
Genome Mining and Biological Engineering of Type III Borosins from Bacteria

Abstract
:1. Introduction
2. Results
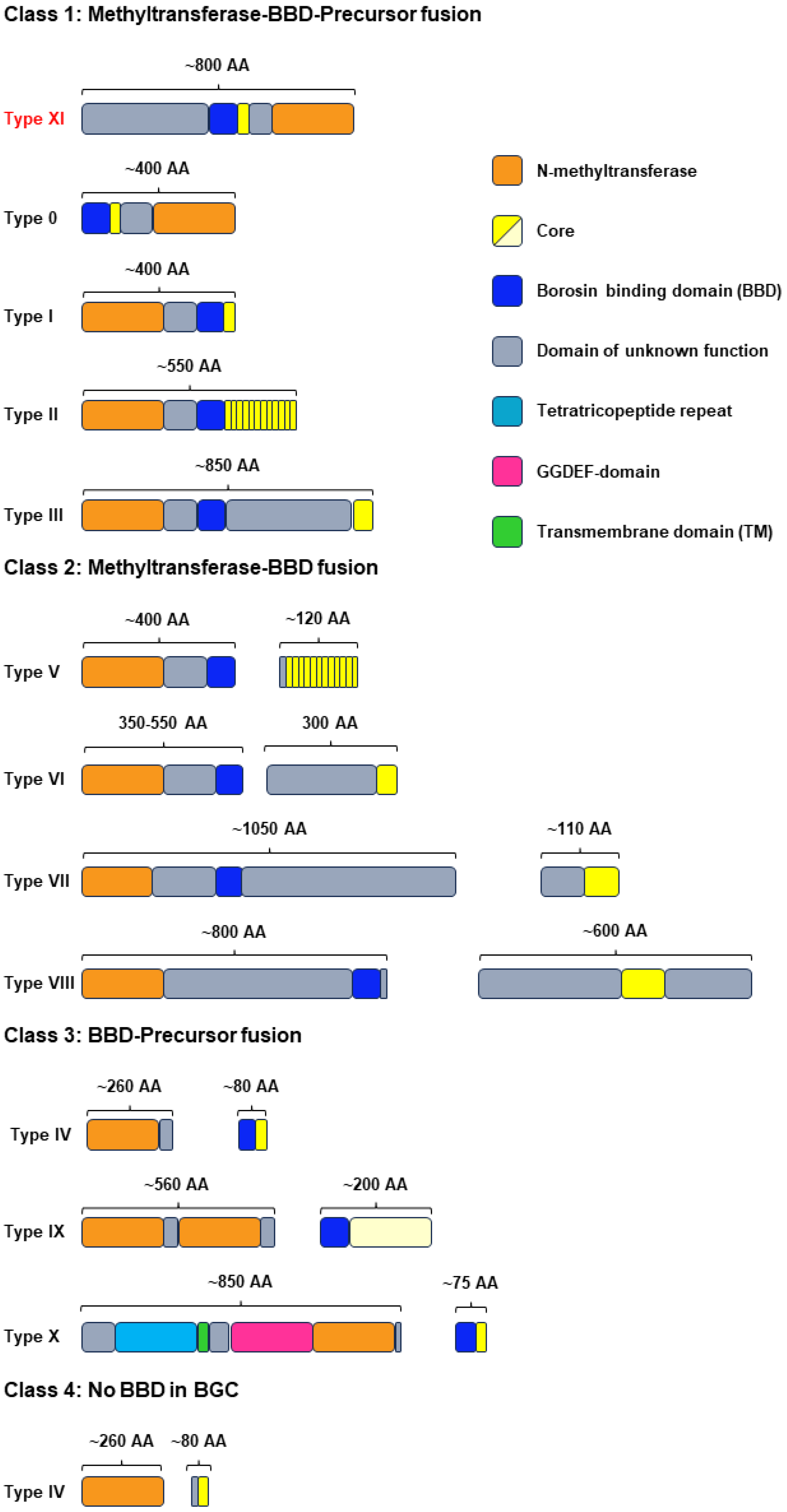
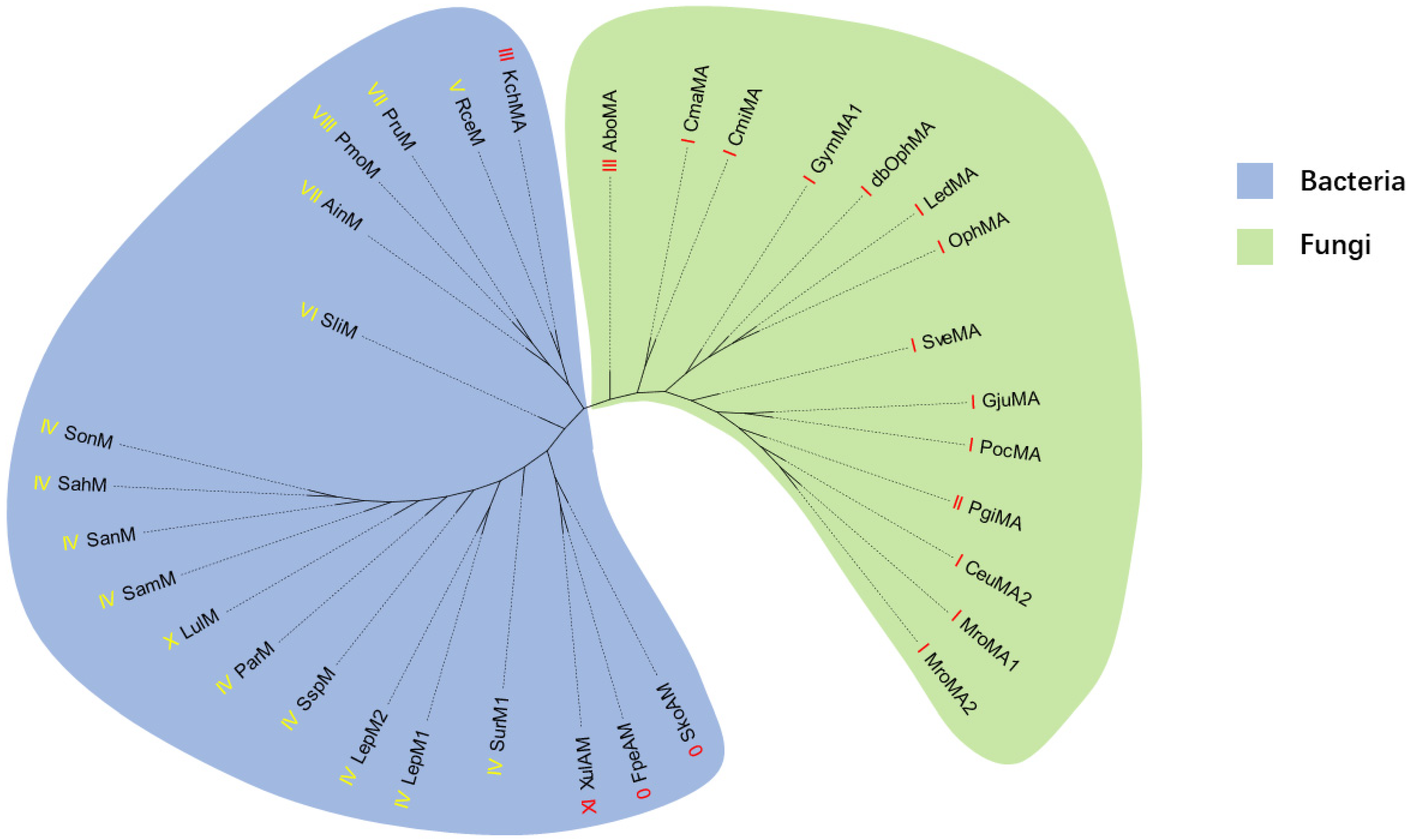
2.1. Genome Mining of New Type III Borosins
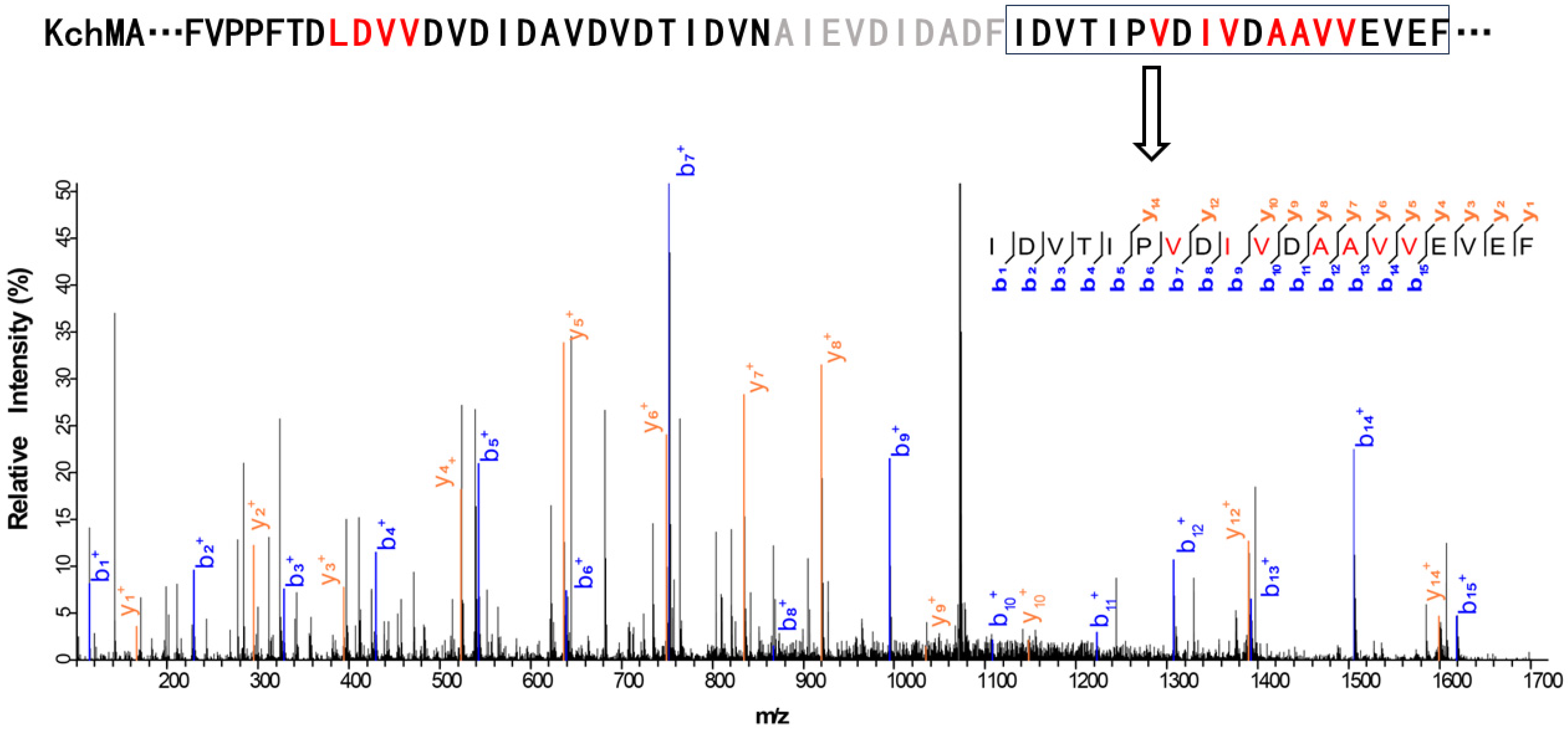
2.2. Validation of the New Type III Borosin KchMA Derived from Bacteria
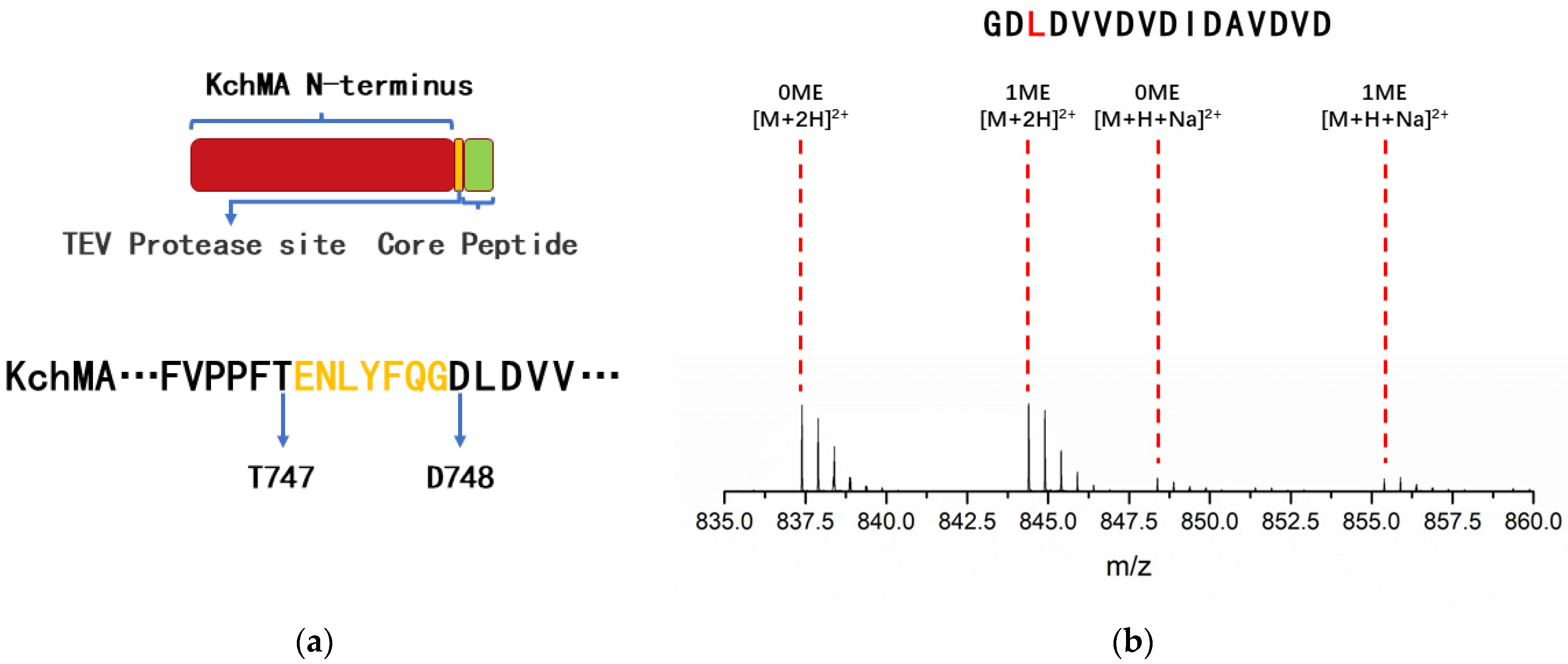
2.3. Introduction of Artificial Protease Sit to Release N-Methylation Peptide
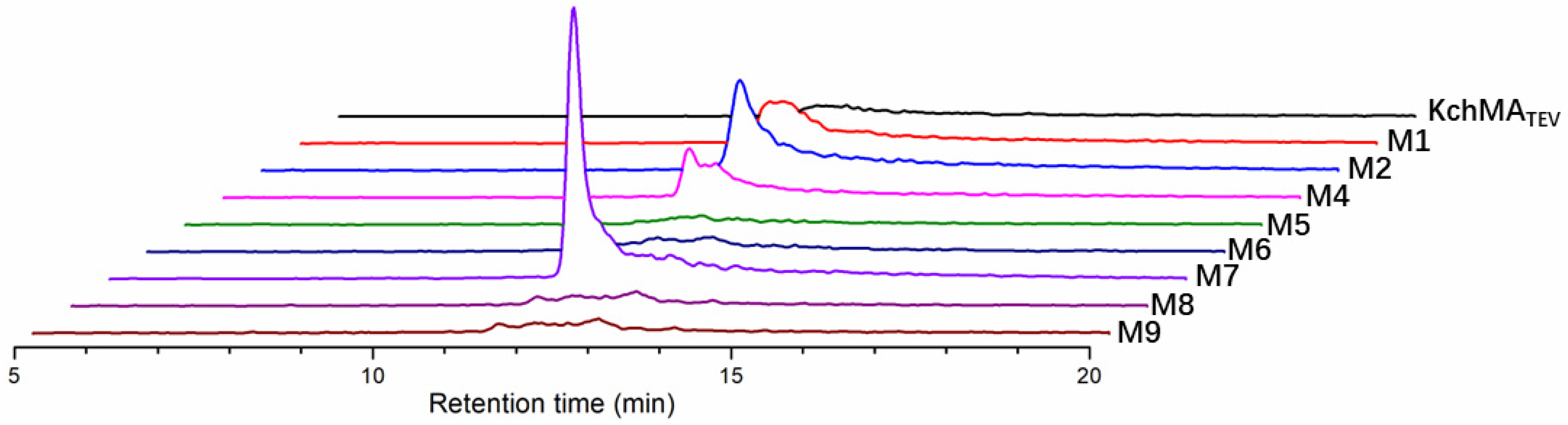
2.4. Rational Evolution of Methyltransferase Domain of KchMATEV
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Genome Mining
4.3. Construction of Phylogenetic Tree
4.4. AI-Assisted Protein Engineering
4.5. Construction of Mutants
4.6. Protein Expression
4.7. Protein Purification
4.8. The Enzymatic Hydrolysis of Borosins
4.9. LC-MS/MS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boulias, K.; Greer, E.L. Means, mechanisms and consequences of adenine methylation in DNA. Nat. Rev. Genet. 2022, 23, 411–428. [Google Scholar] [CrossRef]
- Oerum, S.; Meynier, V.; Catala, M.; Tisné, C. A comprehensive review of mA/mAm RNA methyltransferase structures. Nucleic Acids Res. 2021, 49, 7239–7255. [Google Scholar] [CrossRef] [PubMed]
