ACE2 Knockout Mice Are Resistant to High-Fat Diet-Induced Obesity in an Age-Dependent Manner

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
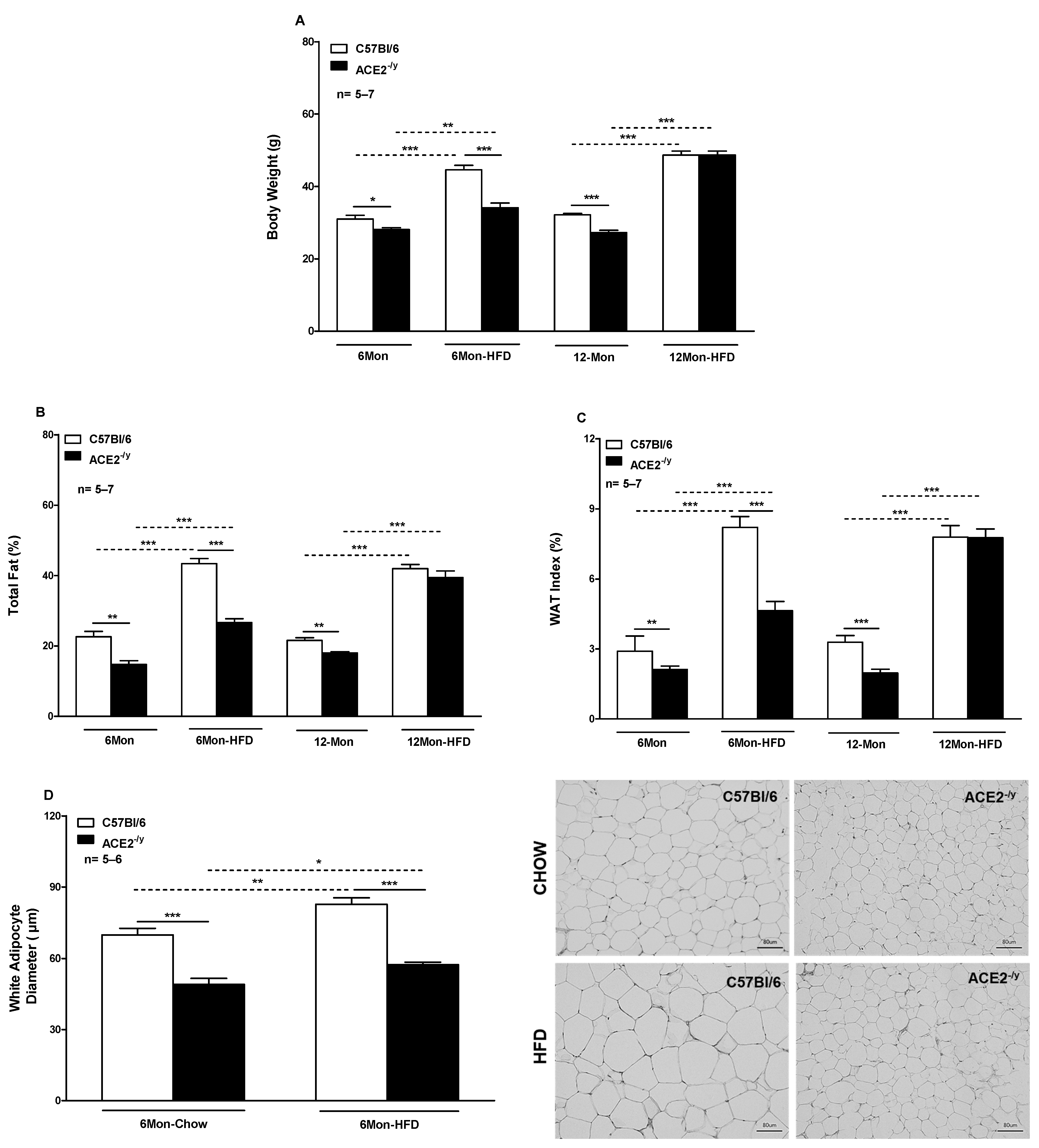
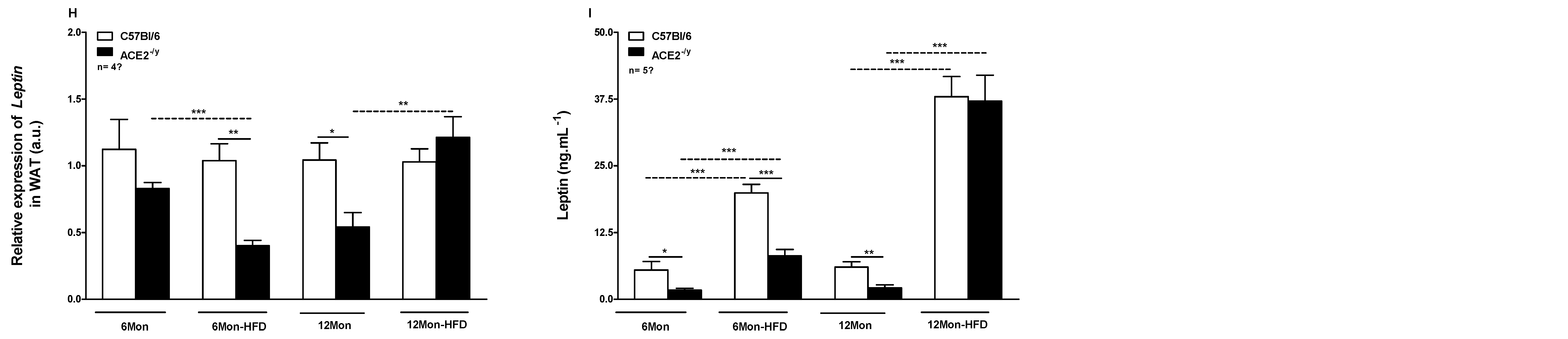
2.1. ACE2 Deficiency Decreases White Fat Depots and the Susceptibility to High-Fat Diet-Induced Obesity
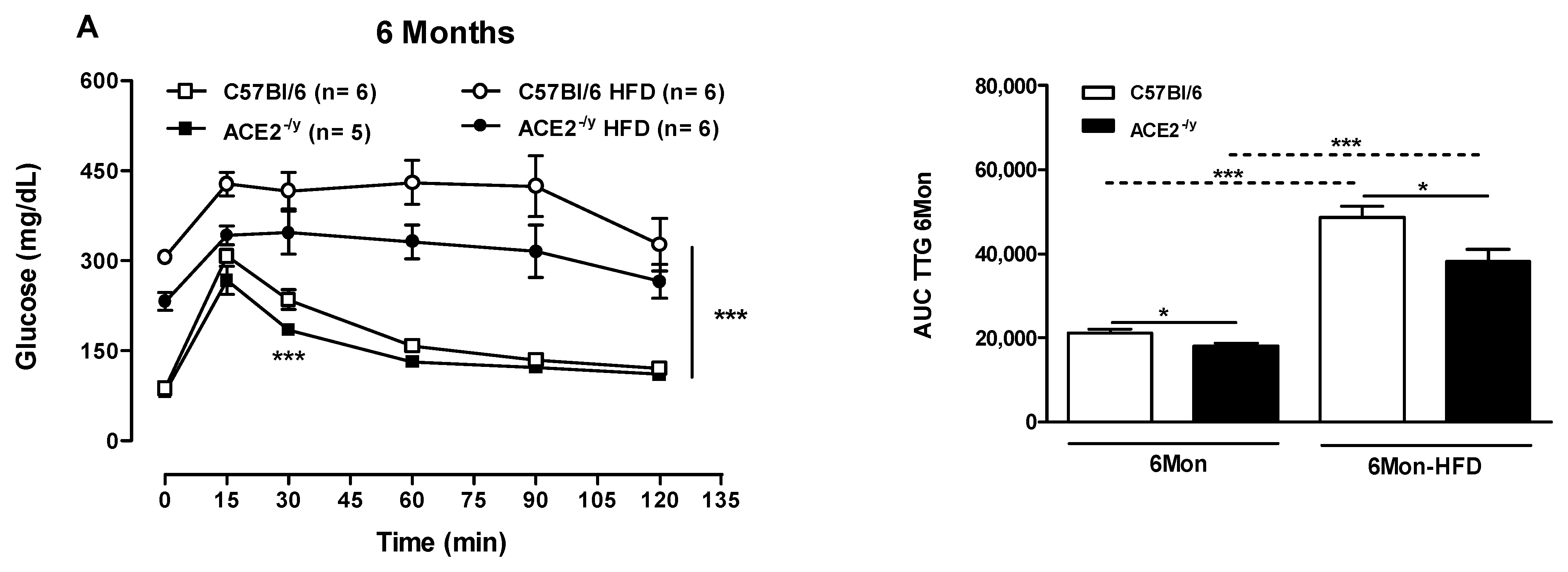
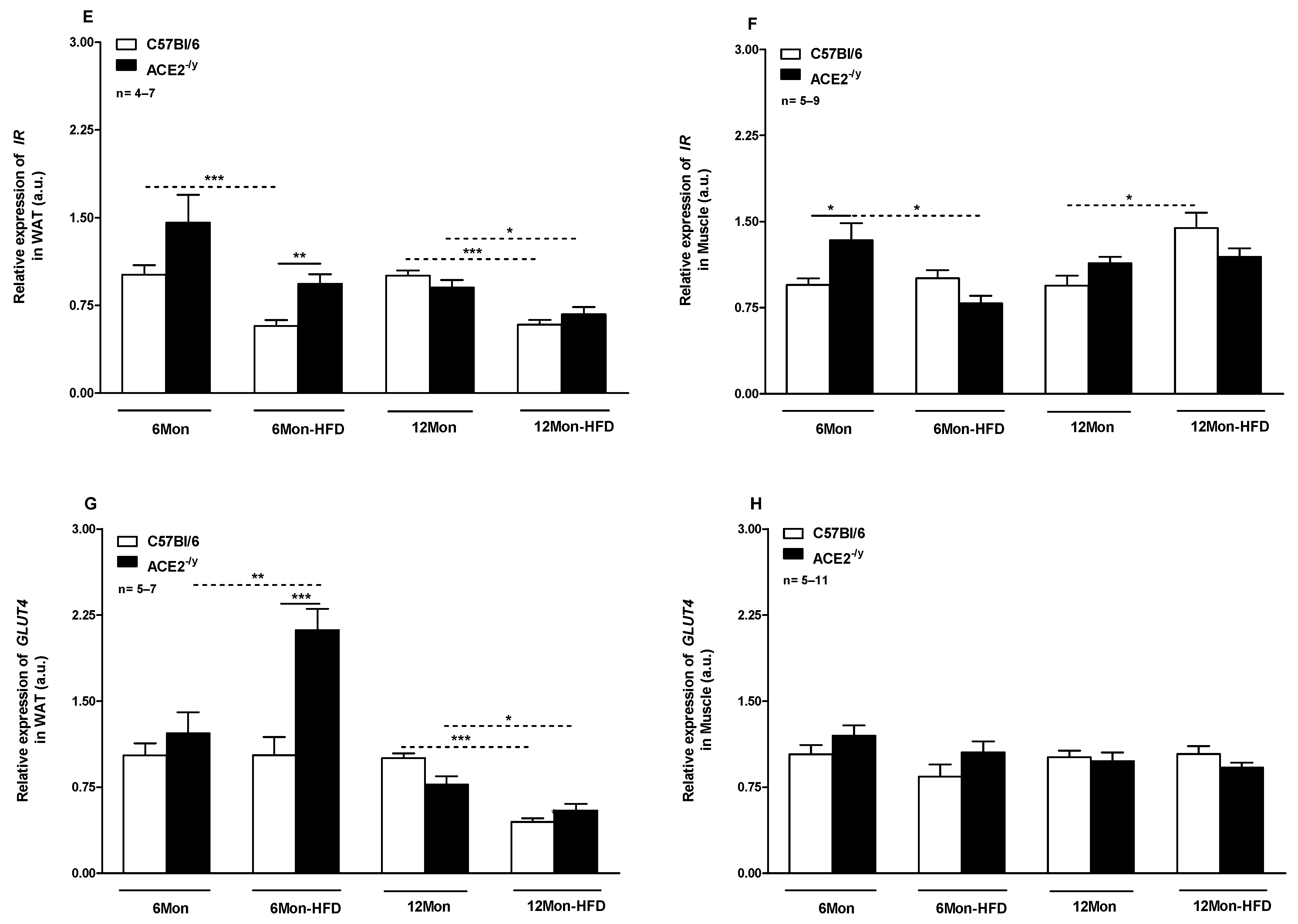
2.2. ACE2 Deficiency Improved Glucose Control and Insulin Signaling
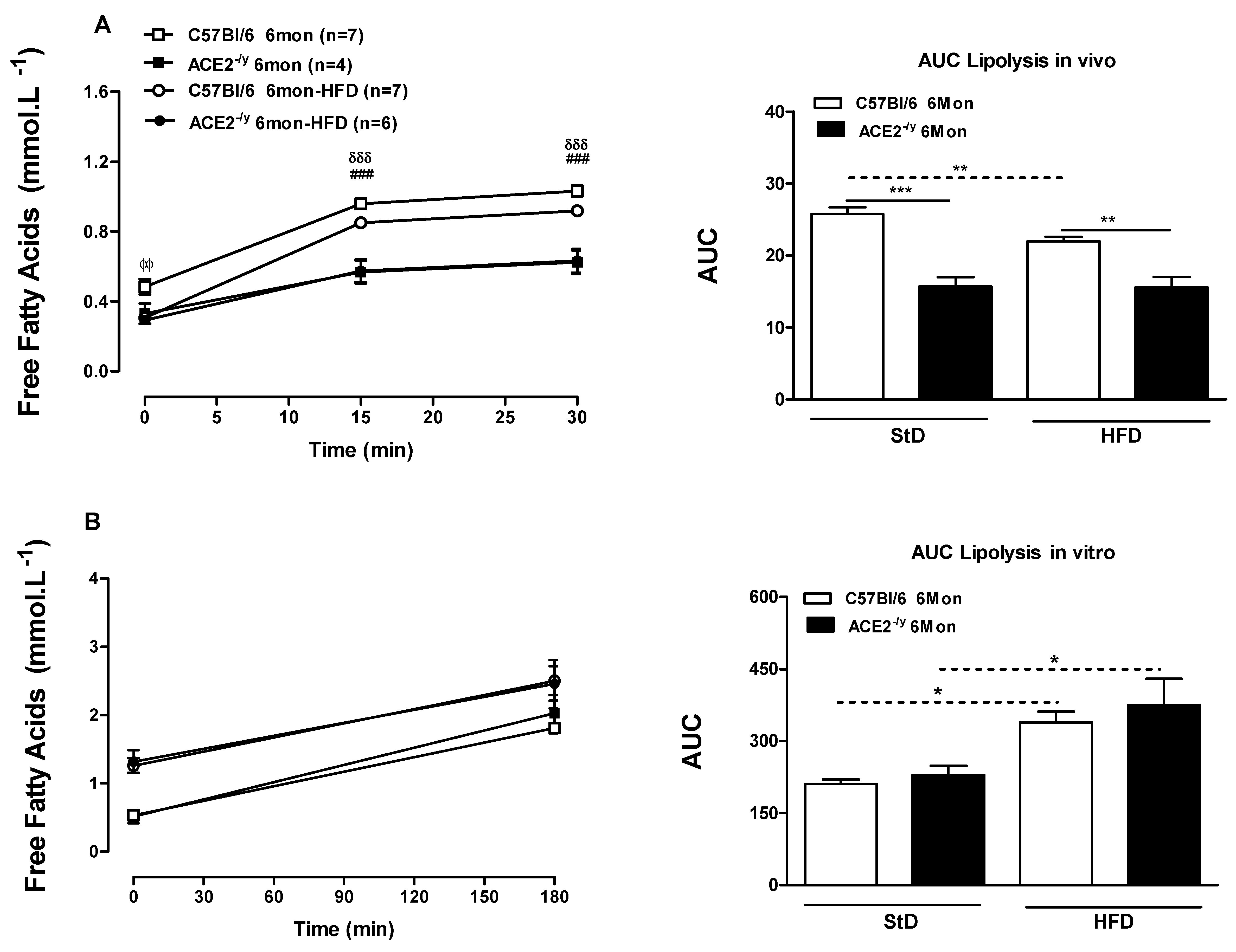
2.3. ACE2 Deficiency Leads to Impairment in Energy Balance
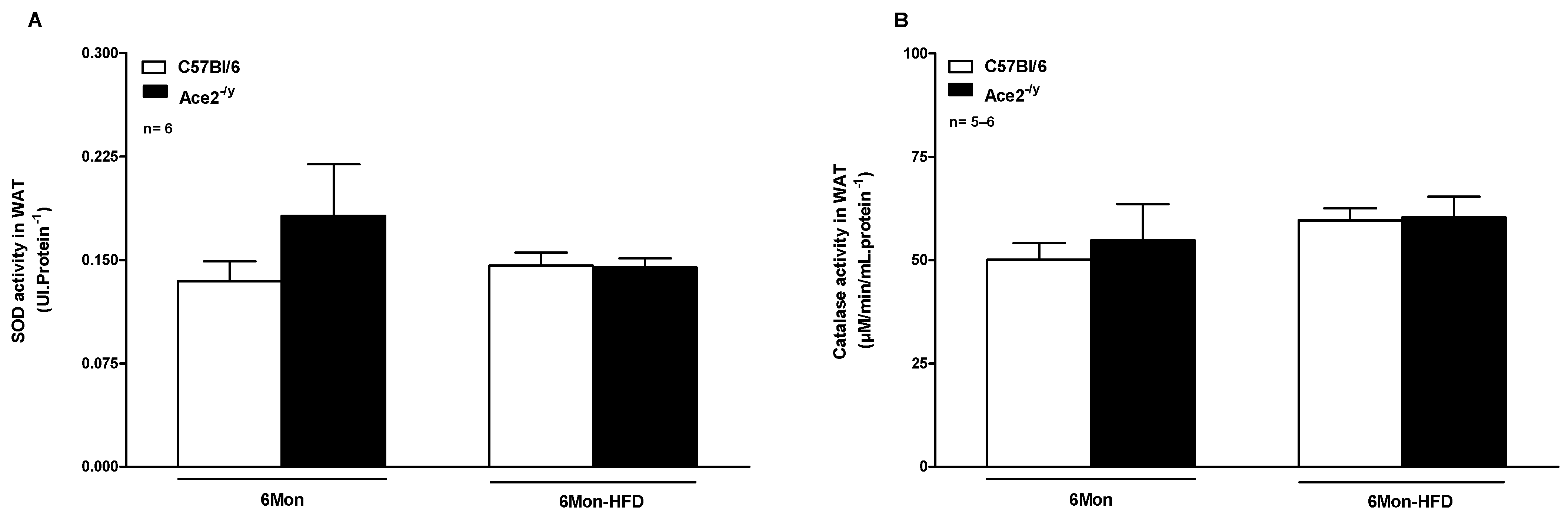
2.4. ACE2 Deficiency Did Not Alter Antioxidant Enzyme Activity in WAT
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Procedures
4.2. Glucose Tolerance and Insulin Sensitivity Tests
4.3. Metabolic Cage System
4.4. Body Composition Analysis
4.5. Lipolysis In Vivo and In Vitro
4.6. Euthanasia and Ex Vivo Experiments
4.7. Biochemical Analyses
4.8. Superoxide Dismutase (SOD) and Catalase (CAT) Activities
4.9. Adipose Tissue Histological Analysis
4.10. Quantitative Real-Time PCR
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, L.A.; Alenina, N.; Bader, M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens. Res. 2011, 34, 154–160. [Google Scholar] [CrossRef]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.-H.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry its immune cross-reactivity with, SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Klang, E.; Kassim, G.; Soffer, S.; Freeman, R.; Levin, M.A.; Reich, D.L. Severe Obesity as an Independent Risk Factor for COVID-19 Mortality in Hospitalized Patients Younger than 50. Obesity 2020, 28, 1595–1599. [Google Scholar] [CrossRef]
