
Receptor Pharmacogenomics: Deciphering Genetic Influence on Drug Response

 , and
, and
Abstract
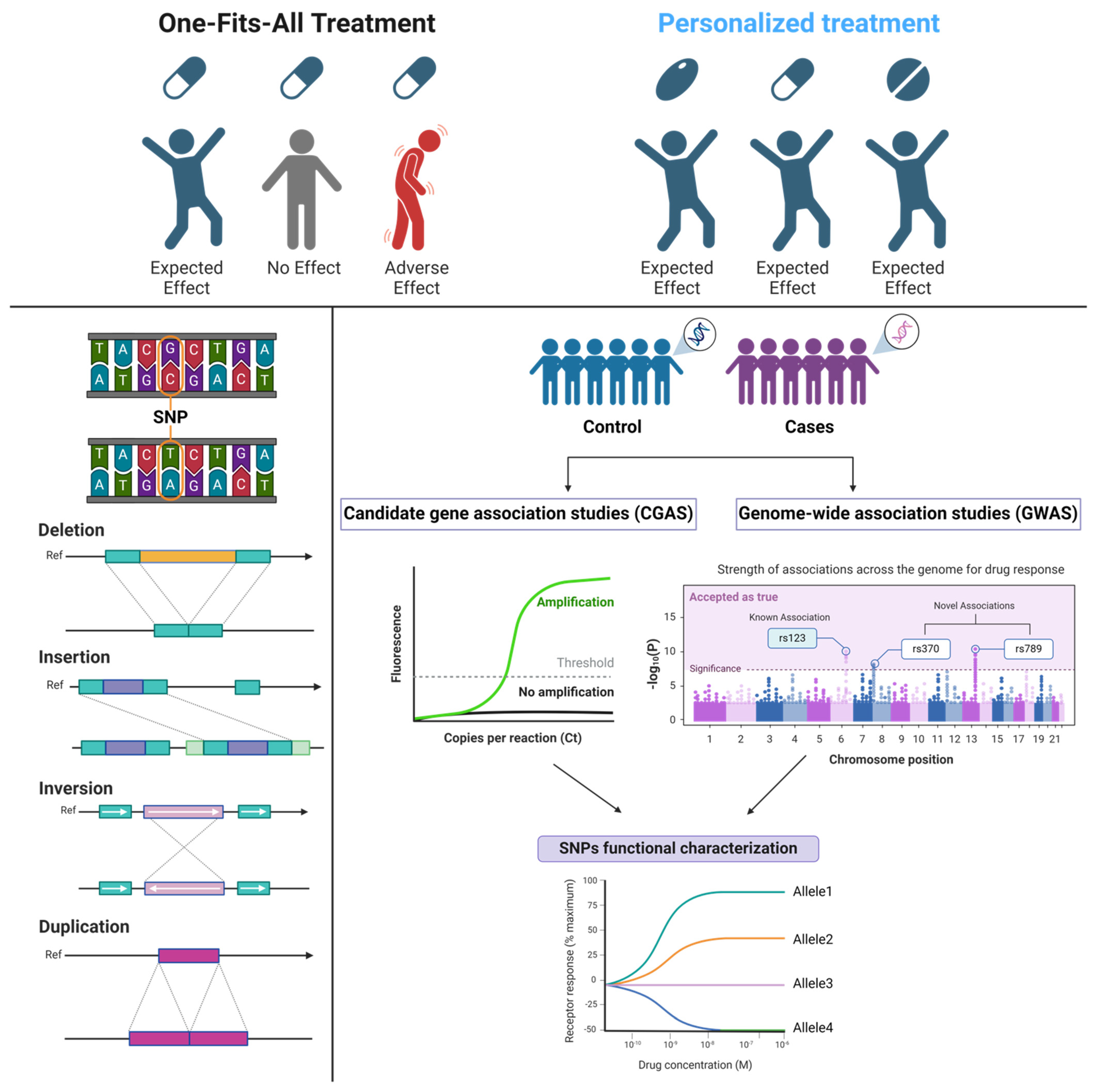
:1. Introduction
2. Pharmacogenomics: Mechanism and Implementation
2.1. An Overview on Pharmacogenes Polymorphism
2.2. Clinical Implementation of Pharmacogenomics
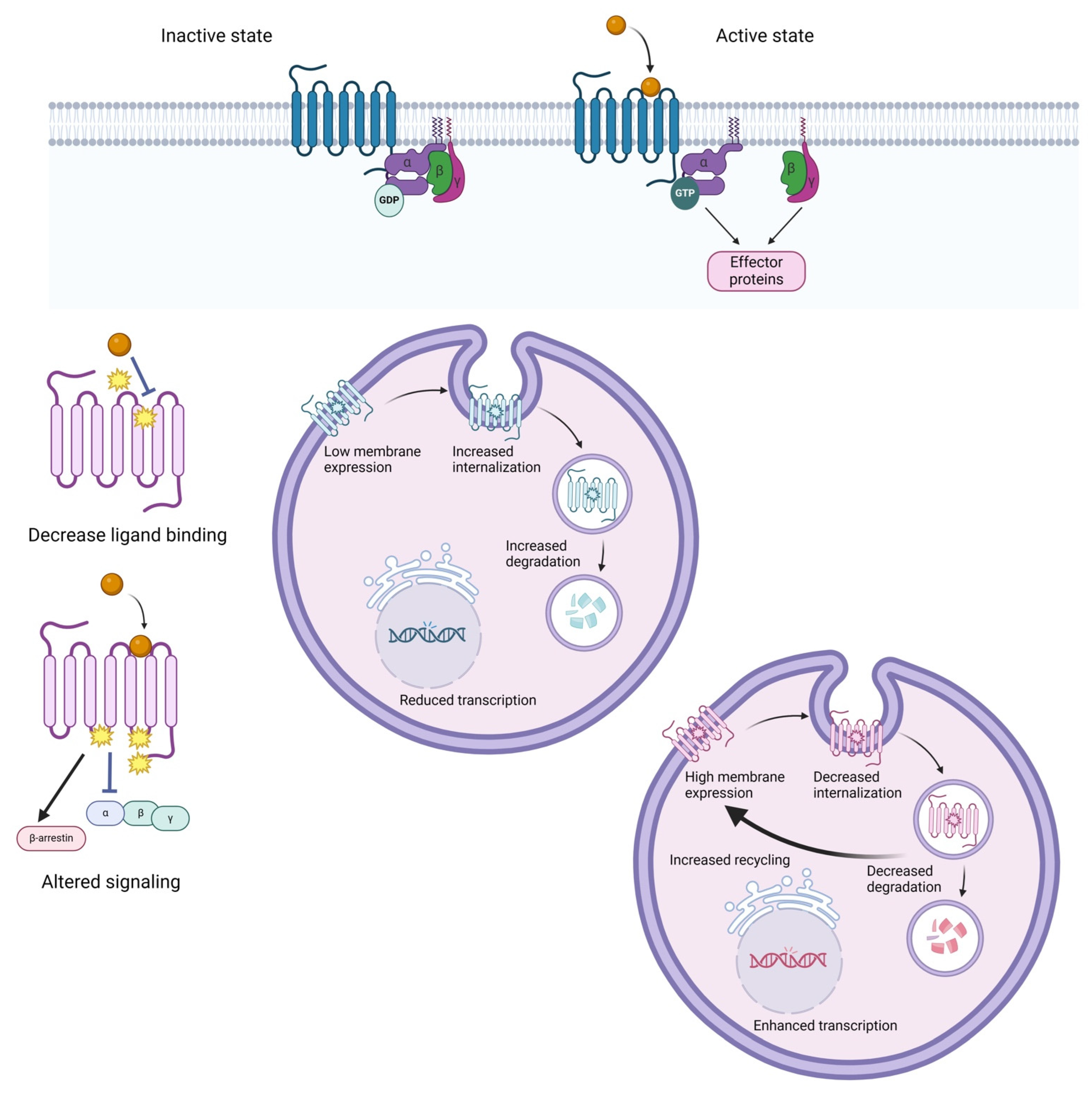
3. The Influence of GPCR Genetic Variation on Drug Response
3.1. GPCR Pharmacogenetics Overview
3.2. Antihypertension Therapy and β1-Adrenoreceptor Polymorphism
3.3. Analgesics Treatment and µ1-Opioid Receptor Polymorphism
3.4. Antipsychotic Drug Treatment and Dopamine/Serotonin Receptor Polymorphism
3.5. Antidepressants and Serotonin Receptors
4. EGFR, a PGx-Guided Therapy Model
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Human Genome Sequencing Consortium, Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [CrossRef]
- Lu, C.; Ahmed, R.; Lamri, A.; Anand, S.S. Use of race, ethnicity, and ancestry data in health research. PLoS Glob. Public Health 2022, 2, e0001060. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; Jamuar, S.S.; Moody, A.R.; Karnes, J.H.; Varga, O.; Hedensted, S.; Spreafico, R.; Hafler, D.A.; McKinney, E.F. From Big Data to Precision Medicine. Front. Med. 2019, 6, 34. [Google Scholar] [CrossRef]
- White, C.; Scott, R.; Paul, C.L.; Ackland, S.P. Pharmacogenomics in the era of personalised medicine. Med. J. Aust. 2022, 217, 510–513. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Table of Pharmacogenetic Associations. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 28 March 2024).
- Klein, T.E.; Chang, J.T.; Cho, M.K.; Easton, K.L.; Fergerson, R.; Hewett, M.; Lin, Z.; Liu, Y.; Liu, S.; Oliver, D.E.; et al. Integrating genotype and phenotype information: An overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base. Pharmacogenom. J. 2001, 1, 167–170. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2021, 110, 563–572. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Pritchard, D.; Patel, J.N.; Stephens, L.E.; McLeod, H.L. Comparison of FDA Table of Pharmacogenetic Associations and Clinical Pharmacogenetics Implementation Consortium guidelines. Am. J. Health Syst. Pharm. 2022, 79, 993–1005. [Google Scholar] [CrossRef]
- Rukov, J.L.; Vinther, J.; Shomron, N. Pharmacogenomics genes show varying perceptibility to microRNA regulation. Pharmacogenet. Genom. 2011, 21, 251–262. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 28 March 2024).
- Jimeno, A.; Hidalgo, M. Pharmacogenomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Biochim. Biophys. Acta 2006, 1766, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Abou Diwan, E.; Zeitoun, R.I.; Abou Haidar, L.; Cascorbi, I.; Khoueiry Zgheib, N. Implementation and obstacles of pharmacogenetics in clinical practice: An international survey. Br. J. Clin. Pharmacol. 2019, 85, 2076–2088. [Google Scholar] [CrossRef]
