
Loss of JCAD/KIAA1462 Protects the Lung from Acute and Chronic Consequences of Chronic Obstructive Pulmonary Disease

 , , ,
, , ,
Abstract
1. Introduction
2. Results
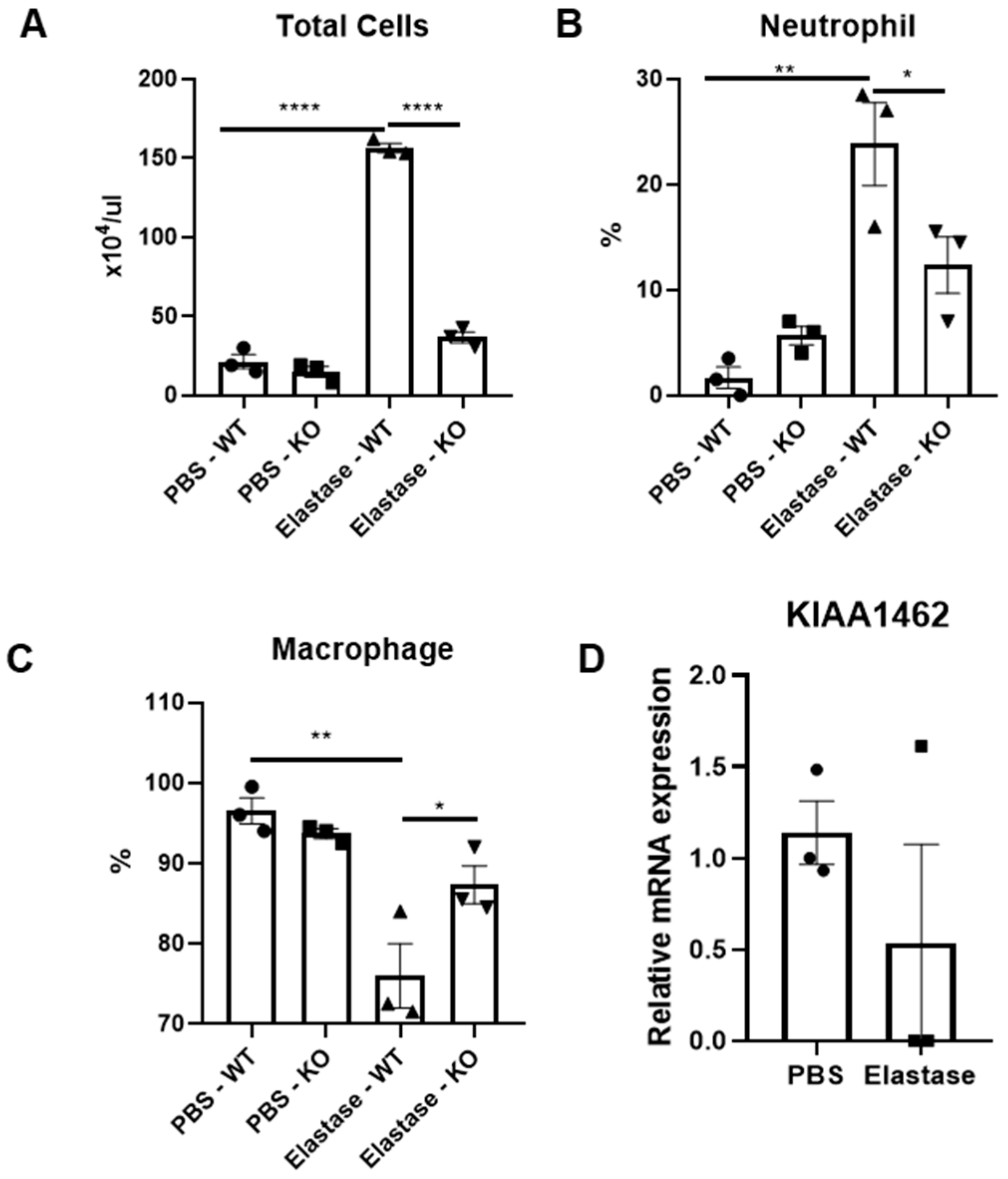
2.1. Amelioration of Acute Inflammatory Response in JCAD-Deficient Mice after Elastase Instillation
2.2. The Structural Integrity of the Lung Was Preserved in JCAD-KO Mice in an Emphysematous Model
2.3. Structural Integrity of the Lung Was Preserved in JCAD-KO Mice in the Emphysematous Model
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.2. Bronchioalveolar Lavage Fluid Analysis
4.3. Histological Analysis and Immunostaining
4.4. In Vitro Study
4.5. Quantitative Real-Time PCR Analysis
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boers, E.; Barrett, M.; Su, J.G.; Benjafield, A.V.; Sinha, S.; Kaye, L.; Zar, H.J.; Vuong, V.; Tellez, D.; Gondalia, R.; et al. Global Burden of Chronic Obstructive Pulmonary Disease through 2050. JAMA Netw. Open 2023, 6, e2346598. [Google Scholar] [CrossRef]
- Berry, C.E.; Wise, R.A. Mortality in COPD: Causes, Risk Factors, and Prevention. COPD J. Chronic Obstr. Pulm. Dis. 2010, 7, 375–382. [Google Scholar] [CrossRef]
- Mirza, S.; Clay, R.D.; Koslow, M.A.; Scanlon, P.D. COPD Guidelines: A Review of the 2018 GOLD Report. Mayo Clin. Proc. 2018, 93, 1488–1502. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Banerjee, R.; Srivastava, S. Molecular Mechanisms and the Interplay of Important Chronic Obstructive Pulmonary Disease Biomarkers Reveals Novel Therapeutic Targets. ACS Omega 2023, 8, 46376–46389. [Google Scholar] [CrossRef]
- Al-Soudi, A.; Kaaij, M.H.; Tas, S.W. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun. Rev. 2017, 16, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Dejana, E. Endothelial Cell-to-Cell Junctions: Molecular Organization and Role in Vascular Homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef]
- Wang, Y.; Malik, A.B.; Sun, Y.; Hu, S.; Reynolds, A.B.; Minshall, R.D.; Hu, G. Innate Immune Function of the Adherens Junction Protein p120-Catenin in Endothelial Response to Endotoxin. J. Immunol. 2011, 186, 3180–3187. [Google Scholar] [CrossRef]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and Immune Response in COPD: Where Do We Stand? Mediators Inflamm. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Poto, R.; Loffredo, S.; Palestra, F.; Marone, G.; Patella, V.; Varricchi, G. Angiogenesis, Lymphangiogenesis, and Inflammation in Chronic Obstructive Pulmonary Disease (COPD): Few Certainties and Many Outstanding Questions. Cells 2022, 11, 1720. [Google Scholar] [CrossRef] [PubMed]