- Ebinger, J.E.; Achamallah, N.; Ji, H.; Claggett, B.L.; Sun, N.; Botting, P.; Nguyen, T.-T.; Luong, E.; Kim, E.H.; Park, E.; et al. Pre-existing traits associated with Covid-19 illness severity. PLoS ONE 2020, 15, e0236240. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Guetta, T.; Barnea, A.; Waldman, M.; Ben-Dor, N.; Barac, Y.D.; Kornowski, R.; Arad, M.; Hochhauser, E.; Aravot, D. Expression of the SARS-CoV-2 receptorACE2 in human heart is associated with uncontrolled diabetes, obesity, and activation of the renin angiotensin system. Cardiovasc. Diabetol. 2021, 20, 90. [Google Scholar] [CrossRef]
- Garreta, E.; Prado, P.; Stanifer, M.L.; Monteil, V.; Marco, A.; Ullate-Agote, A.; Moya-Rull, D.; Vilas-Zornoza, A.; Tarantino, C.; Romero, J.P.; et al. A diabetic milieu increases ACE2 expression and cellular susceptibility to SARS-CoV-2 infections in human kidney organoids and patient cells. Cell Metab. 2022, 34, 857–873.e9. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.T.; Loria, C.M.; Smith, S.C., Jr. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Oikonomou, E.K.; Antoniades, C. The role of adipose tissue in cardiovascular health and disease. Nat. Rev. Cardiol. 2019, 16, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Azushima, K.; Ohki, K.; Wakui, H.; Uneda, K.; Haku, S.; Kobayashi, R.; Haruhara, K.; Kinguchi, S.; Matsuda, M.; Maeda, A.; et al. Adipocyte-Specific Enhancement of Angiotensin II Type 1 Receptor-Associated Protein Ameliorates Diet-Induced Visceral Obesity and Insulin Resistance. J. Am. Hear. Assoc. 2017, 6, e004488. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Costa-Goncalves, A.C.; Todiras, M.; Rabelo, L.A.; Sampaio, W.O.; Moura, M.M.; Santos, S.S.; Luft, F.C.; Bader, M.; Gross, V.; et al. Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension 2008, 51, 574–580. [Google Scholar] [CrossRef]
- Mahmood, I.H.; Abed, M.N.; Merkhan, M.M. Effects of blocking of angiotensin system on the prevalence of metabolic syndrome in type 2 diabetic patients. Pak. J. Med. Sci. 2013, 29, 140–143. [Google Scholar]