- Afjehi-Sadat, L.; Garcia, B.A. Comprehending dynamic protein methylation with mass spectrometry. Curr. Opin. Chem. Biol. 2013, 17, 12–19. [Google Scholar] [CrossRef]
- Abdelraheem, E.; Thair, B.; Varela, R.F.; Jockmann, E.; Popadic, D.; Hailes, H.C.; Ward, J.M.; Iribarren, A.M.; Lewkowicz, E.S.; Andexer, J.N.; et al. Methyltransferases: Functions and Applications. ChemBioChem 2022, 23, e202200212. [Google Scholar] [CrossRef]
- Schönherr, H.; Cernak, T. Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Gu, W.J.; Schmidt, E.W. Applying Promiscuous RiPP Enzymes to Peptide Backbone-Methylation Chemistry. ACS Chem. Biol. 2022, 17, 2165–2178. [Google Scholar] [CrossRef]
- Li, X.F.; Wang, N.C.; Liu, Y.H.; Li, W.P.; Bai, X.Y.; Liu, P.; He, C.Y. Backbone N-methylation of peptides: Advances in synthesis and applications in pharmaceutical drug development. Bioorg. Chem. 2023, 141, 106892. [Google Scholar] [CrossRef]
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738. [Google Scholar] [CrossRef]
- Montalbán-López, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, N.S.; Kälin, N.; Helf, M.J.; Piel, J.; Freeman, M.F.; Künzler, M. Autocatalytic backbone N-methylation in a family of ribosomal peptide natural products. Nat. Chem. Biol. 2017, 13, 833–835. [Google Scholar] [CrossRef]
