
Mechanistic Insights on Metformin and Arginine Implementation as Repurposed Drugs in Glioblastoma Treatment
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
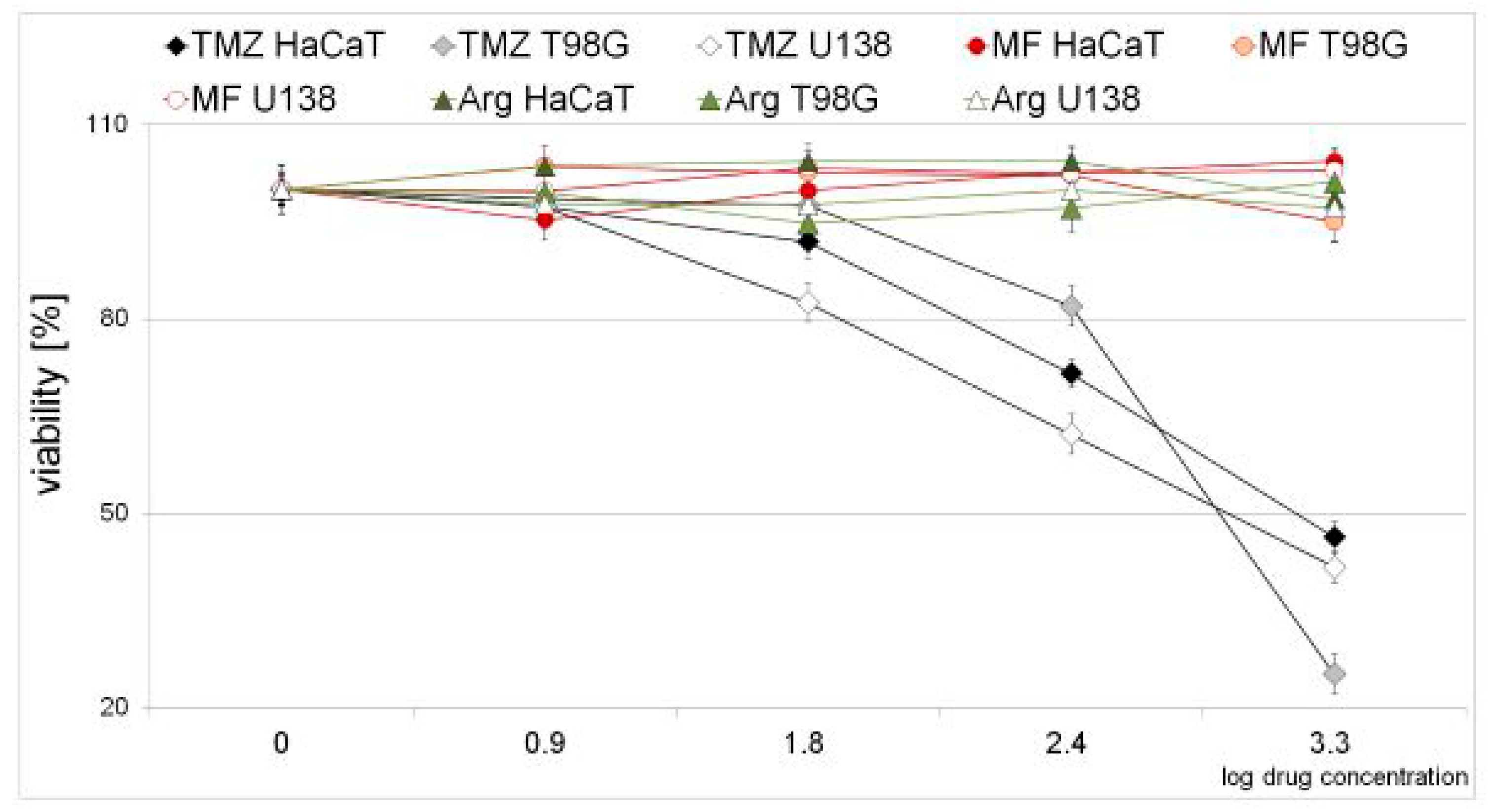
2.1. Cytotoxic Effect of Metformin, Arginine, and Temozolomide in Neoplastic and Normal Cell Lines
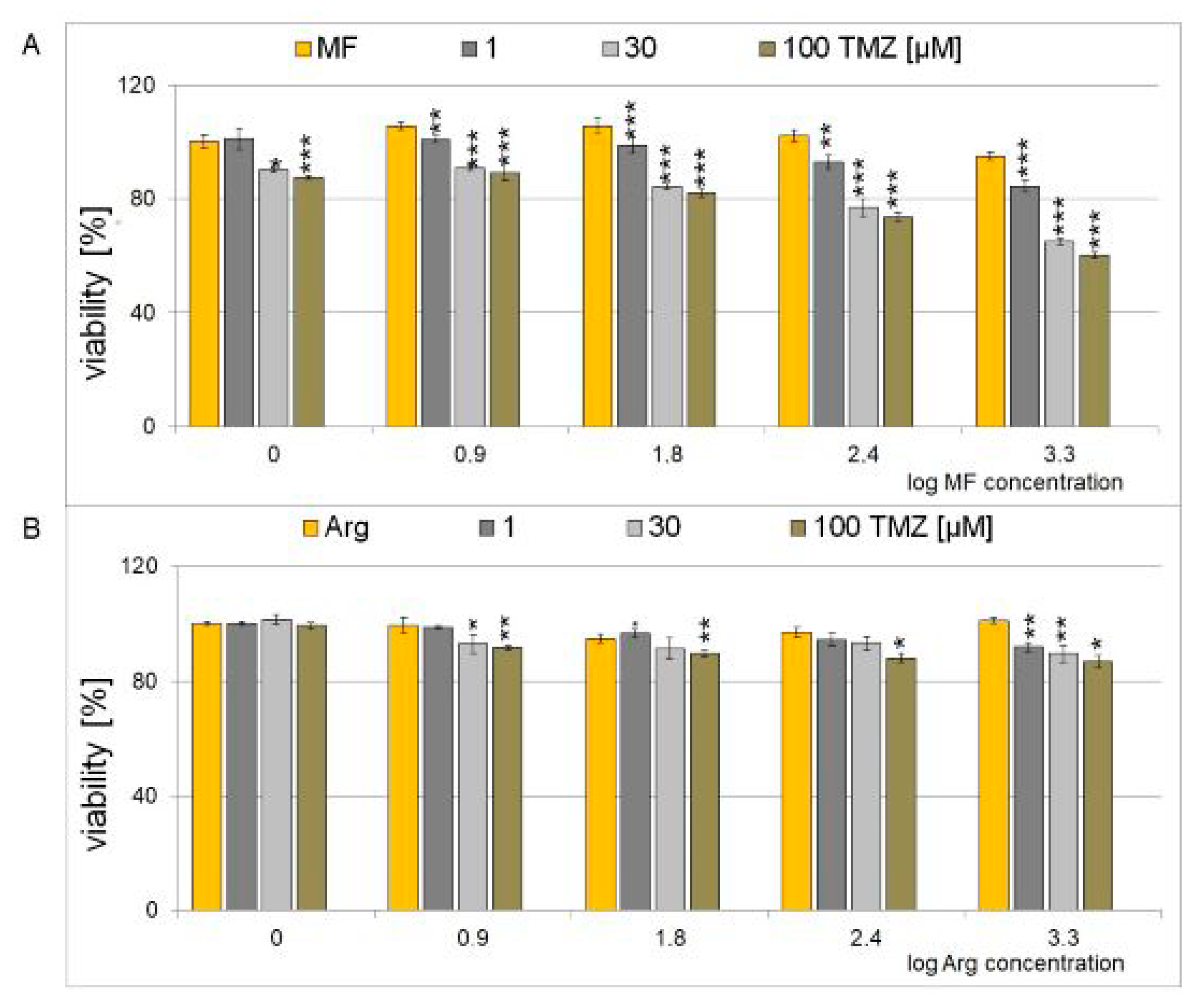
2.2. Cytotoxic Effect of a Combination of Metformin, and Arginine with Temozolomide in Glioblastoma Cell Line
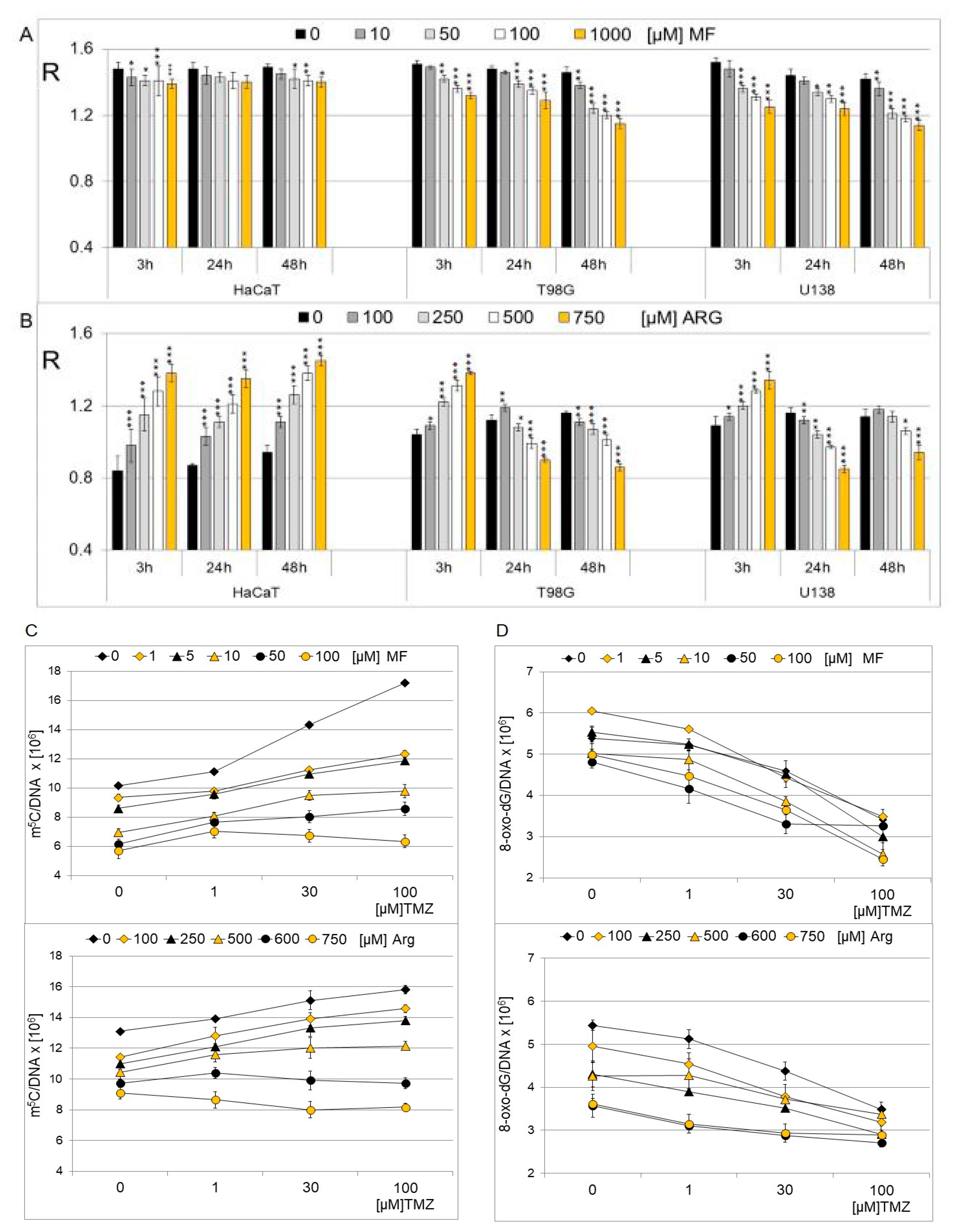
2.3. The Effect of Metformin and Arginine on Genomic DNA Methylation in Cell Lines
2.4. Quantification Estimation of m5C and 8-oxo-dG in DNA of T98G Cell Line Treated with Metformin, Arginine and Temozolomide
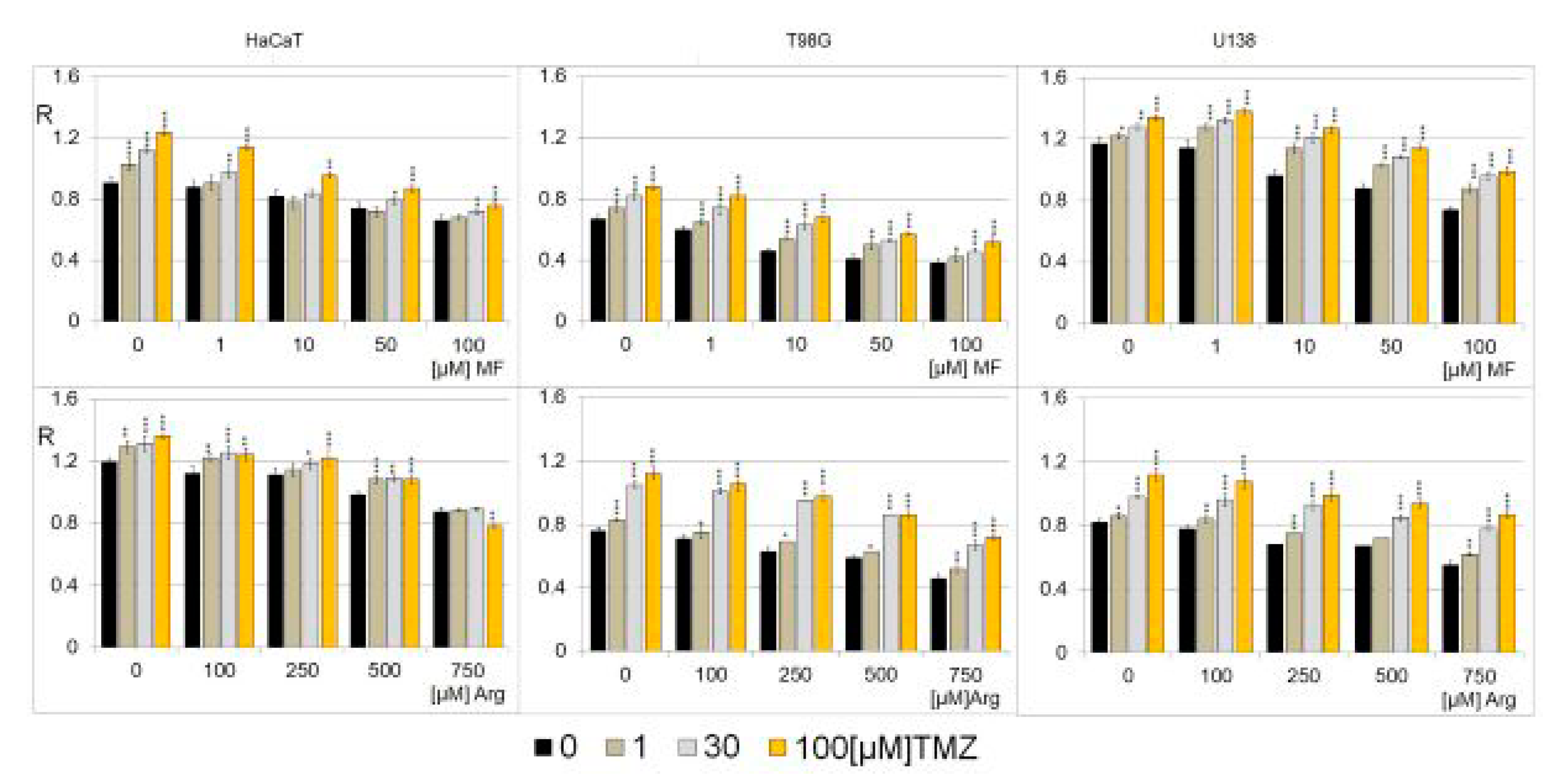
2.5. The Effect of Metformin, Arginine, and Temozolomide on Genomic DNA Methylation
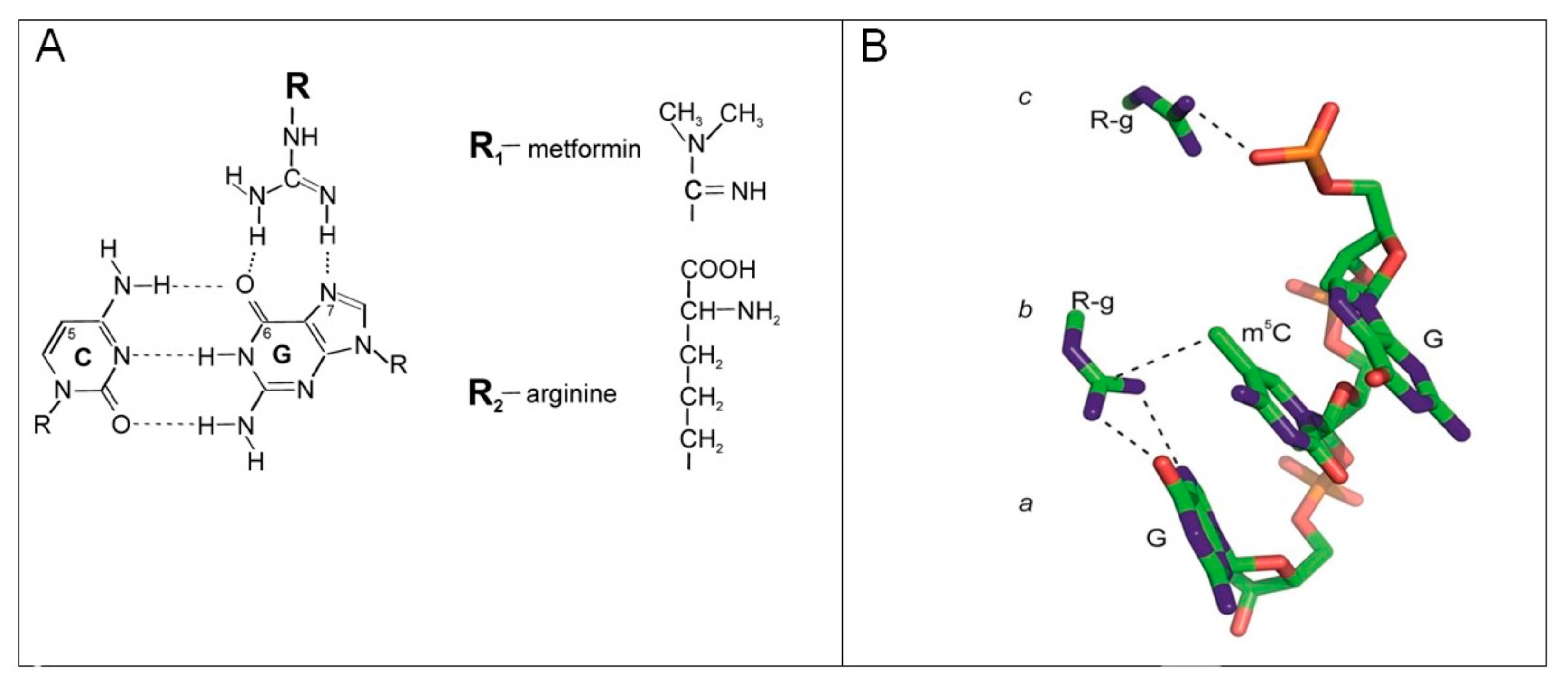
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Line and Culture Conditions
4.3. Cell Viability Assay
4.4. Treatment of Cell Lines with Metformin and Arginine
4.5. Treatment of Cell Lines with the Combination of Metformin, Arginine, and Temozolomide
4.6. Isolation of the DNA
4.7. DNA Hydrolysis, Postlabeling and TLC
4.8. Analysis of 8-oxo-dG Contents in DNA
4.9. Quantification of the Amount of m5C and 8-oxo-dG in Human Genomic DNA
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro-Oncology 2023, 25 (Suppl. S2), iv1–iv99. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Welle, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (≥2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef] [PubMed]