- Silva, A.R.; Aguilar, E.C.; Alvarez-Leite, J.I.; da Silva, R.F.; Arantes, R.M.; Bader, M.; Alenina, N.; Pelli, G.; Lenglet, S.; Galan, K.; et al. Mas receptor deficiency is associated with worsening of lipid profile and severe hepatic steatosis in ApoE-knockout mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R1323–R1330. [Google Scholar] [CrossRef]
- Nunes-Souza, V.; Alenina, N.; Qadri, F.; Penninger, J.M.; Santos, R.A.S.; Bader, M.; Rabelo, L.A. CD36/Sirtuin 1 Axis Impairment Contributes to Hepatic Steatosis in ACE2-Deficient Mice. Oxidative Med. Cell. Longev. 2016, 2016, 6487509. [Google Scholar] [CrossRef]
- Österreicher, C.H.; Taura, K.; De Minicis, S.; Seki, E.; Penz-Österreicher, M.; Kodama, Y.; Kluwe, J.; Schuster, M.; Oudit, G.Y.; Penninger, J.M.; et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology 2009, 50, 929–938. [Google Scholar] [CrossRef]
- Verma, A.; Shan, Z.; Lei, B.; Yuan, L.; Liu, X.; Nakagawa, T.; Grant, M.B.; Lewin, A.S.; Hauswirth, W.W.; Raizada, M.K.; et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 28–36. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, L.A.; Todiras, M.; Nunes-Souza, V.; Qadri, F.; Szijártó, I.A.; Gollasch, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Genetic Deletion of ACE2 Induces Vascular Dysfunction in C57BL/6 Mice: Role of Nitric Oxide Imbalance and Oxidative Stress. PLoS ONE 2016, 11, e0150255. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Rentzsch, B.; Todiras, M.; Iliescu, R.; Popova, E.; Campos, L.A.; Oliveira, M.L.; Baltatu, O.C.; Santos, R.A.; Bader, M. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension 2008, 52, 967–973. [Google Scholar] [CrossRef]
