
Mitochondrial Plasticity and Glucose Metabolic Alterations in Human Cancer under Oxidative Stress—From Viewpoints of Chronic Inflammation and Neutrophil Extracellular Traps (NETs)
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Redox and Reactive Oxygen Species (ROS)
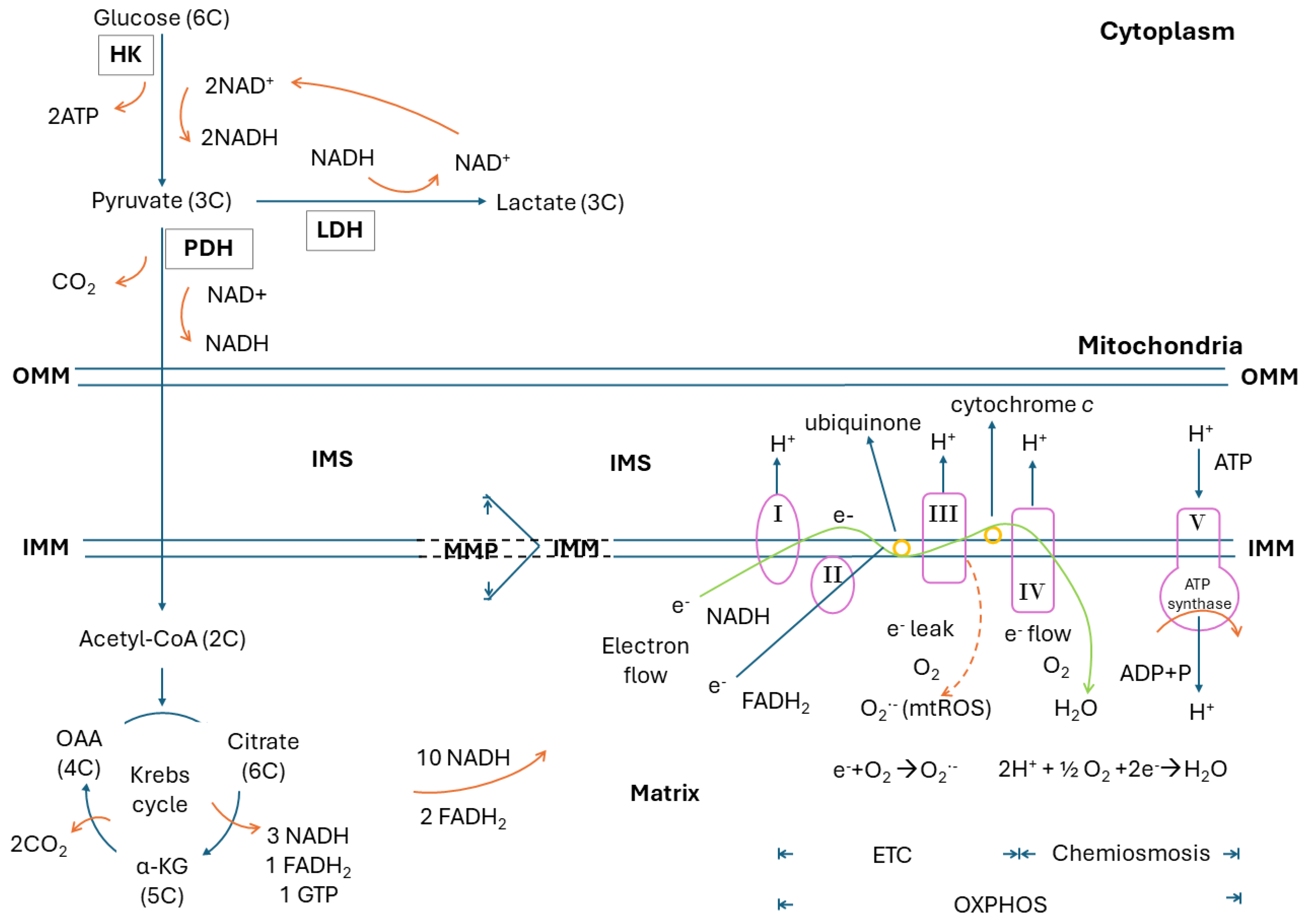
2. Glucose Metabolism, Glycolysis, Krebs Cycle, Electron Transport Chain, Oxidative Phosphorylation, Electron Leak and Mitochondrial ROS
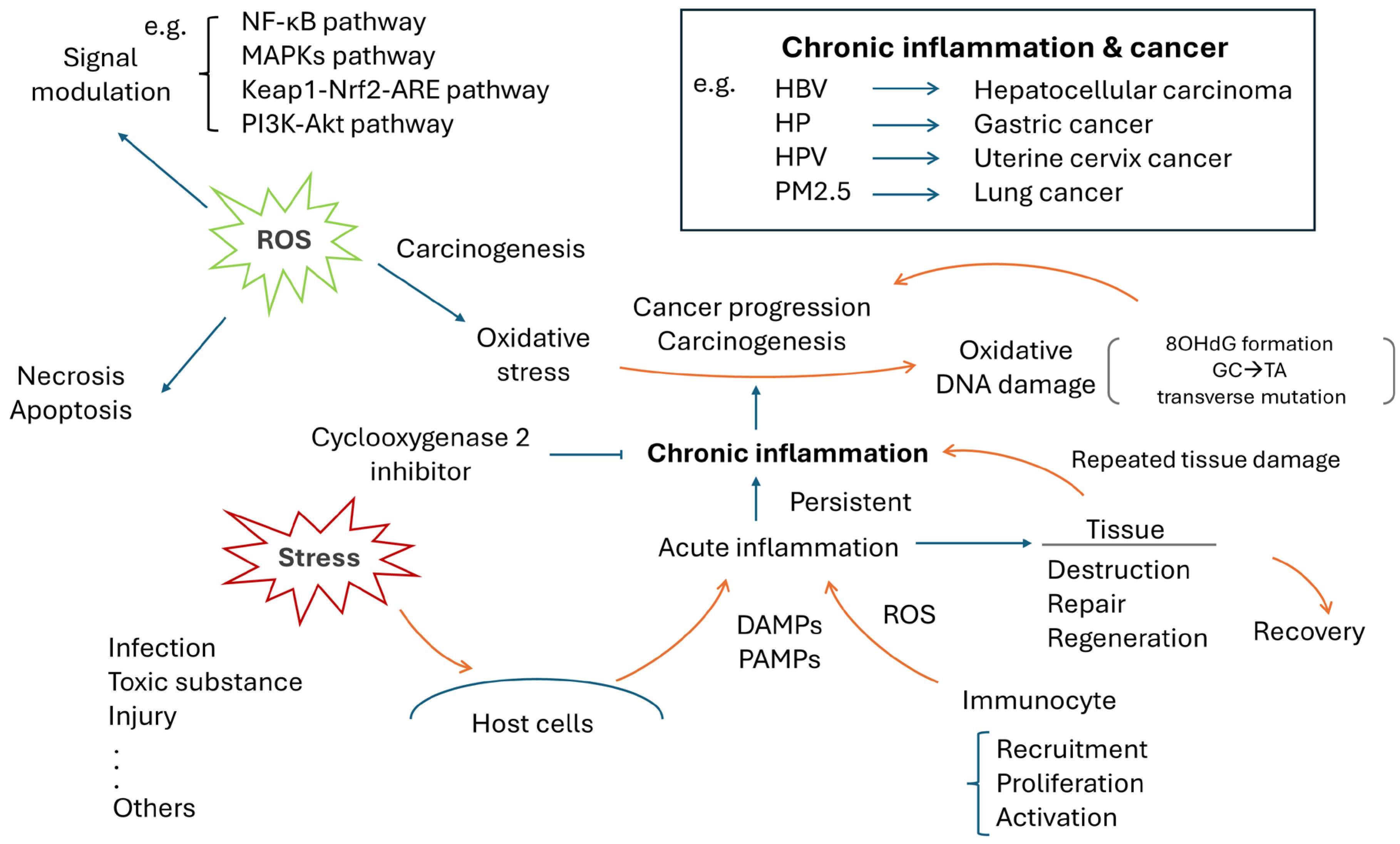
3. Chronic Inflammation and Human Cancer
4. ROS, Oxidative Stress and Cancer
5. NETs, ROS and Host Oxidative Damages
5.1. NOX-Dependent NET Formation
5.2. NOX-Independent NET Formation