- Ramm, S.; Krawczyk, B.; Mühlenweg, A.; Poch, A.; Mösker, E.; Süssmuth, R.D. A Self-Sacrificing-Methyltransferase Is the Precursor of the Fungal Natural Product Omphalotin. Angew. Chem. Int. Ed. 2017, 56, 9994–9997. [Google Scholar] [CrossRef]
- Ongpipattanakul, C.; Nair, S.K. Molecular Basis for Autocatalytic Backbone-Methylation in RiPP Natural Product Biosynthesis. ACS Chem. Biol. 2018, 13, 2989–2999. [Google Scholar] [CrossRef]
- Song, H.; van der Velden, N.S.; Shiran, S.L.; Bleiziffer, P.; Zach, C.; Sieber, R.; Imani, A.S.; Krausbeck, F.; Aebi, M.; Freeman, M.F.; et al. A molecular mechanism for the enzymatic methylation of nitrogen atoms within peptide bonds. Sci. Adv. 2018, 4, eaat2720. [Google Scholar] [CrossRef] [PubMed]
- Quijano, M.R.; Zach, C.; Miller, F.S.; Lee, A.R.; Imani, A.S.; Künzler, M.; Freeman, M.F. Distinct Autocatalytic α-Methylating Precursors Expand the Borosin RiPP Family of Peptide Natural Products. J. Am. Chem. Soc. 2019, 141, 9637–9644. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.S.; Crone, K.K.; Jensen, M.R.; Shaw, S.; Harcombe, W.R.; Elias, M.H.; Freeman, M.F. Conformational rearrangements enable iterative backbone-methylation in RiPP biosynthesis. Nat. Commun. 2021, 12, 5355. [Google Scholar] [CrossRef]
- Imani, A.S.; Lee, A.R.; Vishwanathan, N.; de Waal, F.; Freeman, M.F. Diverse Protein Architectures and α-N-Methylation Patterns Define Split Borosin RiPP Biosynthetic Gene Clusters. ACS Chem. Biol. 2022, 17, 908–917. [Google Scholar] [CrossRef]