- Patel, V.B.; Bodiga, S.; Basu, R.; Das, S.K.; Wang, W.; Wang, Z.; Lo, J.; Grant, M.B.; Zhong, J.; Kassiri, Z.; et al. Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: A critical role of the angiotensin II/AT1 receptor axis. Circ. Res. 2012, 110, 1322–1335. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Liu, G.C.; Zhong, J.; Basu, R.; Chow, F.L.; Zhou, J.; Loibner, H.; Janzek, E.; Schuster, M.; Penninger, J.M.; et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 2010, 59, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Singer, D.; Camargo, S.M.; Ramadan, T.; Schäfer, M.; Mariotta, L.; Herzog, B.; Huggel, K.; Wolfer, D.; Werner, S.; Penninger, J.M.; et al. Defective intestinal amino acid absorption in Ace2 null mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G686–G695. [Google Scholar] [CrossRef]
- Klempin, F.; Mosienko, V.; Matthes, S.; Villela, D.C.; Todiras, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Depletion of angiotensin-converting enzyme 2 reduces brain serotonin and impairs the running-induced neurogenic response. Cell Mol. Life Sci. 2018, 75, 3625–3634. [Google Scholar] [CrossRef]
- Crane, J.D.; Palanivel, R.; Mottillo, E.P.; Bujak, A.L.; Wang, H.; Ford, R.J.; Collins, A.; Blümer, R.M.; Fullerton, M.D.; Yabut, J.M.; et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat. Med. 2015, 21, 166–172. [Google Scholar] [CrossRef]
- Oh, C.M.; Namkung, J.; Go, Y.; Shong, K.E.; Kim, K.; Kim, H.; Park, B.Y.; Lee, H.W.; Jeon, Y.H.; Song, J.; et al. Regulation of systemic energy homeostasis by serotonin in adipose tissues. Nat. Commun. 2015, 6, 6794. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Souza, V.; Cesar-Gomes, C.J.; Da Fonseca, L.J.; Guedes Gda, S.; Smaniotto, S.; Rabelo, L.A. Aging Increases Susceptibility to High Fat Diet-Induced Metabolic Syndrome in C57BL/6 Mice: Improvement in Glycemic and Lipid Profile after Antioxidant Therapy. Oxidative Med. Cell. Longev. 2016, 2016, 1987960. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Piernas, C.; Astbury, N.M.; Hippisley-Cox, J.; O’Rahilly, S.; Aveyard, P.; AJebb, S. Associations between body-mass index and COVID-19 severity in 6.9 million people in England: A prospective, community-based, cohort study. Lancet Diabetes Endocrinol. 2021, 9, 350–359. [Google Scholar] [CrossRef]
