
Activation of G Protein-Coupled Estrogen Receptor 1 (GPER) Attenuates Obesity-Induced Asthma by Switching M1 Macrophages to M2 Macrophages

Abstract
:1. Introduction
2. Results
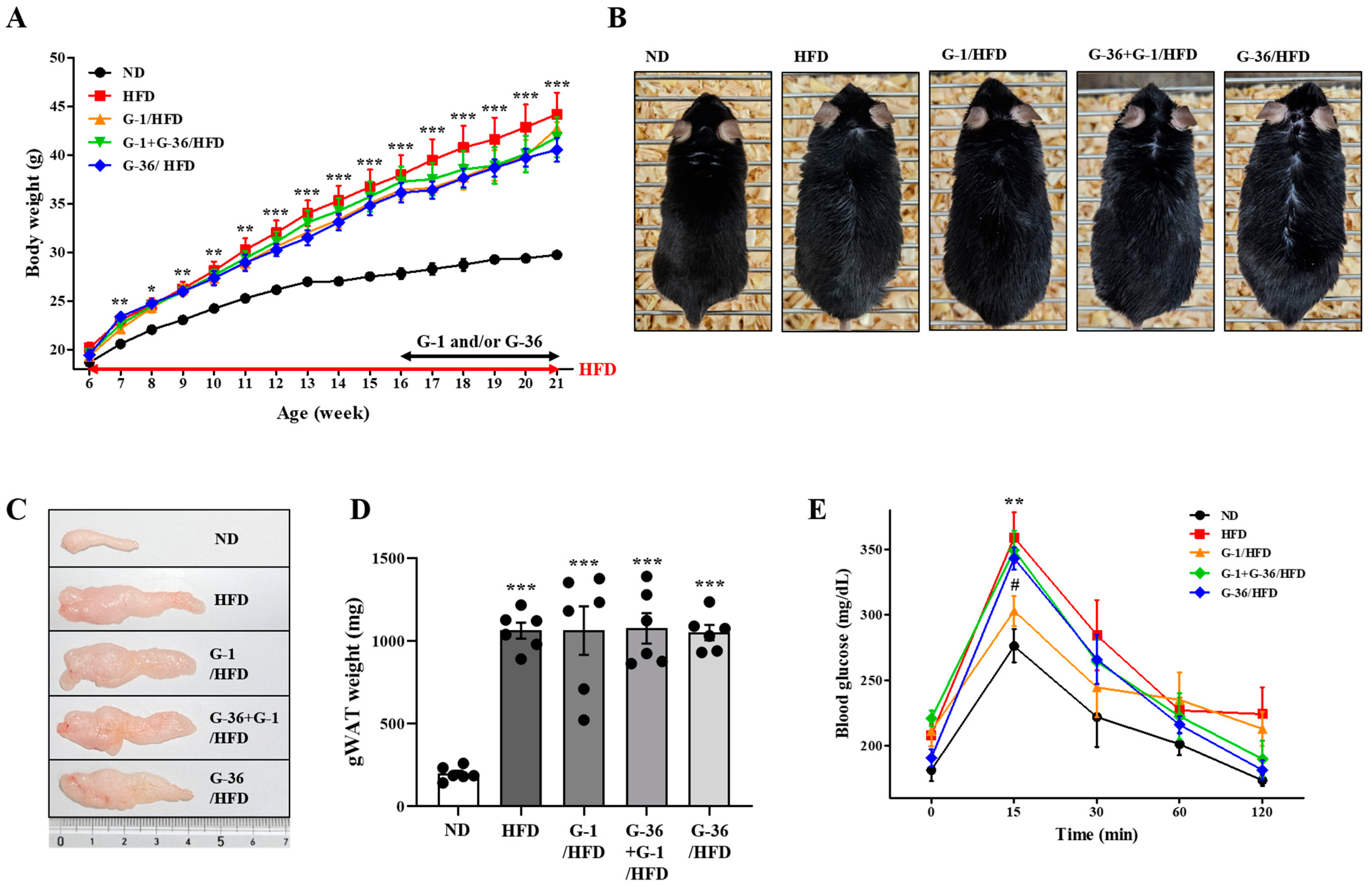