- Cho, H.; Lee, H.; Hong, K.; Chung, H.; Song, I.; Lee, J.S.; Kim, S. Bioinformatic Expansion of Borosins Uncovers Trans-Acting Peptide Backbone-Methyltransferases in Bacteria. Biochemistry 2022, 61, 183–194. [Google Scholar] [CrossRef]
- Lee, A.L.; Carter, R.S.; Imani, A.S.; Dommaraju, S.R.; Hudson, G.A.; Mitchell, D.A.; Freeman, M.F. Discovery of Borosin Catalytic Strategies and Function through Bioinformatic Profiling. ACS Chem. Biol. 2024, 19, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Madhusoodanan, N.; Lee, J.H.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef]
- Khersonsky, O.; Lipsh, R.; Avizemer, Z.; Ashani, Y.; Goldsmith, M.; Leader, H.; Dym, O.; Rogotner, S.; Trudeau, D.L.; Prilusky, J.; et al. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72, 178–186. [Google Scholar] [CrossRef]
- Soucy, S.M.; Huang, J.L.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Song, H.G.; Burton, A.J.; Shirran, S.L.; Fahrig-Kamarauskaite, J.; Kaspar, H.; Muir, T.W.; Künzler, M.; Naismith, J.H. Engineering of a Peptide α-N-Methyltransferase to Methylate Non-Proteinogenic Amino Acids. Angew. Chem. Int. Ed. 2021, 60, 14319–14323. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Ongpipattanakul, C.; Nair, S.K. Bioconjugate Platform for Iterative Backbone N-Methylation of Peptides. ACS Catal. 