- He, Y.; Hoskins, J.M.; McLeod, H.L. Copy number variants in pharmacogenetic genes. Trends Mol. Med. 2011, 17, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.; Kars, M.E.; Wu, Y.; Bayrak, C.S.; Stenson, P.D.; Cooper, D.N.; Schlessinger, A.; Itan, Y. Genome-wide prediction of pathogenic gain- and loss-of-function variants from ensemble learning of a diverse feature set. Genome Med. 2023, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; Van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lauschke, V.M. The genetic landscape of major drug metabolizing cytochrome P450 genes-an updated analysis of population-scale sequencing data. Pharmacogenom. J. 2022, 22, 284–293. [Google Scholar] [CrossRef]
- Pirmohamed, M. Pharmacogenomics: Current status and future perspectives. Nat. Rev. Genet. 2023, 24, 350–362. [Google Scholar] [CrossRef]
- Ramirez-Bello, J.; Jimenez-Morales, M. Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases. Gac. Med. Mex. 2017, 153, 238–250. [Google Scholar]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- David, S. A current guide to candidate gene association studies. Trends Genet. 2021, 37, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Arwood, M.J.; Stocco, G.; Singh, S.; Karnes, J.H.; Ramsey, L.B. Planning and Conducting a Pharmacogenetics Association Study. Clin. Pharmacol. Ther. 2021, 110, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.T.; Chin, Y.M.; Low, S.K. The Roles of Common Variation and Somatic Mutation in Cancer Pharmacogenomics. Oncol. Ther. 2019, 7, 1–32. [Google Scholar] [CrossRef]
- Duarte, R.R.R.; Brentani, H.; Powell, T.R. Ditching candidate gene association studies: Lessons from psychiatric genetics. Braz. J. Psychiatry 2021, 43, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Group, S.C.; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Linskey, D.W.; Linskey, D.C.; McLeod, H.L.; Luzum, J.A. The need to shift pharmacogenetic research from candidate gene to genome-wide association studies. Pharmacogenomics 2021, 22, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Song, L.; Guo, Y.; Chen, C.; Yang, J. From genetic associations to genes: Methods, applications, and challenges. Trends Genet. 2024, 40, 642–667. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Yee, S.W.; Mushiroda, T.; Weinshilboum, R.M.; Ratain, M.J.; Kubo, M. Genome-wide association studies of drug response and toxicity: An opportunity for genome medicine. Nat. Rev. Drug Discov. 2017, 16, 70. [Google Scholar] [CrossRef]
- Shuldiner, A.R.; Relling, M.V.; Peterson, J.F.; Hicks, J.K.; Freimuth, R.R.; Sadee, W.; Pereira, N.L.; Roden, D.M.; Johnson, J.A.; Klein, T.E.; et al. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: Overcoming challenges of real-world implementation. Clin. Pharmacol. Ther. 2013, 94, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, L.H.; Lee, C.R.; Duarte, J.D.; Nutescu, E.A.; Weitzel, K.W.; Stouffer, G.A.; Johnson, J.A. Implementation of inpatient models of pharmacogenetics programs. Am. J. Health Syst. Pharm. 2016, 73, 1944–1954. [Google Scholar] [CrossRef]