5.3. Maladaptive Roles of NET-Related Tissue Damage and Cancer Biology
6. NETs, Oxidative Toxicity, Mitochondrial Dysfunction and Warburg Effect in Human Cancers
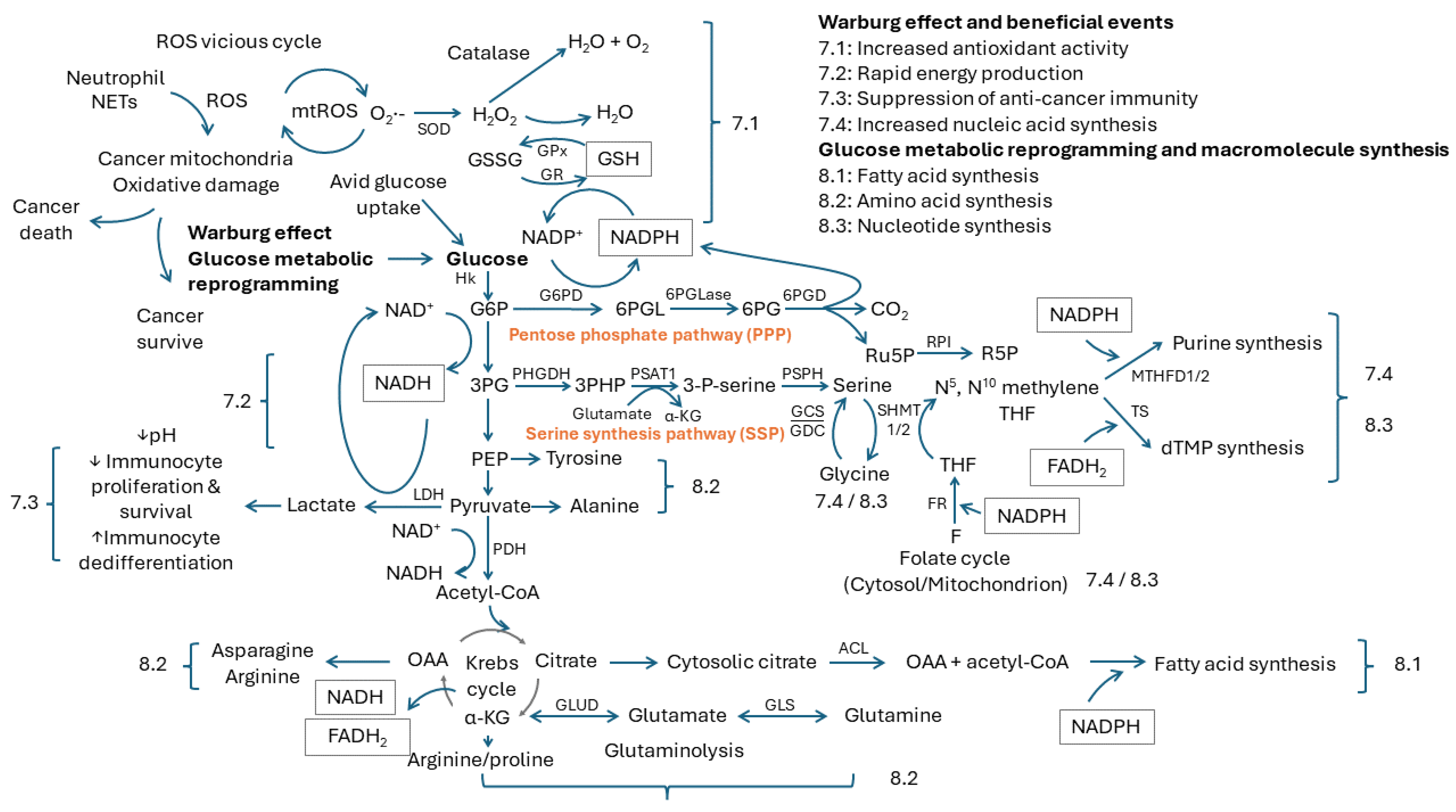
7. Beneficial Events from the Warburg Effect
7.1. Increased Antioxidant Activity
7.2. Rapid Energy Production
7.3. Suppression of Anticancer Immunity
7.4. Increased Nucleic Acid Synthesis
8. Glucose Metabolic Reprogramming and Macromolecule Synthesis
8.1. Fatty Acid Synthesis
8.2. Amino Acid Synthesis
8.3. Nucleotide Synthesis
9. NETs Improve Biogenesis, Dynamics and Mitophagy in Cancer Mitochondria
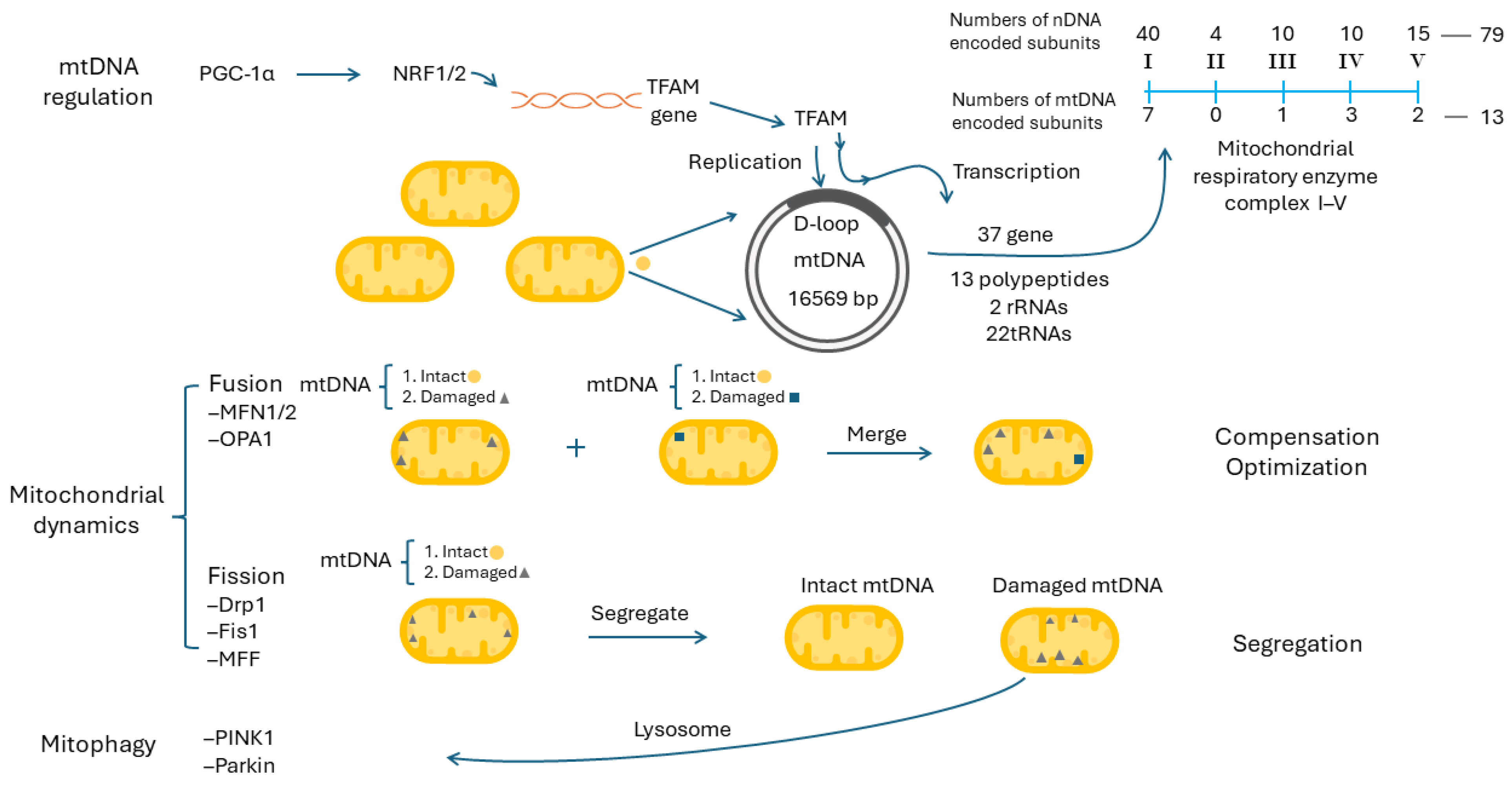
9.1. Mitochondrial DNA Regulation and Mitochondrial Biogenesis
9.2. Mitochondrial Dynamics, Mitophagy and Mitochondrial Biogenesis
9.3. Mitochondrial Dynamics and Mitophagy
9.4. NETs in Cancer Progression and Cancer in NET Formation—Reciprocal Interaction between Cancer and NETs
9.5. NETs Augment mtDNA Biogenesis, Mitochondrial Dynamic and Mitophagy in Cancers to Promote Their Progression
10. Future Perspective
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The golden mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Mechanism of Oxidative Phosphorylation. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9885/ (accessed on 20 August 2024).