2022, 12, 14006–14014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.K.; Gong, J.S.; Feng, D.T.; Su, C.; Li, H.; Rao, Z.M.; Lu, Z.M.; Shi, J.S.; Xu, Z.H. Geometric Remodeling of Nitrilase Active Pocket Based on ALF-Scanning Strategy To Enhance Aromatic Nitrile Substrate Preference and Catalytic Efficiency. Appl. Environ. Microbiol. 2023, 89, e0022023. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.C.; Guo, X.S.; Zhang, H.E.; Dubey, G.K.; Geng, Z.Z.; Fierke, C.A.; Xu, S.Q.; Hampton, J.T.; Liu, W.R. Leveraging a Phage-Encoded Noncanonical Amino Acid: A Novel Pathway to Potent and Selective Epigenetic Reader Protein Inhibitors. Acs Cent. Sci. 2024, 10, 782–792. [Google Scholar] [CrossRef]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef]
- Oberg, N.; Zallot, R.; Gerlt, J.A. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) Web Resou-rce for Genomic Enzymology Tools. J. Mol. Biol. 2023, 435, 168018. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Xie, J.M.; Chen, Y.R.; Cai, G.J.; Cai, R.L.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Gerber, A.S.; Hartl, D.L. Genetic Applications of an Inverse Polymerase Chain-Reaction. Genetics 1988, 120, 621–623. [Google Scholar] [CrossRef]