- van der Wouden, C.H.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; Davila-Fajardo, C.L.; Deneer, V.H.; Dolzan, V.; Ingelman-Sundberg, M.; Jonsson, S.; Karlsson, M.O.; et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2017, 101, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Borobia, A.M.; Dapia, I.; Tong, H.Y.; Arias, P.; Munoz, M.; Tenorio, J.; Hernandez, R.; Garcia Garcia, I.; Gordo, G.; Ramirez, E.; et al. Clinical Implementation of Pharmacogenetic Testing in a Hospital of the Spanish National Health System: Strategy and Experience over 3 Years. Clin. Transl. Sci. 2018, 11, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Wick, J.A.; Schmidlen, T.; Grande, K.; Moretz, C.; Ashcraft, K.; Green, J.; Moyer, N.; Blaxall, B.C. Implementing comprehensive pharmacogenomics in a community hospital-associated primary care setting. J. Am. Pharm. Assoc. 2023, 63, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Munar, M.Y.; Singh, H. Drug dosing adjustments in patients with chronic kidney disease. Am. Fam. Phys. 2007, 75, 1487–1496. [Google Scholar]
- Gloy, V.; Schmitt, A.M.; Dublin, P.; Hirt, J.; Axfors, C.; Kuk, H.; Pereira, T.V.; Locher, C.; Caquelin, L.; Walter-Claudi, M.; et al. The evidence base of US Food and Drug Administration approvals of novel cancer therapies from 2000 to 2020. Int. J. Cancer 2023, 152, 2474–2484. [Google Scholar] [CrossRef] [PubMed]
- Huddart, R.; Sangkuhl, K.; Whirl-Carrillo, M.; Klein, T.E. Are Randomized Controlled Trials Necessary to Establish the Value of Implementing Pharmacogenomics in the Clinic? Clin. Pharmacol. Ther. 2019, 106, 284–286. [Google Scholar] [CrossRef]
- Marrero, R.J.; Cicali, E.J.; Arwood, M.J.; Eddy, E.; DeRemer, D.; Ramnaraign, B.H.; Daily, K.C.; Jones, D., Jr.; Cook, K.J.; Cavallari, L.H.; et al. How to Transition from Single-Gene Pharmacogenetic Testing to Preemptive Panel-Based Testing: A Tutorial. Clin. Pharmacol. Ther. 2020, 108, 557–565. [Google Scholar] [CrossRef]
- Morris, S.A.; Alsaidi, A.T.; Verbyla, A.; Cruz, A.; Macfarlane, C.; Bauer, J.; Patel, J.N. Cost Effectiveness of Pharmacogenetic Testing for Drugs with Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines: A Systematic Review. Clin. Pharmacol. Ther. 2022, 112, 1318–1328. [Google Scholar] [CrossRef]
- van der Wouden, C.H.; Marck, H.; Guchelaar, H.J.; Swen, J.J.; van den Hout, W.B. Cost-Effectiveness of Pharmacogenomics-Guided Prescribing to Prevent Gene-Drug-Related Deaths: A Decision-Analytic Model. Front. Pharmacol. 2022, 13, 918493. [Google Scholar] [CrossRef]
- Chenoweth, M.J.; Giacomini, K.M.; Pirmohamed, M.; Hill, S.L.; van Schaik, R.H.N.; Schwab, M.; Shuldiner, A.R.; Relling, M.V.; Tyndale, R.F. Global Pharmacogenomics Within Precision Medicine: Challenges and Opportunities. Clin. Pharmacol. Ther. 2020, 107, 57–61. [Google Scholar] [CrossRef]
- US Food and Drug Administration Guidance for Industry. Pharmacogenomic Data Submissions. Available online: https://www.fda.gov/media/72420/download (accessed on 9 June 2024).
- US Food and Drug Administration Guidance for Industry. Clinical Pharmacogenomics: Premarket Evaluation in Early-Phase Clinical Studies and Recommendations for Labeling. Available online: https://www.fda.gov/media/84923/download (accessed on 9 June 2024).
- EMA: Guideline on the Use of Pharmacogenetic Methodologies in the Pharmacokinetic Evaluation of Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-pharmacogenetic-methodologies-pharmacokinetic-evaluation-medicinal-products_en.pdf (accessed on 9 June 2024).