- Nakrani, M.N.; Wineland, R.H.; Anjum, F. Physiology, Glucose Metabolism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Köberle, M.; Mrowietz, U.; Bernhardt, I. Proliferating tumor cells mimick glucose metabolism of mature human erythrocytes. Cell Cycle 2019, 18, 1316–1334. [Google Scholar] [CrossRef]
- Peng, S.; Gao, J.; Stojkov, D.; Yousefi, S.; Simon, H.U. Established and emerging roles for mitochondria in neutrophils. Immunol. Rev. 2023, 314, 413–426. [Google Scholar] [CrossRef]
- Reithofer, M.; Karacs, J.; Strobl, J.; Kitzmüller, C.; Polak, D.; Seif, K.; Kamalov, M.; Becker, C.F.W.; Greiner, G.; Schmetterer, K.; et al. Alum triggers infiltration of human neutrophils ex vivo and causes lysosomal destabilization and mitochondrial membrane potential-dependent NET-formation. FASEB J. 2020, 34, 14024–14041. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.I.; Han, J.K.; Hong, S.T.; Lee, K.H. Clonorchiasis and cholangiocarcinoma: Etiologic relationship and imaging diagnosis. Clin. Microbiol. Rev. 2004, 17, 540–552, Table of Contents. [Google Scholar] [CrossRef]
- Okunade, K.S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Merin, K.A.; Shaji, M.; Kameswaran, R. A Review on sun exposure and skin diseases. Indian J. Dermatol. 2022, 67, 625. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhou, R.; Zhang, J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol. Lett. 2018, 15, 7506–7514. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.K.; Zhang, X.; Wu, H.L.; Gan, Y.; Ye, L.; Zheng, H.; Zhu, Z.; Liu, W.J.; Liu, H.F. ROS-ERK pathway as dual mediators of cellular injury and autophagy-associated adaptive response in urinary protein-Irritated renal tubular epithelial cells. J. Diabetes Res. 2021, 1, 6614848. [Google Scholar] [CrossRef]
- Chung, K.S.; Yoo, C.B.; Lee, J.H.; Lee, H.H.; Park, S.E.; Han, H.S.; Lee, S.Y.; Kwon, B.M.; Choi, J.H.; Lee, K.T. Regulation of ROS-dependent JNK pathway by 2′-Hydroxycinnamaldehyde inducing apoptosis in human promyelocytic HL-60 leukemia cells. Pharmaceutics 2021, 13, 1794. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Chi, W.; Su, L.; Li, J.; Zhang, Z.; Yuan, X. ROS-induced oxidative injury involved in pathogenesis of fungal keratitis via p38 MAPK activation. Sci. Rep. 2017, 7, 10421. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 5, 721–733. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Sig Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-kinase/Akt signaling and redox metabolism in cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef]
- Qing, X.; Shi, D.; Lv, X.; Wang, B.; Chen, S.; Shao, Z. Prognostic significance of 8-hydroxy-2′-deoxyguanosine in solid tumors: A meta-analysis. BMC Cancer 2019, 19, 997. [Google Scholar] [CrossRef]
- Klebe, S.; Leigh, J.; Henderson, D.W.; Nurminen, M. Asbestos, smoking and lung cancer: An update. Int. J. Environ. Res. Public Health 2019, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- An, A.R.; Kim, K.M.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, Y.C.; Kim, J.H.; Chae, H.J.; Chung, M.J. Association between expression of 8-OHdG and cigarette smoking in non-small cell lung cancer. J. Pathol. Transl. Med. 2019, 53, 217–224. [Google Scholar] [CrossRef]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. Phagocytosis, degranulation and extracellular traps release by neutrophils—The current knowledge, pharmacological modulation and future prospects. Front. Pharmacol. 2021, 12, 666732. [Google Scholar] [CrossRef]
- Parker, H.; Winterbourn, C. Reactive oxidants and myeloperoxidase and their involvement in neutrophil extracellular traps. Front. Immunol. 2013, 3, 424. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Song, Y.; Wang, R.; He, R.; Wang, T. Neutrophil elastase: From mechanisms to therapeutic potential. J. Pharm. Anal. 2023, 13, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Raad, H.; Paclet, M.H.; Boussetta, T.; Kroviarski, Y.; Morel, F.; Quinn, M.T.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. Regulation of the phagocyte NADPH oxidase activity: Phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J. 2009, 23, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK activation turns on LPS- and gram-negative bacteria-induced NADPH oxidase-dependent suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U. Molecular mechanisms of neutrophil extracellular trap (NETs) degradation. Int. J. Mol. Sci. 2023, 24, 4896. [Google Scholar] [CrossRef] [PubMed]
- Ekaney, M.L.; Otto, G.P.; Sossdorf, M.; Sponholz, C.; Boehringer, M.; Loesche, W.; Rittirsch, D.; Wilharm, A.; Kurzai, O.; Bauer, M.; et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit. Care 2014, 18, 543. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Bonanni, A.; Petretto, A.; Vaglio, A.; Pratesi, F.; Santucci, L.; Migliorini, P.; Bertelli, R.; Galetti, M.; Belletti, S.; et al. Neutrophil extracellular traps profiles in patients with incident systemic lupus erythematosus and lupus nephritis. J. Rheumatol. 2020, 47, 377–386. [Google Scholar] [CrossRef]
- Apel, F.; Zychlinsky, A.; Kenny, E.F. The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 467–475. [Google Scholar] [CrossRef]
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef] [PubMed]