- Ballman, K.V.; Buckner, J.C.; Brown, P.D.; Giannini, C.; Flynn, P.J.; LaPlant, B.R.; Jaeckle, K.A. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro-Oncology 2007, 9, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, F.G.T.; Rose, F.; Pauls, M.; Guay, E.; Ibrahim, M.F.K.; Basulaiman, B.; Tu, M.; Hutton, B.; Nicholas, G.; Ng, T.L. The current landscape of systemic therapy for recurrent glioblastoma: A systematic review of randomized controlled trials. Crit. Rev. Oncol. Hematol. 2022, 169, 103540. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, W.; Shi, Y.; Yan, C.; Kong, Z.; Wang, Y.; Wang, Y.; Ma, W. Imposing phase II and phase III clinical trials of targeted drugs for glioblastoma: Current status and progress. Front. Oncol 2021, 11, 719623. [Google Scholar] [CrossRef]
- Angom, R.S.; Nakka, N.M.R.; Bhattacharya, S. Advances in Glioblastoma Therapy: An Update on Current Approaches. Brain Sci. 2023, 13, 1536. [Google Scholar] [CrossRef]
- Teraiya, M.; Perreault, H.; Chen, V.C. An overview of glioblastoma multiforme and temozolomide resistance: Can LC-MS-based proteomics reveal the fundamental mechanism of temozolomide resistance? Front. Oncol. 2023, 13, 1166207. [Google Scholar] [CrossRef] [PubMed]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Shaw, R.; Basu, M.; Karmakar, S.; Ghosh, M.K. MGMT in TMZ-based glioma therapy: Multifaceted insights and clinical trial perspectives. Biochim. Biophys. Acta Mol. Cell Res. 2024, 71, 119673. [Google Scholar] [CrossRef]
- Alnahhas, I.; Alsawas, M.; Rayi, A.; Palmer, J.D.; Raval, R.; Ong, S.; Giglio, P.; Murad, M.H.; Puduvalli, V. Characterizing benefit from temozolomide in MGMT promoter unmethylated and methylated glioblastoma: A systematic review and meta-analysis. Neuro-Oncol. Adv. 2020, 2, vdaa082. [Google Scholar] [CrossRef] [PubMed]