- Abdullah-Koolmees, H.; van Keulen, A.M.; Nijenhuis, M.; Deneer, V.H.M. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front. Pharmacol. 2020, 11, 595219. [Google Scholar] [CrossRef]
- Chenchula, S.; Atal, S.; Uppugunduri, C.R.S. A review of real-world evidence on preemptive pharmacogenomic testing for preventing adverse drug reactions: A reality for future health care. Pharmacogenom. J. 2024, 24, 9. [Google Scholar] [CrossRef]
- O’Donnell, P.H.; Bush, A.; Spitz, J.; Danahey, K.; Saner, D.; Das, S.; Cox, N.J.; Ratain, M.J. The 1200 patients project: Creating a new medical model system for clinical implementation of pharmacogenomics. Clin. Pharmacol. Ther. 2012, 92, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, O.; Kuivaniemi, H.; Tromp, G.; Faucett, W.A.; Li, R.; Manolio, T.A.; Sanderson, S.C.; Kannry, J.; Zinberg, R.; Basford, M.A.; et al. The Electronic Medical Records and Genomics (eMERGE) Network: Past, present, and future. Genet. Med. 2013, 15, 761–771. [Google Scholar] [CrossRef]
- Gottesman, O.; Scott, S.A.; Ellis, S.B.; Overby, C.L.; Ludtke, A.; Hulot, J.S.; Hall, J.; Chatani, K.; Myers, K.; Kannry, J.L.; et al. The CLIPMERGE PGx Program: Clinical implementation of personalized medicine through electronic health records and genomics-pharmacogenomics. Clin. Pharmacol. Ther. 2013, 94, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.M.; Haidar, C.E.; Wilkinson, M.R.; Crews, K.R.; Baker, D.K.; Kornegay, N.M.; Yang, W.; Pui, C.H.; Reiss, U.M.; Gaur, A.H.; et al. PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166C, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Norris, M.; Dalton, R.; Alam, B.; Eddy, E.; Nguyen, K.A.; Cavallari, L.H.; Sumfest, J.; Wiisanen, K.; Cicali, E.J. Lessons from clinical implementation of a preemptive pharmacogenetic panel as part of a testing pilot program with an employer-sponsored medical plan. Front. Genet. 2023, 14, 1249003. [Google Scholar] [CrossRef]
- Luczak, T.S.; Schillo, P.J.; Renier, C.M.; Waring, S.C.; Friday, B.B. Feasibility of preemptive pharmacogenetic testing in colorectal cancer patients within a community oncology setting. J. Oncol. Pharm. Pract. 2022, 28, 842–849. [Google Scholar] [CrossRef]
- Lteif, C.; Eddy, E.; Terrell, J.; Cavallari, L.H.; Malaty, J.; Duarte, J.D. Feasibility of preemptive pharmacogenetic testing and improvement of medication treatment satisfaction among medically underserved patients. Clin. Transl. Sci. 2024, 17, e13692. [Google Scholar] [CrossRef]
- Maruf, A.A.; Bousman, C.A. Approaches and hurdles of implementing pharmacogenetic testing in the psychiatric clinic. PCN Rep. 2022, 1, e26. [Google Scholar] [CrossRef]
- Kabbani, D.; Akika, R.; Wahid, A.; Daly, A.K.; Cascorbi, I.; Zgheib, N.K. Pharmacogenomics in practice: A review and implementation guide. Front. Pharmacol. 2023, 14, 1189976. [Google Scholar] [CrossRef] [PubMed]
- Luzum, J.A.; Petry, N.; Taylor, A.K.; Van Driest, S.L.; Dunnenberger, H.M.; Cavallari, L.H. Moving Pharmacogenetics Into Practice: It’s All About the Evidence! Clin. Pharmacol. Ther. 2021, 110, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Karas Kuzelicki, N.; Prodan Zitnik, I.; Gurwitz, D.; Llerena, A.; Cascorbi, I.; Siest, S.; Simmaco, M.; Ansari, M.; Pazzagli, M.; Di Resta, C.; et al. Pharmacogenomics education in medical and pharmacy schools: Conclusions of a global survey. Pharmacogenomics 2019, 20, 643–657. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): Advances in structures, mechanisms, and drug discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G Protein-Coupled Receptor Signaling Through beta-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Ruttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Reichel, E.; Hahn, L.B.; Cowley, G.S.; Yandell, D.W.; Sandberg, M.A.; Berson, E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, V.; Canaff, L.; Yun, F.H.; Scillitani, A.; Battista, C.; Muscarella, L.A.; Wong, B.Y.; Notarangelo, A.; D’Agruma, L.; Sacco, M.; et al. Calcium-sensing receptor (CASR) mutations in hypercalcemic states: Studies from a single endocrine clinic over three years. J. Clin. Endocrinol. Metab. 2010, 95, 1819–1829. [Google Scholar] [CrossRef]
- Spanakis, E.; Milord, E.; Gragnoli, C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: Review and missense mutation significance. J. Cell. Physiol. 2008, 217, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sun, P.; Siwko, S.; Liu, M.; Xiao, J. The role of GPCRs in bone diseases and dysfunctions. Bone Res. 2019, 7, 19. [Google Scholar] [CrossRef]
- Liu, T.; Ji, R.L.; Tao, Y.X. Naturally occurring mutations in G protein-coupled receptors associated with obesity and type 2 diabetes mellitus. Pharmacol. Ther. 2022, 234, 108044. [Google Scholar] [CrossRef]
- Fukami, M.; Suzuki, E.; Igarashi, M.; Miyado, M.; Ogata, T. Gain-of-function mutations in G-protein-coupled receptor genes associated with human endocrine disorders. Clin. Endocrinol. 2018, 88, 351–359. [Google Scholar] [CrossRef]
- Yang, L.K.; Hou, Z.S.; Tao, Y.X. Biased signaling in naturally occurring mutations of G protein-coupled receptors associated with diverse human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165973. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef]
- Edward Zhou, X.; Melcher, K.; Eric Xu, H. Structural biology of G protein-coupled receptor signaling complexes. Protein Sci. 2019, 28, 487–501. [Google Scholar] [CrossRef]
- Schoneberg, T.; Liebscher, I. Mutations in G Protein-Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73, 89–119. [Google Scholar] [CrossRef]