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.-T. Neutrophil extracellular traps in inflammatory bowel disease: Pathogenic mechanisms and clinical translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Moorthy, A.N.; Tan, K.B.; Wang, S.; Narasaraju, T.; Chow, V.T. Effect of high-fat diet on the formation of pulmonary neutrophil extracellular traps during Influenza pneumonia in BALB/c mice. Front. Immunol. 2016, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jin, J. Neutrophil extracellular traps: New players in cancer research. Front. Immunol. 2022, 13, 937565. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2022, 22, 173–187. [Google Scholar] [CrossRef]
- Zivkovic, M.; Poljak-Blazi, M.; Zarkovic, K.; Mihaljevic, D.; Schaur, R.J.; Zarkovic, N. Oxidative burst of neutrophils against melanoma B16-F10. Cancer Lett. 2007, 246, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Kloecker, G.; Fleming, C.; Bousamra, M., 2nd; Hansen, R.; Hu, X.; Ding, C.; Cai, Y.; Xiang, D.; Donninger, H.; et al. Human polymorphonuclear neutrophils specifically recognize and kill cancerous cells. Oncoimmunology 2014, 3, e950163. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen species and mitochondrial dynamics: The yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Miles, K.A.; Williams, R.E. Warburg revisited: Imaging tumour blood flow and metabolism. Cancer Imaging 2008, 8, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Duan, Z.; Li, Z.; Ge, F.; Wei, R.; Kong, L. The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 2022, 13, 1091779. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-F.; Tseng, L.-M.; Lee, H.-C. Role of mitochondrial alterations in human cancer progression and cancer immunity. J. Biomed. Sci. 2023, 30, 61. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Liu, C.Y.; Liu, C.C.; Li, A.F.; Hsu, T.W.; Lin, J.H.; Hung, S.C.; Hsu, H.S. Glutathione peroxidase 4 expression predicts poor overall survival in patients with resected lung adenocarcinoma. Sci. Rep. 2022, 12, 20462. [Google Scholar] [CrossRef] [PubMed]
- Brzozowa-Zasada, M.; Piecuch, A.; Bajdak-Rusinek, K.; Janelt, K.; Michalski, M.; Klymenko, O.; Matysiak, N. Immunohistochemical expression of glutathione peroxidase 1 (Gpx-1) as an independent prognostic factor in colon adenocarcinoma patients. Pharmaceuticals 2023, 16, 740. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-León, D.; Monzó-Beltrán, L.; Gómez-Abril, S.Á.; Estañ-Capell, N.; Camarasa-Lillo, N.; Pérez-Ebri, M.L.; Escandón-Álvarez, J.; Alonso-Iglesias, E.; Santaolaria-Ayora, M.L.; Carbonell-Moncho, A.; et al. The effectiveness of glutathione redox status as a possible tumor marker in colorectal cancer. Int. J. Mol. Sci. 2021, 22, 6183. [Google Scholar] [CrossRef]
- Pakfetrat, A.; Dalirsani, Z.; Hashemy, S.I.; Ghazi, A.; Mostaan, L.V.; Anvari, K.; Pour, A.M. Evaluation of serum levels of oxidized and reduced glutathione and total antioxidant capacity in patients with head and neck squamous cell carcinoma. J. Cancer Res. Ther. 2018, 14, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Israel, A.; Schaffer, A.A.; Berkovitch, M.; Ozeri, D.J.; Merzon, E.; Green, I.; Golan-Cohen, A.; Ruppin, E.; Vinker, S.; Magen, E. Glucose-6-phosphate dehydrogenase deficiency and long-term risk of immune-related disorders. Front. Immunol. 2023, 14, 1232560. [Google Scholar] [CrossRef] [PubMed]
- Cocco, P. Does G6PD deficiency protect against cancer? A critical review. J. Epidemiol. Community Health 1987, 41, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, W.; Yang, Y.; Gu, C. Exploring the role of glucose-6-phosphate dehydrogenase in cancer (Review). Oncol. Rep. 2020, 44, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Murthy, D.; Kollala, S.S.; Kosmacek, E.A.; Chatterjee, A.; McDowell, J.A.; Singh, P.K.; Oberley-Deegan, R.E. The central role of NADPH depletion in MnTE-2-PyP-induced prostate cancer cell growth inhibition. Adv. Redox Res. 2021, 3, 100025. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, D.; Jin, L.; Ren, X.; Ouyang, Y.; Zhou, Y.; He, X.; Jia, L.; Tian, Z.; Wu, D.; et al. NADPH selective depletion nanomedicine-mediated radio-immunometabolism regulation for strengthening anti-PDL1 therapy against TNBC. Adv. Sci. 2023, 10, 2203788. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Xu, T.; You, W.; Wang, T.; Zhang, D.; Guo, H.; Zhang, H.; Pan, X.; Wang, Y.; Liu, L.; et al. A mitotic NADPH upsurge promotes chromosome segregation and tumour progression in aneuploid cancer cells. Nat. Metab. 2023, 5, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- DiTullio, D.; Dell’Angelica, E.C. Glucose metabolism. In Fundamentals of Biochemistry: Medical Course & Step 1 Review; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef]
- Hong, S.M.; Hwang, S.W.; Wang, T.; Park, C.W.; Ryu, Y.-M.; Jung, J.-H.; Shin, J.H.; Kim, S.-Y.; Lee, J.L.; Kim, C.W.; et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. 2019, 110, 629–638. [Google Scholar] [CrossRef]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investig. 2013, 123, 1068–1081. [Google Scholar] [CrossRef]