- Boueiz, A.; Lutz, S.M.; Cho, M.H.; Hersh, C.P.; Bowler, R.P.; Washko, G.R.; Halper-Stromberg, E.; Bakke, P.; Gulsvik, A.; Laird, N.M.; et al. Genome-Wide Association Study of the Genetic Determinants of Emphysema Distribution. Am. J. Respir. Crit. Care Med. 2017, 195, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Akashi, M.; Higashi, T.; Masuda, S.; Komori, T.; Furuse, M. A coronary artery disease-associated gene product, JCAD/KIAA1462, is a novel component of endothelial cell–cell junctions. Biochem. Biophys. Res. Commun. 2011, 413, 224–229. [Google Scholar] [CrossRef]
- Hara, T.; Monguchi, T.; Iwamoto, N.; Akashi, M.; Mori, K.; Oshita, T.; Okano, M.; Toh, R.; Irino, Y.; Shinohara, M.; et al. Targeted Disruption of JCAD (Junctional Protein Associated With Coronary Artery Disease)/KIAA1462, a Coronary Artery Disease–Associated Gene Product, Inhibits Angiogenic Processes In Vitro and In Vivo. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Xu, Y.; Liu, P.; Zhang, S.; Liu, H.; Slavin, S.; Kumar, S.; Koroleva, M.; Luo, J.; Wu, X.; et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur. Heart J. 2019, 40, 2398–2408. [Google Scholar] [CrossRef]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, S.; Natali, G.; Nogara, A.M.; Trevisani, M.; Lagrasta, C.A.M.; Pontis, S. The role of pulmonary vascular endothelium in chronic obstructive pulmonary disease (COPD): Does endothelium play a role in the onset and progression of COPD? Explor. Med. 2023, 4, 1116–1134. [Google Scholar] [CrossRef]
- Wang, L.; Xu, Z.; Chen, B.; He, W.; Hu, J.; Zhang, L.; Liu, X.; Chen, F. The Role of Vascular Endothelial Growth Factor in Small-airway Remodelling in a Rat Model of Chronic Obstructive Pulmonary Disease. Sci. Rep. 2017, 7, 41202. [Google Scholar] [CrossRef] [PubMed]
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity 2021, 54, 1989–2004.e9. [Google Scholar] [CrossRef]
- Gilowska, I. CXCL8 (interleukin 8)—The key inflammatory mediator in chronic obstructive pulmonary disease? Postepy Hig. Med. Dosw. 2014, 68, 842–850. [Google Scholar] [CrossRef]
- Cambier, S.; Gouwy, M.; Proost, P. The chemokines CXCL8 and CXCL12: Molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell. Mol. Immunol. 2023, 20, 217–251. [Google Scholar] [CrossRef]
- Rincon, M.; Irvin, C.G. Role of IL-6 in Asthma and Other Inflammatory Pulmonary Diseases. Int. J. Biol. Sci. 2012, 8, 1281–1290. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 Overexpression Induces Pulmonary Hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Suraya, R.; Nagano, T.; Ryanto, G.R.T.; Effendi, W.I.; Hazama, D.; Katsurada, N.; Yamamoto, M.; Tachihara, M.; Emoto, N.; Nishimura, Y.; et al. Budesonide/glycopyrronium/formoterol fumarate triple therapy prevents pulmonary hypertension in a COPD mouse model via NFκB inactivation. Respir. Res. 2022, 23, 173. [Google Scholar] [CrossRef]