- Fuentes, A.V.; Pineda, M.D.; Venkata, K.C.N. Comprehension of Top 200 Prescribed Drugs in the US as a Resource for Pharmacy Teaching, Training and Practice. Pharmacy 2018, 6, 43. [Google Scholar] [CrossRef]
- Johnson, J.A.; Zineh, I.; Puckett, B.J.; McGorray, S.P.; Yarandi, H.N.; Pauly, D.F. Beta 1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin. Pharmacol. Ther. 2003, 74, 44–52. [Google Scholar] [CrossRef]
- Si, D.; Wang, J.; Xu, Y.; Chen, X.; Zhang, M.; Zhou, H. Association of common polymorphisms in beta1-adrenergic receptor with antihypertensive response to carvedilol. J. Cardiovasc. Pharmacol. 2014, 64, 306–309. [Google Scholar] [CrossRef]
- Fayed, M.S.; Saleh, M.A.; Sabri, N.A.; Elkholy, A.A. beta1-adrenergic receptor polymorphisms: A possible genetic predictor of bisoprolol response in acute coronary syndrome. Future Sci. OA 2023, 9, FSO895. [Google Scholar] [CrossRef]
- Magvanjav, O.; McDonough, C.W.; Gong, Y.; McClure, L.A.; Talbert, R.L.; Horenstein, R.B.; Shuldiner, A.R.; Benavente, O.R.; Mitchell, B.D.; Johnson, J.A.; et al. Pharmacogenetic Associations of beta1-Adrenergic Receptor Polymorphisms With Cardiovascular Outcomes in the SPS3 Trial (Secondary Prevention of Small Subcortical Strokes). Stroke 2017, 48, 1337–1343. [Google Scholar] [CrossRef]
- Turner, S.; Francis, B.; Vijverberg, S.; Pino-Yanes, M.; Maitland-van der Zee, A.H.; Basu, K.; Bignell, L.; Mukhopadhyay, S.; Tavendale, R.; Palmer, C.; et al. Childhood asthma exacerbations and the Arg16 beta2-receptor polymorphism: A meta-analysis stratified by treatment. J. Allergy Clin. Immunol. 2016, 138, 107–113.e105. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, S.; Alvala, M.; Nayarisseri, A.; Sharda, S.; Goud, H.; Mundluru, H.P.; Singh, S.K. Molecular dynamic simulations reveal suboptimal binding of salbutamol in T164I variant of beta2 adrenergic receptor. PLoS ONE 2017, 12, e0186666. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Lin, H.C.; Lee, W.P.; Chu, C.J.; Lin, M.W.; Lee, F.Y.; Hou, M.C.; Jap, J.S.; Lee, S.D. Association of the G-protein and alpha2-adrenergic receptor gene and plasma norepinephrine level with clonidine improvement of the effects of diuretics in patients with cirrhosis with refractory ascites: A randomised clinical trial. Gut 2010, 59, 1545–1553. [Google Scholar] [CrossRef]
- Bond, C.; LaForge, K.S.; Tian, M.; Melia, D.; Zhang, S.; Borg, L.; Gong, J.; Schluger, J.; Strong, J.A.; Leal, S.M.; et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: Possible implications for opiate addiction. Proc. Natl. Acad. Sci. USA 1998, 95, 9608–9613. [Google Scholar] [CrossRef]
- Mura, E.; Govoni, S.; Racchi, M.; Carossa, V.; Ranzani, G.N.; Allegri, M.; van Schaik, R.H. Consequences of the 118A>G polymorphism in the OPRM1 gene: Translation from bench to bedside? J. Pain Res. 2013, 6, 331–353. [Google Scholar] [CrossRef]
- Knapman, A.; Santiago, M.; Connor, M. Buprenorphine signalling is compromised at the N40D polymorphism of the human mu opioid receptor in vitro. Br. J. Pharmacol. 2014, 171, 4273–4288. [Google Scholar] [CrossRef]
- Zhang, J.P.; Lencz, T.; Malhotra, A.K. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: A meta-analysis. Am. J. Psychiatry 2010, 167, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, X.; Xiang, Q.; Zhou, S.; Zhao, N.; Xie, Q.; Zhao, X.; Zhou, Y.; Cui, Y. Association between dopamine receptor gene polymorphisms and effects of risperidone treatment: A systematic review and meta-analysis. Basic Clin. Pharmacol. Toxicol. 2019, 124, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Robinson, D.G.; Napolitano, B.; Sevy, S.; Kane, J.M.; Goldman, D.; Malhotra, A.K. DRD2 promoter region variation predicts antipsychotic-induced weight gain in first episode schizophrenia. Pharmacogenet. Genom. 2010, 20, 569–572. [Google Scholar] [CrossRef]
- Bosia, M.; Lorenzi, C.; Pirovano, A.; Guglielmino, C.; Cocchi, F.; Spangaro, M.; Bramanti, P.; Smeraldi, E.; Cavallaro, R. COMT Val158Met and 5-HT1A-R -1019 C/G polymorphisms: Effects on the negative symptom response to clozapine. Pharmacogenomics 2015, 16, 35–44. [Google Scholar] [CrossRef]
- Hong, C.J.; Chen, T.J.; Yu, Y.W.; Tsai, S.J. Response to fluoxetine and serotonin 1A receptor (C-1019G) polymorphism in Taiwan Chinese major depressive disorder. Pharmacogenom. J. 2006, 6, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Fukuda, T.; Wakeno, M.; Okugawa, G.; Takekita, Y.; Watanabe, S.; Yamashita, M.; Hosoi, Y.; Azuma, J.; Kinoshita, T.; et al. Effect of 5-HT1A gene polymorphisms on antidepressant response in major depressive disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 115–123. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, R.; Cabaleiro, T.; Ochoa, D.; Roman, M.; Borobia, A.M.; Carcas, A.J.; Ayuso, C.; Novalbos, J.; Abad-Santos, F. Pharmacodynamic genetic variants related to antipsychotic adverse reactions in healthy volunteers. Pharmacogenomics 2013, 14, 1203–1214. [Google Scholar] [CrossRef]
- Sanguesa, E.; Fernandez-Egea, E.; Concha, J.; Garcia, C.B.; Ribate, M.P. Impact of Pharmacogenetic Testing on Clozapine Treatment Efficacy in Patients with Treatment-Resistant Schizophrenia. Biomedicines 2024, 12, 597. [Google Scholar] [CrossRef]
- Blasi, G.; Selvaggi, P.; Fazio, L.; Antonucci, L.A.; Taurisano, P.; Masellis, R.; Romano, R.; Mancini, M.; Zhang, F.; Caforio, G.; et al. Variation in Dopamine D2 and Serotonin 5-HT2A Receptor Genes is Associated with Working Memory Processing and Response to Treatment with Antipsychotics. Neuropsychopharmacology 2015, 40, 1600–1608. [Google Scholar] [CrossRef]