- Murray, C.M.; Hutchinson, R.; Bantick, J.R.; Belfield, G.P.; Benjamin, A.D.; Brazma, D.; Bundick, R.V.; Cook, I.D.; Craggs, R.I.; Edwards, S.; et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 2005, 1, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-associated lactic acid production blunts tumor Immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Boue, S.; Belmonte, J.C.I. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, M.; Gupta, R.; Kumar, V.; Kumar, V.; Rani, R. Intervention on lactate in cancer: A promising approach for the development of cancer therapeutics. Adv. Cancer Biol. Metastasis 2022, 5, 100058. [Google Scholar] [CrossRef]
- Lv, J.; Zhou, Z.; Wang, J.; Yu, H.; Lu, H.; Yuan, B.; Han, J.; Zhou, R.; Zhang, X.; Yang, X.; et al. Prognostic value of lactate dehydrogenase expression in different cancers: A meta-analysis. Am. J. Med. Sci. 2019, 358, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, L.; Wang, Z.; He, H.; Chen, H.; Duan, Z.; Li, Y.; Si, Q.; Chuang, T.H.; Chen, C.; et al. Lactate Dehydrogenase-A (LDH-A) Preserves Cancer Stemness and Recruitment of Tumor-Associated Macrophages to Promote Breast Cancer Progression. Front. Oncol. 2021, 11, 654452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef]
- Reid, M.A.; Allen, A.E.; Liu, S.; Liberti, M.V.; Liu, P.; Liu, X.; Dai, Z.; Gao, X.; Wang, Q.; Liu, Y.; et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 2018, 9, 5442. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cantley, L.C. Toward a better understanding of folate metabolism in health and disease. J. Exp. Med. 2019, 216, 253–266. [Google Scholar] [CrossRef]
- Chou, Y.-T.; Jiang, J.-K.; Yang, M.-H.; Lu, J.-W.; Lin, H.-K.; Wang, H.-D.; Yuh, C.-H. Identification of a noncanonical function for ribose-5-phosphate isomerase A promotes colorectal cancer formation by stabilizing and activating β-catenin via a novel C-terminal domain. PLoS Biol. 2018, 16, e2003714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, W.; Zhao, X.; Xie, Y.; Luo, D.; Chen, S. Comprehensive analysis of PHGDH for predicting prognosis and immunotherapy response in patients with endometrial carcinoma. BMC Med. Genom. 2023, 16, 29. [Google Scholar] [CrossRef]
- Chandrika, M.; Chua, P.J.; Muniasamy, U.; Huang, R.Y.J.; Thike, A.A.; Ng, C.T.; Tan, P.H.; Yip, G.W.; Bay, B.H. Prognostic significance of phosphoglycerate dehydrogenase in breast cancer. Breast Cancer Res. Treat. 2021, 186, 655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zheng, A.; Hydbring, P.; Ambroise, G.; Ouchida, A.T.; Goiny, M.; Vakifahmetoglu-Norberg, H.; Norberg, E. PHGDH defines a metabolic subtype in lung adenocarcinomas with poor prognosis. Cell Rep. 2017, 19, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Huang, Y.; Jing, S.; Pei, Y.; Qian, Y.; Zeng, Y. Serine hydroxymethyltransferase 2 predicts unfavorable outcomes in multiple cancer: A systematic review and meta-analysis. Transl. Cancer Res. 2022, 11, 444–455. [Google Scholar] [CrossRef]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—Hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166016. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A. Chapter 5—Fatty acid biosynthesis. In Sweet Biochemistry; Kumari, A., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 21–24. [Google Scholar]
- Qian, X.; Hu, J.; Zhao, J.; Chen, H. ATP citrate lyase expression is associated with advanced stage and prognosis in gastric adenocarcinoma. Int. J. Clin. Exp. Med. 2015, 8, 7855–7860. [Google Scholar]
- Wang, Y.; Wang, Y.; Shen, L.; Pang, Y.; Qiao, Z.; Liu, P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol. Rep. 2012, 27, 1156–1162. [Google Scholar] [CrossRef]
- Wang, J.; Ye, W.; Yan, X.; Guo, Q.; Ma, Q.; Lin, F.; Huang, J.; Jin, J. Low expression of ACLY associates with favorable prognosis in acute myeloid leukemia. J. Transl. Med. 2019, 17, 149. [Google Scholar] [CrossRef]
- Csanadi, A.; Kayser, C.; Donauer, M.; Gumpp, V.; Aumann, K.; Rawluk, J.; Prasse, A.; Hausen, A.z.; Wiesemann, S.; Werner, M.; et al. Prognostic value of malic enzyme and ATP-citrate lyase in non-small cell lung cancer of the young and the elderly. PLoS ONE 2015, 10, e0126357. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.-J.; Zhou, X.-F.; Long, H.; Li, C.-Y.; Wei, J.; Yu, X.-Q.; Guo, Z.-Y.; Zhou, Y.-Q.; Deng, Z.-S. Recent advance of ATP citrate lyase inhibitors for the treatment of cancer and related diseases. Bioorg. Chem. 2024, 142, 106933. [Google Scholar] [CrossRef]
- Nguyen, J.H.; Chung, J.D.; Lynch, G.S.; Ryall, J.G. The microenvironment is a critical regulator of muscle stem cell activation and proliferation. Front. Cell Dev. Biol. 2019, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hoppe, T. Role of amino acid metabolism in mitochondrial homeostasis. Front. Cell Dev. Biol. 2023, 11, 1127618. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yang, M.; Gaur, U.; Xu, H.; Yao, Y.; Li, D. Alpha-ketoglutarate: Physiological functions and applications. Biomol. Ther. 2016, 24, 1–8. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Kim, J.Y.; Heo, S.-H.; Choi, S.K.; Song, I.H.; Park, I.A.; Kim, Y.-A.; Park, H.S.; Park, S.Y.; Bang, W.S.; Gong, G.; et al. Glutaminase expression is a poor prognostic factor in node-positive triple-negative breast cancer patients with a high level of tumor-infiltrating lymphocytes. Virchows Arch. 2017, 470, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Su, K.; Liu, Q.; Li, B.; Wang, Y.; Cheng, C.; Li, Y.; Xu, C.; Chen, J.; Wu, H.; et al. Kidney-type glutaminase is a biomarker for the diagnosis and prognosis of hepatocellular carcinoma: A prospective study. BMC Cancer 2023, 23, 1081. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef]