- Zhao, J.F.; Ji, W.J.; Ji, X.J.; Zhang, Q. Biochemical Characterization of an Arginine 2,3-Aminomutase with Dual Substrate Specificity. Chin. J. Chem. 2020, 38, 959–962. [Google Scholar] [CrossRef]
- Wang, L.H.; Li, D.Q.; Fu, Y.; Wang, H.P.; Zhang, J.F.; Yuan, Z.F.; Sun, R.X.; Zeng, R.; He, S.M.; Gao, W. PFind 2.0: A software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2985–2991. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants Name | Position 49 | Position 188 |
|---|---|---|
| KchMATEV | C | F |
| M1 | V | Y |
| M2 | T | F |
| M3 | A | W |
| M4 | M | M |
| M5 | S | V |
| M6 | C | L |
| M7 | L | H |
| M8 | G | T |
| M9 | I | I |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Guo, S.; Zhang, W.; Deng, Z.; Zhang, Q.; Ding, W. Genome Mining and Biological Engineering of Type III Borosins from Bacteria. Int. J. Mol. Sci. 2024, 25, 9350. https://doi.org/10.3390/ijms25179350
Xu K, Guo S, Zhang W, Deng Z, Zhang Q, Ding W. Genome Mining and Biological Engineering of Type III Borosins from Bacteria. International Journal of Molecular Sciences. 2024; 25(17):9350. https://doi.org/10.3390/ijms25179350
Chicago/Turabian StyleXu, Kuang, Sijia Guo, Wei Zhang, Zixin Deng, Qi Zhang, and Wei Ding. 2024. "Genome Mining and Biological Engineering of Type III Borosins from Bacteria" International Journal of Molecular Sciences 25, no. 17: 9350. https://doi.org/10.3390/ijms25179350