- Gupte, M.; Boustany-Kari, C.M.; Bharadwaj, K.; Police, S.; Thatcher, S.; Gong, M.C.; English, V.L.; Cassis, L.A. ACE2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R781–R788. [Google Scholar] [CrossRef]
- Cai, Z.; Fang, L.; Jiang, Y.; Liang, M.; Wang, J.; Shen, Y.; Wang, Z.; Liang, F.; Huo, H.; Pan, C.; et al. Angiotensin II Promotes White Adipose Tissue Browning and Lipolysis in Mice. Oxidative Med. Cell. Longev. 2022, 2022, 6022601. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Souza, V.; Dias-Júnior, N.M.; Eleutério-Silva, M.A.; Ferreira-Neves, V.P.; Moura, F.A.; Alenina, N.; Bader, M.; Rabelo, L.A. 3-Amino-1,2,4-Triazole Induces Quick and Strong Fat Loss in Mice with High Fat-Induced Metabolic Syndrome. Oxidative Med. Cell. Longev. 2020, 2020, 3025361. [Google Scholar] [CrossRef]
- Rodriguez, A.; Ezquerro, S.; Mendez-Gimenez, L.; Becerril, S.; Fruhbeck, G. Revisiting the adipocyte: A model for integration of cytokine signaling in the regulation of energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E691–E714. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef]
- Airaksinen, K.; Jokkala, J.; Ahonen, I.; Auriola, S.; Kolehmainen, M.; Hanhineva, K.; Tiihonen, K. High-Fat Diet, Betaine, and Polydextrose Induce Changes in Adipose Tissue Inflammation and Metabolism in C57BL/6J Mice. Mol. Nutr. Food Res. 2018, 62, e1800455. [Google Scholar] [CrossRef]
- Gavrila, A.; Chan, J.L.; Yiannakouris, N.; Kontogianni, M.; Miller, L.C.; Orlova, C.; Mantzoros, C.S. Serum adiponectin levels are inversely associated with overall and central fat distribution but are not directly regulated by acute fasting or leptin administration in humans: Cross-sectional and interventional studies. J. Clin. Endocrinol. Metab. 2003, 88, 4823–4831. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Combs, T.P.; Pajvani, U.B.; Berg, A.H.; Lin, Y.; Jelicks, L.A.; Laplante, M.; Nawrocki, A.R.; Rajala, M.W.; Parlow, A.F.; Cheeseboro, L.; et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 2004, 145, 367–383. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Nicolson, M.; Hirsch, J.; Heymsfield, S.B.; Gallagher, D.; Chu, F.; Leibel, R.L. Effects of gender, body composition, and menopause on plasma concentrations of leptin. J. Clin. Endocrinol. Metab. 1996, 81, 3424–3427. [Google Scholar] [PubMed]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. Narrative review: The role of leptin in human physiology: Emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, J.T. Angiotensin, thirst, and sodium appetite. Physiol. Rev. 1998, 78, 583–686. [Google Scholar] [CrossRef]
- Jayasooriya, A.P.; Mathai, M.L.; Walker, L.L.; Begg, D.P.; Denton, D.A.; Cameron-Smith, D.; Egan, G.F.; McKinley, M.J.; Rodger, P.D.; Sinclair, A.J.; et al. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc. Natl. Acad. Sci. USA 2008, 105, 6531–6536. [Google Scholar] [CrossRef]
- Bailey-Downs, L.C.; Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: A paracrine mechanism contributing to vascular redox dysregulation and inflammation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 780–792. [Google Scholar] [CrossRef]
- Silva RA, P.; Chu, Y.; Miller, J.D.; Mitchell, I.J.; Penninger, J.M.; Faraci, F.M.; Heistad, D.D. Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke 2012, 43, 3358–3363. [Google Scholar] [CrossRef]
- Takeda, M.; Yamamoto, K.; Takemura, Y.; Takeshita, H.; Hongyo, K.; Kawai, T.; Hanasaki-Yamamoto, H.; Oguro, R.; Takami, Y.; Tatara, Y.; et al. Loss of ACE2 exaggerates high-calorie diet-induced insulin resistance by reduction of GLUT4 in mice. Diabetes 2013, 62, 223–233. [Google Scholar] [CrossRef]