- Craze, M.L.; El-Ansari, R.; Aleskandarany, M.A.; Cheng, K.W.; Alfarsi, L.; Masisi, B.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; et al. Glutamate dehydrogenase (GLUD1) expression in breast cancer. Breast Cancer Res. Treat. 2019, 174, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl. Med. 2015, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Mukha, D.; Fokra, M.; Feldman, A.; Sarvin, B.; Sarvin, N.; Nevo-Dinur, K.; Besser, E.; Hallo, E.; Aizenshtein, E.; Schug, Z.T.; et al. Glycine decarboxylase maintains mitochondrial protein lipoylation to support tumor growth. Cell Metab. 2022, 34, 775–782.e9. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, M.; Magnelli, V.; Renò, F. NETosis in wound healing: When enough is enough. Cells 2021, 10, 494. [Google Scholar] [CrossRef]
- Busch, K.B.; Kowald, A.; Spelbrink, J.N. Quality matters: How does mitochondrial network dynamics and quality control impact on mtDNA integrity? Philos. Trans. R. Soc. B Biol. 2014, 369, 20130442. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef]
- Lee, H.-T.; Lin, C.-S.; Pan, S.-C.; Wu, T.-H.; Lee, C.-S.; Chang, D.-M.; Tsai, C.-Y.; Wei, Y.-H. Alterations of oxygen consumption and extracellular acidification rates by glutamine in PBMCs of SLE patients. Mitochondrion 2019, 44, 65–74. [Google Scholar] [CrossRef]
- Moraes, C.T. What regulates mitochondrial DNA copy number in animal cells? Trends Genet. 2001, 17, 199–205. [Google Scholar] [CrossRef]
- Kozhukhar, N.; Alexeyev, M.F. 35 years of TFAM research: Old protein, new puzzles. Biology 2023, 12, 823. [Google Scholar] [CrossRef] [PubMed]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1alpha in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Lin, C.-S.; Lee, H.-T.; Lee, M.-H.; Pan, S.-C.; Ke, C.-Y.; Chiu, A.W.-H.; Wei, Y.-H. Role of mitochondrial DNA copy number alteration in human renal cell carcinoma. Int. J. Mol. Sci. 2016, 17, 814. [Google Scholar] [CrossRef]
- Lin, C.-S.; Yeh, Y.-C.; Pan, S.-C.; Lu, S.-Y.; Chen, Y.-J.; Chueh, W.-Y.; Wei, Y.-H. Role of mitochondrial DNA copy number alteration in non-small cell lung cancer. Formos. J. Surg. 2020, 53, 165–176. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Tu, H.-F.; Yang, M.-H.; Chen, Y.-F.; Lan, X.-Y.; Huang, C.-L.; Chen, H.-M.; Li, W.-C. Mitochondrial genome and its regulator TFAM modulates head and neck tumourigenesis through intracellular metabolic reprogramming and activation of oncogenic effectors. Cell Death Dis. 2021, 12, 961. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-C.; Lee, H.-T.; Pan, S.-C.; Cho, S.-H.; Cheng, C.; Ou, L.-H.; Lin, C.-I.; Lin, C.-S.; Wei, Y.-H. Metabolic reprogramming in response to alterations of mitochondrial DNA and mitochondrial dysfunction in gastric adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 1857. [Google Scholar] [CrossRef]
- Lin, C.-S.; Lee, H.-T.; Lee, S.-Y.; Shen, Y.-A.; Wang, L.-S.; Chen, Y.-J.; Wei, Y.-H. High mitochondrial DNA copy number and bioenergetic function are associated with tumor Invasion of esophageal squamous cell carcinoma cell lines. Int. J. Mol. Sci. 2012, 13, 11228–11246. [Google Scholar] [CrossRef]
- Lin, C.-S.; Liu, L.-T.; Ou, L.-H.; Pan, S.-C.; Lin, C.-I.; Wei, Y.-H. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 2018, 39, 316–330. [Google Scholar] [CrossRef]
- Wu, C.-H.; Hsieh, P.-F.; Lee, Y.-H.; Kuo, W.-W.; Wu, R.-C.; Lin, Y.-Y.; Hung, C.-H.; Hsieh, M.-L.; Pang, S.-T.; Yang, Y.-L.; et al. Nuclear Respiratory Factor 1 Overexpression Inhibits Proliferation and Migration of PC3 Prostate Cancer Cells. Cancer Genom. Proteom. 2022, 19, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Yin, C.; Zhao, P.; Guo, B.; Ke, W.; Zheng, X.; Xie, D.; Wang, Y.; Wang, G.; Jia, Y.; et al. Nuclear respiratory factor 1 drives hepatocellular carcinoma progression by activating LPCAT1-ERK1/2-CREB axis. Biol. Direct 2023, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Chen, L.; Song, Z.; He, H. The fate of damaged mitochondrial DNA in the cell. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119233. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Hausenloy, D.J. Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014, 171, 1890–1906. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, Y.; Liu, F. Prognostic impact of mitofusin 2 expression in colon cancer. Transl. Cancer Res. 2022, 11, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Zamberlan, M.; Boeckx, A.; Muller, F.; Vinelli, F.; Ek, O.; Vianello, C.; Coart, E.; Shibata, K.; Christian, A.; Grespi, F.; et al. Inhibition of the mitochondrial protein Opa1 curtails breast cancer growth. J. Exp. Clin. Cancer Res. 2022, 41, 95. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fan, Y.; Song, Z.; Han, B.; Meng, Y.; Cao, P.; Tan, K. Identification of DRP1 as a prognostic factor correlated with immune infiltration in breast cancer. Int. Immunopharmacol. 2020, 89, 107078. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef]
- Lu, X.; Yao, Y.; Ma, Y.; Zhang, X.; Peng, H.; Pei, Y.; Lu, Y.; Wang, L. Low expression of PINK1 and PARK2 predicts poor prognosis in patients with esophageal squamous cell carcinoma. World J. Surg. Oncol. 2023, 21, 321. [Google Scholar] [CrossRef]