- Barciszewska, A.M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef] [PubMed]
- Belter, A.; Barciszewski, J.; Barciszewska, A.M. Revealing the epigenetic effect of temozolomide on glioblastoma cell lines in therapeutic conditions. PLoS ONE 2020, 15, e0229534. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl. S3), 245–254. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Ovalle, R. A History of the Fenton Reactions. (Fenton Chemistry for Beginners); Ahmad, R., Ed.; Reactive Oxygen Species; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis. DNA Repair 2017, 56, 75–83. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Zukiel, R.; Nowak, S.; Barciszewska, A.M.; Gawronska, I.; Keith, G.; Barciszewska, M.Z. A simple epigenetic method for the diagnosis and classification of brain tumors. Mol. Cancer Res. 2004, 2, 196–202. [Google Scholar] [CrossRef]
- Cadieux, B.; Ching, T.T.; VandenBerg, S.R.; Costello, J.F. Genome-wide Hypomethylation in Human Glioblastomas Associated with Specific Copy Number Alteration, Methylenetetrahydrofolate Reductase Allele Status, and Increased Proliferation. Cancer Res. 2006, 66, 8469–8476. [Google Scholar] [CrossRef]
- Boot, J.; Rosser, G.; Kancheva, D.; Vinel, C.; Lim, Y.M.; Pomella, N.; Zhang, X.; Guglielmi, L.; Sheer, D.; Barnes, M.; et al. Global hypo-methylation in a proportion of glioblastoma enriched for an astrocytic signature is associated with increased invasion and altered immune landscape. Elife 2022, 11, e77335. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Giel-Pietraszuk, M.; Perrigue, P.M.; Naskręt-Barciszewska, M. Total DNA Methylation Changes Reflect Random Oxidative DNA Damage in Gliomas. Cells 2019, 8, 1065. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. The role of DNA hypermethylation and demethylation in cancer and cancer therapy. Curr. Oncol. 2008, 15, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 5, a026948. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Chavez-Blanco, A.; Revilla-Vazquez, A.; Benitez-Bribiesca, L.; Duenas-González, A. Global DNA hypermethylation-associated cancer chemotherapy resistance and its reversion with the demethylating agent hydralazine. J. Transl. Med. 2006, 4, 32. [Google Scholar] [CrossRef]
- Jiang, H.; Zuo, J.; Li, B.; Chen, R.; Luo, K.; Xiang, X.; Lu, S.; Huang, C.; Liu, L.; Tang, J.; et al. Drug-induced oxidative stress in cancer treatments: Angel or devil? Redox Biol. 2023, 63, 102754. [Google Scholar] [CrossRef]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef]
- Sharma, S.; Zhang, Y.; Akter, K.A.; Nozohouri, S.; Archie, S.R.; Patel, D.; Villalba, H.; Abbruscato, T. Permeability of Metformin across an In Vitro Blood–Brain Barrier Model during Normoxia and Oxygen-Glucose Deprivation Conditions: Role of Organic Cation Transporters (Octs). Pharmaceutics 2023, 15, 1357. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Gong, T.; Du, Y.; Wang, Y.; Ge, T.; Liu, J. Mechanism of metformin regulation in central nervous system: Progression and future perspectives. Biomed. Pharmacother. 2022, 156, 113686. [Google Scholar] [CrossRef]
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Gulgas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Takhwifa, F.; Aninditha, T.; Setiawan, H.; Sauriasari, R. The potential of metformin as an antineoplastic in brain tumors: A systematic review. Heliyon 2021, 7, e06558. [Google Scholar] [CrossRef]