- Basu, A.; Chadda, R.K.; Sood, M.; Kaur, H.; Kukreti, R. Association of serotonin transporter (SLC6A4) and receptor (5HTR1A, 5HTR2A) polymorphisms with response to treatment with escitalopram in patients with major depressive disorder: A preliminary study. Indian J. Med. Res. 2015, 142, 40–45. [Google Scholar] [CrossRef]
- Horstmann, S.; Lucae, S.; Menke, A.; Hennings, J.M.; Ising, M.; Roeske, D.; Muller-Myhsok, B.; Holsboer, F.; Binder, E.B. Polymorphisms in GRIK4, HTR2A, and FKBP5 show interactive effects in predicting remission to antidepressant treatment. Neuropsychopharmacology 2010, 35, 727–740. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, L.; Zhao, S. Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules 2021, 11, 936. [Google Scholar] [CrossRef]
- Zhang, F.; Steinberg, S.F. S49G and R389G polymorphisms of the beta(1)-adrenergic receptor influence signaling via the cAMP-PKA and ERK pathways. Physiol. Genom. 2013, 45, 1186–1192. [Google Scholar] [CrossRef]
- Parvez, B.; Chopra, N.; Rowan, S.; Vaglio, J.C.; Muhammad, R.; Roden, D.M.; Darbar, D. A common beta1-adrenergic receptor polymorphism predicts favorable response to rate-control therapy in atrial fibrillation. J. Am. Coll. Cardiol. 2012, 59, 49–56. [Google Scholar] [CrossRef]
- Pacanowski, M.A.; Gong, Y.; Cooper-Dehoff, R.M.; Schork, N.J.; Shriver, M.D.; Langaee, T.Y.; Pepine, C.J.; Johnson, J.A.; Investigators, I. beta-adrenergic receptor gene polymorphisms and beta-blocker treatment outcomes in hypertension. Clin. Pharmacol. Ther. 2008, 84, 715–721. [Google Scholar] [CrossRef]
- Sobczak, M.; Salaga, M.; Storr, M.A.; Fichna, J. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: Current concepts and future perspectives. J. Gastroenterol. 2014, 49, 24–45. [Google Scholar] [CrossRef]
- Halikere, A.; Popova, D.; Scarnati, M.S.; Hamod, A.; Swerdel, M.R.; Moore, J.C.; Tischfield, J.A.; Hart, R.P.; Pang, Z.P. Addiction associated N40D mu-opioid receptor variant modulates synaptic function in human neurons. Mol. Psychiatry 2020, 25, 1406–1419. [Google Scholar] [CrossRef]
- Taqi, M.M.; Faisal, M.; Zaman, H. OPRM1 A118G Polymorphisms and Its Role in Opioid Addiction: Implication on Severity and Treatment Approaches. Pharmacogenom. Pers. Med. 2019, 12, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, I.; Boku, S.; Takahashi, Y. Psychopharmacology of atypical antipsychotic drugs: From the receptor binding profile to neuroprotection and neurogenesis. Psychiatry Clin. Neurosci. 2015, 69, 243–258. [Google Scholar] [CrossRef]
- Pan, Y.; Yao, J.; Wang, B. Association of dopamine D1 receptor gene polymorphism with schizophrenia: A meta-analysis. Neuropsychiatr. Dis. Treat. 2014, 10, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Basile, V.S.; Jin, Y.; Masellis, M.; Badri, F.; Keator, D.; Wu, J.C.; Alva, G.; Carreon, D.T.; Bunney, W.E., Jr.; et al. D1 receptor alleles predict PET metabolic correlates of clinical response to clozapine. Mol. Psychiatry 2003, 8, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Ota, V.K.; Spindola, L.N.; Gadelha, A.; dos Santos Filho, A.F.; Santoro, M.L.; Christofolini, D.M.; Bellucco, F.T.; Ribeiro-dos-Santos, A.K.; Santos, S.; Mari Jde, J.; et al. DRD1 rs4532 polymorphism: A potential pharmacogenomic marker for treatment response to antipsychotic drugs. Schizophr. Res. 2012, 142, 206–208. [Google Scholar] [CrossRef]
- de Matos, L.P.; Santana, C.V.; Souza, R.P. Meta-analysis of dopamine receptor D1 rs4532 polymorphism and susceptibility to antipsychotic treatment response. Psychiatry Res. 2015, 229, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C.; Chen, S.F.; Chen, C.H.; Lin, C.C.; Chen, S.J.; Chen, Y.J.; Luu, S.U. Effects of DRD2/ANKK1 gene variations and clinical factors on aripiprazole efficacy in schizophrenic patients. J. Psychiatr. Res. 2009, 43, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Robinson, D.G.; Gallego, J.A.; John, M.; Yu, J.; Addington, J.; Tohen, M.; Kane, J.M.; Malhotra, A.K.; Lencz, T. Association of a Schizophrenia Risk Variant at the DRD2 Locus With Antipsychotic Treatment Response in First-Episode Psychosis. Schizophr. Bull. 2015, 41, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Magistrelli, L.; Ferrari, M.; Furgiuele, A.; Milner, A.V.; Contaldi, E.; Comi, C.; Cosentino, M.; Marino, F. Polymorphisms of Dopamine Receptor Genes and Parkinson’s Disease: Clinical Relevance and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 3781. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, Y.; Lv, Q.; Sheng, Y.H.; Chen, L.; Li, M.; Shen, L.; Huai, C.; Yi, Z.; Cui, D.; et al. Genetic Association of Olanzapine Treatment Response in Han Chinese Schizophrenia Patients. Front. Pharmacol. 2019, 10, 177. [Google Scholar] [CrossRef]
- Naveen, M.; Patil, A.N.; Pattanaik, S.; Kaur, A.; Banerjee, D.; Grover, S. ABCB1 and DRD3 polymorphism as a response predicting biomarker and tool for pharmacogenetically guided clozapine dosing in Asian Indian treatment resistant schizophrenia patients. Asian J. Psychiatr. 2020, 48, 101918. [Google Scholar] [CrossRef]
- Zhou, W.; Chang, W.; Yan, Y.; Shen, L.; Li, W.; Yi, Z.; Qin, S. Pharmacogenetics analysis of serotonin receptor gene variants and clinical response to risperidone in Han Chinese schizophrenic patients. Neurosci. Lett. 2018, 683, 202–206. [Google Scholar] [CrossRef]
- Gupta, M.; Jain, S.; Moily, N.S.; Kaur, H.; Jajodia, A.; Purushottam, M.; Kukreti, R. Genetic studies indicate a potential target 5-HTR(3B) for drug therapy in schizophrenia patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159b, 1006–1008. [Google Scholar] [CrossRef]