- Celis-Pinto, J.C.; Fernández-Velasco, A.A.; Corte-Torres, M.D.; Santos-Juanes, J.; Blanco-Agudín, N.; Piña Batista, K.M.; Merayo-Lloves, J.; Quirós, L.M.; Fernández-Vega, I. PINK1 Immunoexpression predicts survival in patients undergoing hepatic resection for colorectal liver metastases. Int. J. Mol. Sci. 2023, 24, 6506. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.G.; Tolkach, Y.; Esser, L.K.; Ellinger, J.; Stöhr, C.; Ritter, M.; Wach, S.; Taubert, H.; Stephan, C.; Hartmann, A.; et al. Mitophagy-associated genes PINK1 and PARK2 are independent prognostic markers of survival in papillary renal cell carcinoma and associated with aggressive tumor behavior. Sci. Rep. 2020, 10, 18857. [Google Scholar] [CrossRef]
- Masuda, S.; Nakazawa, D.; Shida, H.; Miyoshi, A.; Kusunoki, Y.; Tomaru, U.; Ishizu, A. NETosis markers: Quest for specific, objective, and quantitative markers. Clin. Chim. Acta 2016, 459, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Thålin, C.; Lundström, S.; Seignez, C.; Daleskog, M.; Lundström, A.; Henriksson, P.; Helleday, T.; Phillipson, M.; Wallén, H.; Demers, M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 2018, 13, e0191231. [Google Scholar] [CrossRef]
- Grilz, E.; Mauracher, L.M.; Posch, F.; Konigsbrugge, O.; Zochbauer-Muller, S.; Marosi, C.; Lang, I.; Pabinger, I.; Ay, C. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br. J. Haematol. 2019, 186, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Cervena, K.; Vodicka, P.; Vymetalkova, V. Diagnostic and prognostic impact of cell-free DNA in human cancers: Systematic review. Mutat. Res. Rev. Mutat. Res. 2019, 781, 100–129. [Google Scholar] [CrossRef]
- Cisneros-Villanueva, M.; Hidalgo-Pérez, L.; Rios-Romero, M.; Cedro-Tanda, A.; Ruiz-Villavicencio, C.A.; Page, K.; Hastings, R.; Fernandez-Garcia, D.; Allsopp, R.; Fonseca-Montaño, M.A.; et al. Cell-free DNA analysis in current cancer clinical trials: A review. Br. J. Cancer 2022, 126, 391–400. [Google Scholar] [CrossRef]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Akizuki, M.; Fukutomi, T.; Takasugi, M.; Takahashi, S.; Sato, T.; Harao, M.; Mizumoto, T.; Yamashita, J. Prognostic significance of immunoreactive neutrophil elastase in human breast cancer: Long-term follow-up results in 313 patients. Neoplasia 2007, 9, 260–264. [Google Scholar] [CrossRef]
- Qu, Z.; Han, Y.; Zhu, Q.; Ding, W.; Wang, Y.; Zhang, Y.; Wei, W.; Lei, Y.; Li, M.; Jiao, Y.; et al. A novel neutrophil extracellular traps signature for overall survival prediction and tumor microenvironment identification in gastric cancer. J. Inflamm. Res. 2023, 16, 3419–3436. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef]
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Hypoxia and inflammation are two sides of the same coin. Proc. Natl. Acad. Sci. USA 2013, 110, 18351–18352. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodríguez, R.A.; Trejo-Solís, C.; Cabrera-Cano, A.; Gómez-Manzo, S.; Dávila-Borja, V.M. Hypoxia as a Modulator of Inflammation and Immune Response in Cancer. Cancers 2022, 14, 2291. [Google Scholar] [CrossRef] [PubMed]
- Castaño, M.; Tomás-Pérez, S.; González-Cantó, E.; Aghababyan, C.; Mascarós-Martínez, A.; Santonja, N.; Herreros-Pomares, A.; Oto, J.; Medina, P.; Götte, M.; et al. Neutrophil extracellular traps and cancer: Trapping our attention with their involvement in ovarian cancer. Int. J. Mol. Sci. 2023, 24, 5995. [Google Scholar] [CrossRef]
- Ng, M.S.F.; Kwok, I.; Tan, L.; Shi, C.; Cerezo-Wallis, D.; Tan, Y.; Leong, K.; Calvo, G.F.; Yang, K.; Zhang, Y.; et al. Deterministic reprogramming of neutrophils within tumors. Science 2024, 383, 12. [Google Scholar] [CrossRef]
- Kim, M.M.; Clinger, J.D.; Masayesva, B.G.; Ha, P.K.; Zahurak, M.L.; Westra, W.H.; Califano, J.A. Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin. Cancer Res. 2004, 10, 8512–8515. [Google Scholar] [CrossRef]
- Lin, C.S.; Chang, S.C.; Wang, L.S.; Chou, T.Y.; Hsu, W.H.; Wu, Y.C.; Wei, Y.H. The role of mitochondrial DNA alterations in esophageal squamous cell carcinomas. J. Thorac. Cardiovasc. Surg. 2010, 139, 189–197. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capeloa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-T.; Lin, C.-S.; Liu, C.-Y.; Chen, P.; Tsai, C.-Y.; Wei, Y.-H. Mitochondrial Plasticity and Glucose Metabolic Alterations in Human Cancer under Oxidative Stress—From Viewpoints of Chronic Inflammation and Neutrophil Extracellular Traps (NETs). Int. J. Mol. Sci. 2024, 25, 9458. https://doi.org/10.3390/ijms25179458
Lee H-T, Lin C-S, Liu C-Y, Chen P, Tsai C-Y, Wei Y-H. Mitochondrial Plasticity and Glucose Metabolic Alterations in Human Cancer under Oxidative Stress—From Viewpoints of Chronic Inflammation and Neutrophil Extracellular Traps (NETs). International Journal of Molecular Sciences. 2024; 25(17):9458. https://doi.org/10.3390/ijms25179458
Chicago/Turabian StyleLee, Hui-Ting, Chen-Sung Lin, Chao-Yu Liu, Po Chen, Chang-Youh Tsai, and Yau-Huei Wei. 2024. "Mitochondrial Plasticity and Glucose Metabolic Alterations in Human Cancer under Oxidative Stress—From Viewpoints of Chronic Inflammation and Neutrophil Extracellular Traps (NETs)" International Journal of Molecular Sciences 25, no. 17: 9458. https://doi.org/10.3390/ijms25179458