry Investigation of Some ATP-Binding Cassette. Medicina 2024, 60, 200. [Google Scholar] [CrossRef]
- Crowley, E.; McDevitt, C.A.; Callaghan, R. Generating Inhibitors of P-Glycoprotein: Where to, now? Multi-Drug Resist. Cancer 2010, 596, 405–432. [Google Scholar]
- Ruf, P.; Lindhofer, H. Induction of a long- lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001, 98, 2526–2534. [Google Scholar] [CrossRef]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Neijssen, J.; Cardoso, R.M.; Chevalier, K.M.; Wiegman, L.; Valerius, T.; Anderson, G.M.; Moores, S.L.; Schuurman, J.; Parren, P.W.; Strohl, W.R.; et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J. Biol. Chem. 2021, 296, 100641. [Google Scholar] [CrossRef]
- Zhu, Y.; Peng, B.J.; Kumar, S.; Stover, L.; Chang, J.Y.; Lyu, J.; Zhang, T.; Schrecke, S.; Azizov, D.; Russell, D.H.; et al. Polyamine detergents tailored for native mass spectrometry studies of membrane proteins. Nat. Commun. 2023, 14, 5676. [Google Scholar] [CrossRef]
- Levesque, I.; Juliano, B.R.; Parson, K.F.; Ruotolo, B.T. A Critical evaluation of detergent exchange methodologies for membrane protein native mass Spectrometry. J. Am. Soc. Mass Spectrom. 2023, 34, 2662–2671. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Reference | Method of Analysis | (a) Tumor, (b) Therapy | Potential Biomarkers in Response to ICI Therapy |
|---|---|---|---|
| [23] | 1. Trapped ion mobility spectrometry coupled with tandem mass spectrometry (MS/MS). 2. RNA-sequence analysis. Both analyses in 1 and 2 were supported with machine learning algorithms. |
| Gene expression profile: MOXD1, PHAF1, KRT7, ANKRD30A, TMEM184A, KIR3DL1, and KCNK4 According to the authors, the above profile predicted a durable response to anti-PD-1/PD-L1 |
| [35] | LC-MS/MS). |
| A metabolite panel consisting of hypoxanthine and histidine, identified in serum samples. |
| [36] | LC-MS/MS |
| Lipid and ketone body metabolism proteins in cancer cells |
| [37] | LC-MS/MS |
| A high abundance of activated CD8 T cells. Using machine learning, a set of 10 proteins was identified as potential biomarkers: COL15A1, SAMHD1, DHX15, PTDSS1, CFI, ORM2, VWF, APOA1, EMC2, and COL6A2 |
| [38] | High-resolution isoelectric focusing liquid chromatography–tandem mass spectrometry (HiRIEF LC-MS/MS) |
| 1. An increase in circulating PD-1 was observed during anti-PD-1 treatment. 2. Anti-PD-1 responders had an increase in plasma proteins involved in the T cell response, neutrophil degranulation, inflammation, cell adhesion, and immune suppression. 3. An association between plasma proteins and progression-free survival (PFS). The proteins included interleukin 6; interleukin 10; proline-rich acidic protein 1; desmocollin 3; C-C motif chemokine ligands 2, 3 and 4; and vascular endothelial growth factor A |
| Trade Name and Year of Approval | Indications | Approved in |
|---|---|---|
| Removab (2009) | Ovarian intraperitoneal ascites | Withdrawn in 2017. |
| Blincyto (2014) | To treat Philadelphia chromosome-negative relapsed or refractory B cell precursor acute lymphoblastic leukemia | USA, EU, Japan |
| Rybrevant (2021) | To treat locally advanced or metastatic non-small cell lung cancer with certain mutations | USA, EU |
| KImmtrak (2022) | To treat a form of unresectable or metastatic uveal melanoma | USA, EU |
| Lunsumio (2022) | To treat relapsed or refractory follicular lymphoma | USA, EU |
| Kaltanni (2022) | Hepatocellular carcinoma | China |
| Tecvayli (2022) | To treat relapsed or refractory multiple myeloma | USA, EU |
| Columvi (2023) | To treat relapsed or refractory diffuse large B cell lymphoma or large B cell lymphoma | USA, EU |
| Epkinly (2023) | To treat relapsed or refractory diffuse large B cell lymphoma | USA, EU, Japan |
| Talvey (2023) | Relapsed/refractory multiple myeloma | USA |
| Elrexfio (2023) | Relapsed/refractory multiple myeloma | USA, EU |
| Hemlibra (2017) | To prevent or reduce the frequency of bleeding episodes in hemophilia A with factor VIII inhibitors | USA, EU, Japan |
| Vabysmo (2022) | To treat neovascular (wet) age-related macular degenerated and diabetic macular edema | USA, EU, Japan |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agostini, M.; Traldi, P.; Hamdan, M. Proteomic Investigation of Immune Checkpoints and Some of Their Inhibitors. Int. J. Mol. Sci. 2024, 25, 9276. https://doi.org/10.3390/ijms25179276
Agostini M, Traldi P, Hamdan M. Proteomic Investigation of Immune Checkpoints and Some of Their Inhibitors. International Journal of Molecular Sciences. 2024; 25(17):9276. https://doi.org/10.3390/ijms25179276
Chicago/Turabian StyleAgostini, Marco, Pietro Traldi, and Mahmoud Hamdan. 2024. "Proteomic Investigation of Immune Checkpoints and Some of Their Inhibitors" International Journal of Molecular Sciences 25, no. 17: 9276. https://doi.org/10.3390/ijms25179276