- Li, J.Z.; Li, Y.R. Cardiovascular Protection by Metformin: Latest Advances in Basic and Clinical Research. Cardiology 2023, 148, 374–384. [Google Scholar] [CrossRef]
- Bu, Y.; Peng, M.; Tang, X.; Xu, X.; Wu, Y.; Chen, A.F.; Yang, X. Protective effects of metformin in various cardiovascular diseases: Clinical evidence and AMPK-dependent mechanisms. J. Cell. Mol. Med. 2022, 26, 4886–4903. [Google Scholar] [CrossRef]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef]
- Valtorta, S.; Dico, A.L.; Raccagni, I.; Martelli, C.; Pieri, V.; Rainone, P.; Todde, S.; Zinnhardt, B.; De Bernardi, E.; Coliva, A.; et al. Imaging Metformin Efficacy as Add-On Therapy in Cells and Mouse Models of Human EGFR Glioblastoma. Front. Oncol. 2021, 11, 664149. [Google Scholar] [CrossRef]
- Krajcer, A.; Grzywna, E.; Lewandowska-Łańcucka, J. Strategies increasing the effectiveness of temozolomide at various levels of anti-GBL therapy. Biomed. Pharmacother. 2023, 165, 115174. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.B. Mechanism of excessive purine biosynthesis in hypoxanthine-guanine phosphoribosyltransferase deficiency. J. Clin. Investig. 1970, 49, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef] [PubMed]
- Bobak, Y.P.; Vynnytska, B.O.; Kurlishchuk, Y.V.; Sibirny, A.A.; Stasyk, O.V. Cancer cell sensitivity to arginine deprivation in vitro is not determined by endogenous levels of arginine metabolic enzymes. Cell Biol. Int. 2010, 34, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.; Lamb, J.; Smith, S.; Wheatley, D.N. Single amino acid (arginine) deprivation: Rapid and selective death of cultured transformed and malignant cells. Br. J. Cancer 2000, 83, 800–810. [Google Scholar] [CrossRef]