- Melkersson, K.I.; Gunes, A.; Dahl, M.L. Impact of serotonin receptor 2A gene haplotypes on C-peptide levels in clozapine- and olanzapine-treated patients. Hum. Psychopharmacol. 2010, 25, 347–352. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef]
- Commons, K.G.; Linnros, S.E. Delayed Antidepressant Efficacy and the Desensitization Hypothesis. ACS Chem. Neurosci. 2019, 10, 3048–3052. [Google Scholar] [CrossRef]
- Chauhan, M.; Parry, R.; Bobo, W.V. Vilazodone for Major Depression in Adults: Pharmacological Profile and an Updated Review for Clinical Practice. Neuropsychiatr. Dis. Treat. 2022, 18, 1175–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, Y.; Li, J.; Ma, H.; Jin, Q.; Wang, Y.; Wu, L.; Zhu, G. Association between the 5-HT1A receptor gene polymorphism (rs6295) and antidepressants: A meta-analysis. Int. Clin. Psychopharmacol. 2012, 27, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Q.; Li, X.R.; He, L.; He, G.; Yu, T.; Sun, X.L. 5-HTR1A and 5-HTR2A genetic polymorphisms and SSRI antidepressant response in depressive Chinese patients. Neuropsychiatr. Dis. Treat. 2016, 12, 1623–1629. [Google Scholar] [CrossRef]
- Scutt, G.; Overall, A.; Scott, R.; Patel, B.; Hachoumi, L.; Yeoman, M.; Wright, J. Does the 5-HT(1A) rs6295 polymorphism influence the safety and efficacy of citalopram therapy in the oldest old? Ther. Adv. Drug Saf. 2018, 9, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Bousman, C.A.; Stevenson, J.M.; Ramsey, L.B.; Sangkuhl, K.; Hicks, J.K.; Strawn, J.R.; Singh, A.B.; Ruano, G.; Mueller, D.J.; Tsermpini, E.E.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6, CYP2C19, CYP2B6, SLC6A4, and HTR2A Genotypes and Serotonin Reuptake Inhibitor Antidepressants. Clin. Pharmacol. Ther. 2023, 114, 51–68. [Google Scholar] [CrossRef]
- Wan, Y.-S.; Zhai, X.-J.; Tan, H.-A.; Ai, Y.-S.; Zhao, L.-B. Associations between the 1438A/G, 102T/C, and rs7997012G/A polymorphisms of HTR2A and the safety and efficacy of antidepressants in depression: A meta-analysis. Pharmacogenom. J. 2021, 21, 200–215. [Google Scholar] [CrossRef]
- Lin, J.Y.; Jiang, M.Y.; Kan, Z.M.; Chu, Y. Influence of 5-HTR2A genetic polymorphisms on the efficacy of antidepressants in the treatment of major depressive disorder: A meta-analysis. J. Affect. Disord. 2014, 168, 430–438. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, L.; Sun, Y.H.; Ren, C.; Chen, J.; Wu, Z.Q.; Jiang, Y.H.; Lv, X.L. Association of HTR2A T102C and A-1438G polymorphisms with susceptibility to major depressive disorder: A meta-analysis. Neurol. Sci. 2014, 35, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Yuan, F.; Ren, D.; Zhu, Y.; Bi, Y.; Hu, J.; Guo, Z.; Xu, F.; Niu, W.; Wu, X.; et al. HTR1A and HTR2A variants may not predict venlafaxine treatment response in China Han population with major depressive disorder. Psychiatry Res. 2018, 270, 1179–1180. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Borgeaud, M.; Parikh, K.; Banna, G.L.; Kim, F.; Olivier, T.; Le, X.; Addeo, A. Unveiling the Landscape of Uncommon EGFR Mutations in NSCLC-A Systematic Review. J. Thorac. Oncol. 2024, 19, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liu, X.; Zheng, L.; Wang, S.; Zhang, J.; Xiong, S.; Zhang, P.; Jiao, Z.; Zhao, G.; Zhou, C.; et al. Comprehensive profiling of EGFR mutation subtypes reveals genomic-clinical associations in non-small-cell lung cancer patients on first-generation EGFR inhibitors. Neoplasia 2023, 38, 100888. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Das, R.; Endres, N.F.; Eastwood, M.P.; Wemmer, D.E.; Kuriyan, J.; Shaw, D.E. Architecture and membrane interactions of the EGF receptor. Cell 2013, 152, 557–569. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Amelia, T.; Kartasasmita, R.E.; Ohwada, T.; Tjahjono, D.H. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022, 27, 819. [Google Scholar] [CrossRef]
- Thomas, R.; Weihua, Z. Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Leek, R.; Fox, S.B.; Moore, J.W.; Taylor, C.; Chambers, P.; Harris, A.L. EGFR mutations in exons 18–21 in sporadic breast cancer. Ann. Oncol. 2007, 18, 203–205. [Google Scholar] [CrossRef]
- Metzger, B.; Chambeau, L.; Begon, D.Y.; Faber, C.; Kayser, J.; Berchem, G.; Pauly, M.; Boniver, J.; Delvenne, P.; Dicato, M.; et al. The human epidermal growth factor receptor (EGFR) gene in European patients with advanced colorectal cancer harbors infrequent mutations in its tyrosine kinase domain. BMC Med. Genet. 2011, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Gonzalez, J.P.; Chaves, J.J.; Parra-Medina, R. Multiple mutations in the EGFR gene in lung cancer: A systematic review. Transl. Lung Cancer Res. 2022, 11, 2148–2163. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Nagasaka, M.; Zhu, V.W.; Lim, S.M.; Greco, M.; Wu, F.; Ou, S.I. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors For Advanced EGFR+ NSCLC. J. Thorac. Oncol. 2021, 16, 740–763. [Google Scholar] [CrossRef]
- Hanley, M.J.; Camidge, D.R.; Fram, R.J.; Gupta, N. Mobocertinib: Mechanism of action, clinical, and translational science. Clin. Transl. Sci. 2024, 17, e13766. [Google Scholar] [CrossRef]
- Cai, W.Q.; Zeng, L.S.; Wang, L.F.; Wang, Y.Y.; Cheng, J.T.; Zhang, Y.; Han, Z.W.; Zhou, Y.; Huang, S.L.; Wang, X.W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- PharmGKB Database. EGFR. Clinical Annotations. Available online: https://www.pharmgkb.org (accessed on 20 June 2024).