- Niu, M.J.; Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 2008, 34, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Blume, C.; Jackson, C.L.; Spalluto, C.M.; Legebeke, J.; Nazlamova, L.; Conforti, F.; Perotin, J.-M.; Frank, M.; Butler, J.; Crispin, M.; et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet 2021, 53, 205–214. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 6 Months | 12 Months | ||||||
|---|---|---|---|---|---|---|---|---|
| Standard Diet | High-Fat Diet | Standard Diet | High-Fat Diet | |||||
| C57Bl/6 | ACE2-/y | C57Bl/6 | ACE2-/y | C57Bl/6 | ACE2-/y | C57Bl/6 | ACE2-/y | |
| Muscle % | 67.77 ± 1.54 | 72.85 ± 1.78 | 48.11 ± 1.40 c | 64.85 ± 1.12 bd | 69.50 ± 0.74 | 72.84 ± 0.27 a | 49.92 ± 1.17 c | 51.85 ± 1.88 b |
| Water % | 7.15 ± 0.11 | 7.34 ± 0.4 | 6.47 ± 0.13 c | 6.43 ± 0.18 b | 6.54 ± 0.19 | 6.71 ± 0.12 | 6.16 ± 0.11 | 6.54 ± 0.14 |
| Triglyceride (mg·dL−1) | 22.02 ± 1.9 | 16.57 ± 2.3 | 26.9 ± 2.2 | 29.0 ± 1.7 b | 33.9 ± 3.3 | 32.9 ± 7.4 | 45.8 ± 6.0 | 44.9 ± 3.9 |
| Cholesterol (mg·dL−1) | 36.97 ± 2.1 | 45.23 ± 4.4 | 126.3 ± 4.7 c | 140.7 ± 10.2 b | 68.1 ± 5.7 | 73.3 ± 12.6 | 173.6 ± 11.1 c | 210.3 ± 27.6 b |
| NEFA (mmol·L−1) | 0.29 ± 0.03 | 0.14 ± 0.01 a | 0.49 ± 0.05 c | 0.49 ± 0.05 b | 0.48 ± 0.1 | 0.27 ± 0.07 | 0.42 ± 0.04 | 0.42 ± 0.09 |
| Glucose (mg·dL−1) | 121.6 ± 8.8 | 103.5 ± 7.0 | 153.0 ± 9.3 c | 165.2 ± 7.8 b | 138.8 ± 9.0 | 130.6 ± 6.6 | 187.8 ± 6.7 c | 176.8 ± 6.9 b |
| Insulin (pg·dL−1) | 179.2 ± 12.2 | 175.7 ± 4.9 | 238.7 ± 25.8 | 184.1 ± 4.4 | 303.5 ± 6.8 | 301.2 ± 9.2 | 333.0 ± 17.5 | 331.1 ± 17.3 |
| Food intake (g·day−1) | 5.12 ± 0.44 | 4.64 ± 0.15 | 4.57 ± 0.37 | 3.34 ± 0.18 bd | ND | ND | ND | ND |
| Liquid intake (g·day−1) | 3.00 ± 0.3 | 4.27 ± 0.1 a | 2.61 ± 0.2 | 4.68 ± 0.4 d | ND | ND | ND | ND |
| Total Energy Expenditure (kcal·h−1) | 0.55 ± 0.03 | 0.49 ± 0.01 | 0.58 ± 0.01 | 0.48 ± 0.01 d | ND | ND | ND | ND |
| Primers | Sequence Forward and Reverse (5′–3′) and Length (bp) |
|---|---|