- Feun, L.; You, M.; Wu, C.J.; Kuo, M.T.; Wangpaichitr, M.; Spector, S.; Savaraj, N. Arginine deprivation as a targeted therapy for cancer. Curr. Pharm. Des. 2008, 14, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.J.; Rieckmann, C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-Arginine Availability Regulates T-Lymphocyte Cell-Cycle Progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef]
- Field, G.C.; Pavlyk, I.; Szlosarek, P.W. Bench-to-Bedside Studies of Arginine Deprivation in Cancer. Molecules. 2023, 28, 2150. [Google Scholar] [CrossRef]
- Schnöller, L.E.; Piehlmaier, D.; Weber, P.; Brix, N.; Fleischmann, D.F.; Nieto, A.E.; Selmansberger, M.; Heider, T.; Hess, J.; Niyazi, M.; et al. Systematic in vitro analysis of therapy resistance in glioblastoma cell lines by integration of clonogenic survival data with multi-level molecular data. Radiat. Oncol. 2023, 18, 51. [Google Scholar] [CrossRef]
- Zhao, H.; Swanson, K.D.; Zheng, B. Therapeutic repurposing of biguanides in cancer. Trends Cancer 2021, 7, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Zheng, Y.; Yao, Y.; Jia, R.; Ge, S.; Zhuang, A. Metformin and cancer hallmarks: Shedding new lights on therapeutic repurposing. J. Transl. Med. 2023, 21, 403. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Samajdar, R.N.; Mukherjee, S.; Bhattacharyya, A.J.; Bagchi, B. Unique features of metformin: A combined experimental, theoretical, and simulation study of its structure, dynamics, and interaction energetics with DNA grooves. J. Phys. Chem. 2018, 122, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, W.W.; Huan, X.; Wu, G.; Li, G.; Zhou, Y.H.; Najafi, M. Mechanisms of cancer cell killing by metformin: A review on different cell death pathways. Mol. Cell. Biochem. 