- Costa, D.B. Kinase inhibitor-responsive genotypes in EGFR mutated lung adenocarcinomas: Moving past common point mutations or indels into uncommon kinase domain duplications and rearrangements. Transl. Lung Cancer Res. 2016, 5, 331–337. [Google Scholar] [CrossRef]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal growth factor receptor in pancreatic cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, X.; Ou, K.; Zhang, M.; Gao, L.; Yang, L. Advanced pancreatic cancer with KRAS wild-type and EGFR-sensitive mutation respond favorably to furmonertinib: A case report. Front. Oncol. 2023, 13, 1151178. [Google Scholar] [CrossRef]
- Mody, J.; Kamgar, M. Pancreatic Adenocarcinoma with Co-Occurrence of KRAS and EGFR Mutations: Case Report and Literature Review. Case Rep. Oncol. 2024, 17, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Doleschal, B.; Petzer, A.; Rumpold, H. Current concepts of anti-EGFR targeting in metastatic colorectal cancer. Front. Oncol. 2022, 12, 1048166. [Google Scholar] [CrossRef]
- Garcia-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Taberna, M.; Oliva, M.; Mesia, R. Cetuximab-Containing Combinations in Locally Advanced and Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 383. [Google Scholar] [CrossRef]
- Rehmani, H.S.; Issaeva, N. EGFR in head and neck squamous cell carcinoma: Exploring possibilities of novel drug combinations. Ann. Transl. Med. 2020, 8, 813. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Chen, W.; Cote, M.L.; Bollig-Fischer, A.; Land, S.; Schwartz, A.G.; Bepler, G. Fibroblast growth factor receptor 1 amplification in non-small cell lung cancer by quantitative real-time PCR. PLoS ONE 2013, 8, e79820. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Croucher, D.R.; Kennedy, S.P.; An, Q.; Pilkington, G.J.; Hafizi, S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis 2016, 5, e266. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tang, N.; Thompson, R.C.; Mobley, B.C.; Clark, S.W.; Sarkaria, J.N.; Wang, J. InsR/IGF1R Pathway Mediates Resistance to EGFR Inhibitors in Glioblastoma. Clin. Cancer Res. 2016, 22, 1767–1776. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | Ligands | Effects | Ref. |
|---|---|---|---|---|
| ADRB1 | rs1801253:R389G | Metoprolol Carvedilol Bisoprolol | ↓ Antihypertensive response | [76,77,78] |
| rs1801252:S49G | Atenolol | ↑ Adverse cardiovascular events | [79] | |
| ADRB2 | rs1042713:R16G | LABA | ↑ Risk of asthma exacerbation | [80] |
| rs1800888:T164I | Salbutamol | ↓ Ligand binding | [81] | |
| ADRA2C | rs11269124: Del 322–325 | Clonidine | ↑ Efficacy | [82] |
| OPM1 | rs1799971:N40D | Endogenous opioids Buprenorphine | ↑ β-endorphin binding affinity ↓ Expression ↓ Efficacy for buprenorphine | [83,84,85] |
| DRD2 | rs1799732 DRD2 PROM −141C Ins/Del | Antipsychotics * | ↓ Efficacy ↑ Weight gain | [86,87,88] |
| HTR1A | rs6295 | Clozapine | ↑ Efficacy | [89] |
| Fluoxetine Paroxetine Milnacipran | ↑ Efficacy | [90,91] | ||
| HTR2A | rs6313 | Risperidone | ↑ Adverse cardiovascular events | [92] |
| rs6314 | Clozapine | ↑ Efficacy | [93] | |
| Olanzapine | ↑ Efficacy | [94] | ||
| rs6311 | Escitalopram | ↑Memory loss | [95] | |
| rs7997012 | Antidepressants | ↑ Treatment outcome ↑ Remission | [96] |
| Drug | Drug Class | Labeling Sections | Type of Cancer | Targeted Mutation |
|---|---|---|---|---|
| Erlotinib | TKI (I) | Indications and usage, dosage and administration, adverse reactions, and clinical studies | NSCLC Pancreatic cancer | Exon 19 deletion Exon 21 L858R |
| Gefitinib | TKI (I) | Indications and usage, dosage and administration, and clinical studies | NSCLC | Exon 19 deletion Exon 21 L858R |
| Afatinib | TKI (II) | Indications and usage, dosage and administration, adverse reactions, and clinical studies | NSCLC | Exon 19 deletion Exon 21 L858R |
| Dacomitinib | TKI (II) | Indications and usage, dosage, and administration, adverse reactions, use in specific populations, and clinical studies | NSCLC | Exon 19 deletion Exon 21 L858R |
| Osimertinib | TKI (III) | Indications and usage, dosage and administration, adverse reactions, and clinical studies | NSCLC | Exon 19 deletion Exon 21 L858R T790M |
| Mobocertinib | TKI | Indications and usage, dosage and administration, adverse reactions, and clinical studies | NSCLC | Exon 20 insertion |
| Amivantamab-vmjw | Mab | Indications and usage, dosage and administration, adverse reactions, and clinical studies | NSCLC | Exon 20 insertion |
| Cetuximab | Mab | Indications and usage, dosage and administration, adverse reactions, and clinical studies | CRC SCCHN | |
| Panitumumab | Mab | Adverse reactions, clinical pharmacology, and clinical studies | CRC |
| Drug | EGFR Variant | Level of Evidence | Pharmacology | Phenotype |
|---|---|---|---|---|
| Gefitinib | rs121434568 | 1A | Efficacy | Carcinoma, NSCLC |
| rs121434569 | 2B | Efficacy | Carcinoma, NSCLC, drug resistance | |
| rs2293347 | 3 | Efficacy | Carcinoma, NSCLC | |
| rs712829 | 3 | Efficacy | Neoplasms | |
| rs11568315 | 3 | Efficacy | Carcinoma, NSCLC | |
| Erlotinib | rs121434569 | 2B | Efficacy | Adenocarcinoma, carcinoma, NSCLC, drug resistance, lung neoplasms |
| rs712829 | 3 | Efficacy | Neoplasms | |
| rs712829 | 3 | Toxicity | Neoplasms | |
| rs2227983 | 3 | Toxicity | Carcinoma, NSCLC, colorectal neoplasms, neoplasms, pancreatic neoplasms | |
| Cetuximab | rs2227983 | 3 | Efficacy | Head and neck neoplasms |
| rs712829 | 3 | Efficacy | Colorectal neoplasms | |
| rs712830 | 3 | Toxicity | Colorectal neoplasms | |
| Panitumumab | rs712829 | 3 | Efficacy | Colorectal neoplasms |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anghel, S.A.; Dinu-Pirvu, C.-E.; Costache, M.-A.; Voiculescu, A.M.; Ghica, M.V.; Anuța, V.; Popa, L. Receptor Pharmacogenomics: Deciphering Genetic Influence on Drug Response. Int. J. Mol. Sci. 2024, 25, 9371. https://doi.org/10.3390/ijms25179371
Anghel SA, Dinu-Pirvu C-E, Costache M-A, Voiculescu AM, Ghica MV, Anuța V, Popa L. Receptor Pharmacogenomics: Deciphering Genetic Influence on Drug Response. International Journal of Molecular Sciences. 2024; 25(17):9371. https://doi.org/10.3390/ijms25179371
Chicago/Turabian StyleAnghel, Sorina Andreea, Cristina-Elena Dinu-Pirvu, Mihaela-Andreea Costache, Ana Maria Voiculescu, Mihaela Violeta Ghica, Valentina Anuța, and Lăcrămioara Popa. 2024. "Receptor Pharmacogenomics: Deciphering Genetic Influence on Drug Response" International Journal of Molecular Sciences 25, no. 17: 9371. https://doi.org/10.3390/ijms25179371