- Umezawa, K.; Nagano, T.; Kobayashi, K.; Dokuni, R.; Katsurada, M.; Yamamoto, M.; Yoshikawa, Y.; Kataoka, T.; Nishimura, Y. Phospholipase Cε plays a crucial role in neutrophilic inflammation accompanying acute lung injury through augmentation of CXC chemokine production from alveolar epithelial cells. Respir. Res. 2019, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Crowley, G.; Kwon, S.; Caraher, E.J.; Haider, S.H.; Lam, R.; Batra, P.; Melles, D.; Liu, M.; Nolan, A. Quantitative lung morphology: Semi-automated measurement of mean linear intercept. BMC Pulm. Med. 2019, 19, 206. [Google Scholar] [CrossRef]
- Marien, K.M.; Croons, V.; Waumans, Y.; Sluydts, E.; De Schepper, S.; Andries, L.; Waelput, W.; Fransen, E.; Vermeulen, P.B.; Kockx, M.M.; et al. Development and Validation of a Histological Method to Measure Microvessel Density in Whole-Slide Images of Cancer Tissue. PLoS ONE 2016, 11, e0161496. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| GAPDH-Forward | 5′-GCACCGTCAAGGCTGAGAAC-3′ |
| GAPDH-Reverse | 5′-ATGGTGGTGAAGACGCCAGT-3′ |
| IL-6 Forward | 5′-GGTACATCCTCGACGGCATCT -3′ |
| IL-6 Reverse | 5′-GTGCCTCTTTGCTGCTTTCAC-3′ |
| RANTES-Forward | 5′-AGCTTCCTTGAACCATTATGCTG-3′ |
| RANTES-Reverse | 5′-AGGTCTTCATTGGTGACCTGCT-3′ |
| TNFα-Forward | 5′-GCTTGTTCCTCAGCCTCTTC-3′ |
| TNFα-Reverse | 5′-GGTTATCTCTCAGCTCCACGC-3′ |
| CXCL8-Forward | 5′-GCATAAAGACATACTCCAAACC-3′ |
| CXCL8-Reverse | 5′ACTTCTCCACAACCCTCTG-3′ |
| KIAA1462-Forward | 5′-CCTGGAACTGGGAATGAGTATG-3′ |
| KIAA1462-Reverse | 5′-GTACTGAACGAAGCCGTCATAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suraya, R.; Nagano, T.; Yumura, M.; Hara, T.; Akashi, M.; Yamamoto, M.; Tachihara, M.; Nishimura, Y.; Kobayashi, K. Loss of JCAD/KIAA1462 Protects the Lung from Acute and Chronic Consequences of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2024, 25, 9492. https://doi.org/10.3390/ijms25179492
Suraya R, Nagano T, Yumura M, Hara T, Akashi M, Yamamoto M, Tachihara M, Nishimura Y, Kobayashi K. Loss of JCAD/KIAA1462 Protects the Lung from Acute and Chronic Consequences of Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences. 2024; 25(17):9492. https://doi.org/10.3390/ijms25179492
Chicago/Turabian StyleSuraya, Ratoe, Tatsuya Nagano, Masako Yumura, Tetsuya Hara, Masaya Akashi, Masatsugu Yamamoto, Motoko Tachihara, Yoshihiro Nishimura, and Kazuyuki Kobayashi. 2024. "Loss of JCAD/KIAA1462 Protects the Lung from Acute and Chronic Consequences of Chronic Obstructive Pulmonary Disease" International Journal of Molecular Sciences 25, no. 17: 9492. https://doi.org/10.3390/ijms25179492
APA StyleSuraya, R., Nagano, T., Yumura, M., Hara, T., Akashi, M., Yamamoto, M., Tachihara, M., Nishimura, Y., & Kobayashi, K. (2024). Loss of JCAD/KIAA1462 Protects the Lung from Acute and Chronic Consequences of Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences, 25(17), 9492. https://doi.org/10.3390/ijms25179492