2023, 478, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Kripichnikov, D.; McFarlane, S.I.; Sowers, J.R. Metformin: An update. Ann. Intern. Med. 2002, 137, E25–E33. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichsel, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.R.; Harris, A.L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic. Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef]
- Garcia-Calzon, S.; Schrader, S.; Perfilyev, A.; Martinell, M.; Ahlqvist, E.; Ling, C. DNA methylation partially mediates antidiabetic effects of metformin on HbA1c levels in individuals with type 2 diabetes. Diabetes Res. Clin. Pract. 2023, 202, 110807. [Google Scholar] [CrossRef]
- Forzano, I.; Avvisato, R.; Varzideh, F.; Jankauskas, S.S.; Cioppa, A.; Mone, P.; Salemme, L.; Kansakar, U.; Tesorio, T.; Trimarco, V.; et al. L-Arginine in diabetes: Clinical and preclinical evidence. Cardiovasc. Diabetol. 2023, 22, 89. [Google Scholar] [CrossRef]
- Yarus, M. A specific amino acid binding site composed of RNA. Science 1988, 240, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Michel, F.; Hanna, M.; Green, R.; Bartel, D.P.; Szostak, J.W. The guanosine binding site of the tetrahymena ribozyme. Nature 1989, 342, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Kribelbauer, J.; Lu, X.J.; Rohs, R.; Mann, R.S.; Bussemaker, H.J. Toward a mechanistic understanding of DNA methylation readout by transcription factors. J. Mol. Biol. 2020, 432, 1801–1815. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Blumenthal, R.M.; Cheng, X. A common mode of recognition for methylated CpG. Trends Biochem. Sci. 2013, 38, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.D.; Landweber, L.F. Rhyme or reason: RNA-arginine interactions and the genetic code. Chem. Biol. 1998, 5, R215–R220. [Google Scholar] [CrossRef]
- Chavali, S.S.; Cavender, C.E.; Mathewa, D.H.; Wedekind, J.E. Arginine forks are a widespread motif to recognize phosphate backbones and guanine nucleobases in the RNA major groove. J. Am. Chem. Soc. 2020, 142, 19835–19839. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barciszewska, A.-M.; Belter, A.; Barciszewski, J.F.; Gawrońska, I.; Giel-Pietraszuk, M.; Naskręt-Barciszewska, M.Z. Mechanistic Insights on Metformin and Arginine Implementation as Repurposed Drugs in Glioblastoma Treatment. Int. J. Mol. Sci. 2024, 25, 9460. https://doi.org/10.3390/ijms25179460
Barciszewska A-M, Belter A, Barciszewski JF, Gawrońska I, Giel-Pietraszuk M, Naskręt-Barciszewska MZ. Mechanistic Insights on Metformin and Arginine Implementation as Repurposed Drugs in Glioblastoma Treatment. International Journal of Molecular Sciences. 2024; 25(17):9460. https://doi.org/10.3390/ijms25179460
Chicago/Turabian StyleBarciszewska, Anna-Maria, Agnieszka Belter, Jakub F. Barciszewski, Iwona Gawrońska, Małgorzata Giel-Pietraszuk, and Mirosława Z. Naskręt-Barciszewska. 2024. "Mechanistic Insights on Metformin and Arginine Implementation as Repurposed Drugs in Glioblastoma Treatment" International Journal of Molecular Sciences 25, no. 17: 9460. https://doi.org/10.3390/ijms25179460