| Leptin | F: CGTGTGTGAAATGTCATTGATCCT (24) R: GACACCAAAACCCTCATCAAGAC (23) |
| Adiponectin | F: GGAACTTGTGCAGGTTGGAT (20) R: CCTTCAGCTCCTGTCATTCC (20) |
| Lipoprotein lipase | F: AGTGGCCGAGAGCGAGAAC (19) R: CCACCTCCGTGTAAATCAAGAAG (23) |
| Hormone-sensitive lipase | F: ACGGATACCGTAGTTTGGTGC (21) R: TCCAGAAGTGCACATCCAGGT (21) |
| Adrenergic β3 receptor | F: GCTGACTTGGTAGTGGGACTC (21) R: TAGAAGGAGACGGAGGAGGAG (21) |
| Glucose transporter type 4 | F: TGATTCTGCTGCCCTTCTGT (20) R: GGACATTGGACGCTCTCTCT (20) |
| Insulin receptor | F: CCACCAATACGTCATTCACAAC (22) R: GGGCAGATGTCACAGAATCAA (21) |
| Glyceraldehyde 3-phosphate dehydrogenase | F: CCATCACCATCTTCCAGGAG (20) R: GTGGTTCACACCCATCACAA (20) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes-Souza, V.; Alenina, N.; Qadri, F.; Mosienko, V.; Santos, R.A.S.; Bader, M.; Rabelo, L.A. ACE2 Knockout Mice Are Resistant to High-Fat Diet-Induced Obesity in an Age-Dependent Manner. Int. J. Mol. Sci. 2024, 25, 9515. https://doi.org/10.3390/ijms25179515
Nunes-Souza V, Alenina N, Qadri F, Mosienko V, Santos RAS, Bader M, Rabelo LA. ACE2 Knockout Mice Are Resistant to High-Fat Diet-Induced Obesity in an Age-Dependent Manner. International Journal of Molecular Sciences. 2024; 25(17):9515. https://doi.org/10.3390/ijms25179515
Chicago/Turabian StyleNunes-Souza, Valéria, Natalia Alenina, Fatimunnisa Qadri, Valentina Mosienko, Robson Augusto Souza Santos, Michael Bader, and Luiza Antas Rabelo. 2024. "ACE2 Knockout Mice Are Resistant to High-Fat Diet-Induced Obesity in an Age-Dependent Manner" International Journal of Molecular Sciences 25, no. 17: 9515. https://doi.org/10.3390/ijms25179515
APA StyleNunes-Souza, V., Alenina, N., Qadri, F., Mosienko, V., Santos, R. A. S., Bader, M., & Rabelo, L. A. (2024). ACE2 Knockout Mice Are Resistant to High-Fat Diet-Induced Obesity in an Age-Dependent Manner. International Journal of Molecular Sciences, 25(17), 9515. https://doi.org/10.3390/ijms25179515