2.1. G-1 Improved the Glucose Tolerance Capacity; However, It Did Not Suppress the HFD-Induced Obesity in C57BL/6 Mice
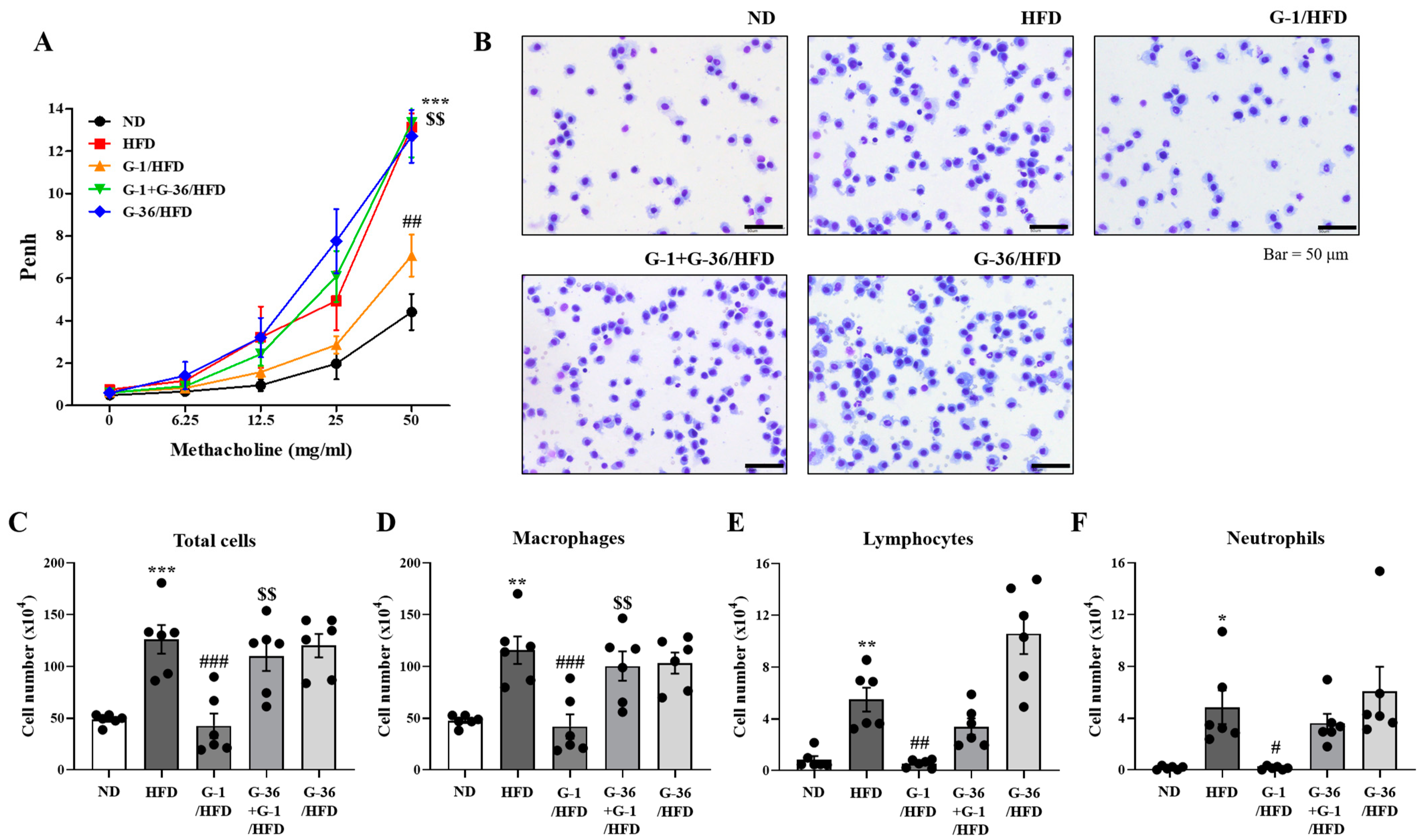
2.2. G-1 Suppressed Airway Hyperresponsiveness (AHR) and Immune Cell Infiltration in the Bronchoalveolar Lavage Fluid (BALF) of C57BL/6 Mice
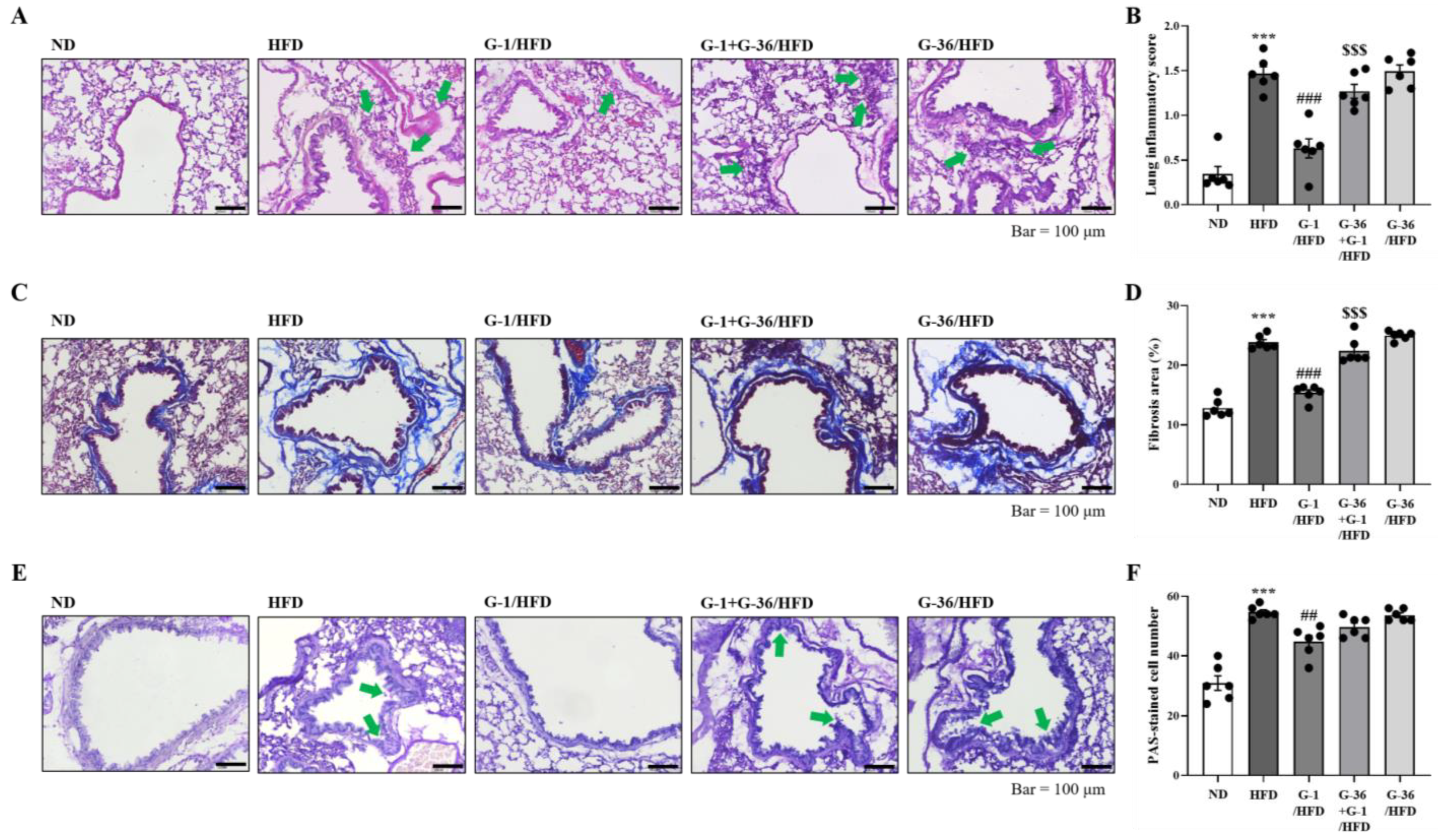
2.3. G-1 Suppressed Histopathological Changes in the Lungs of C57BL/6 Mice
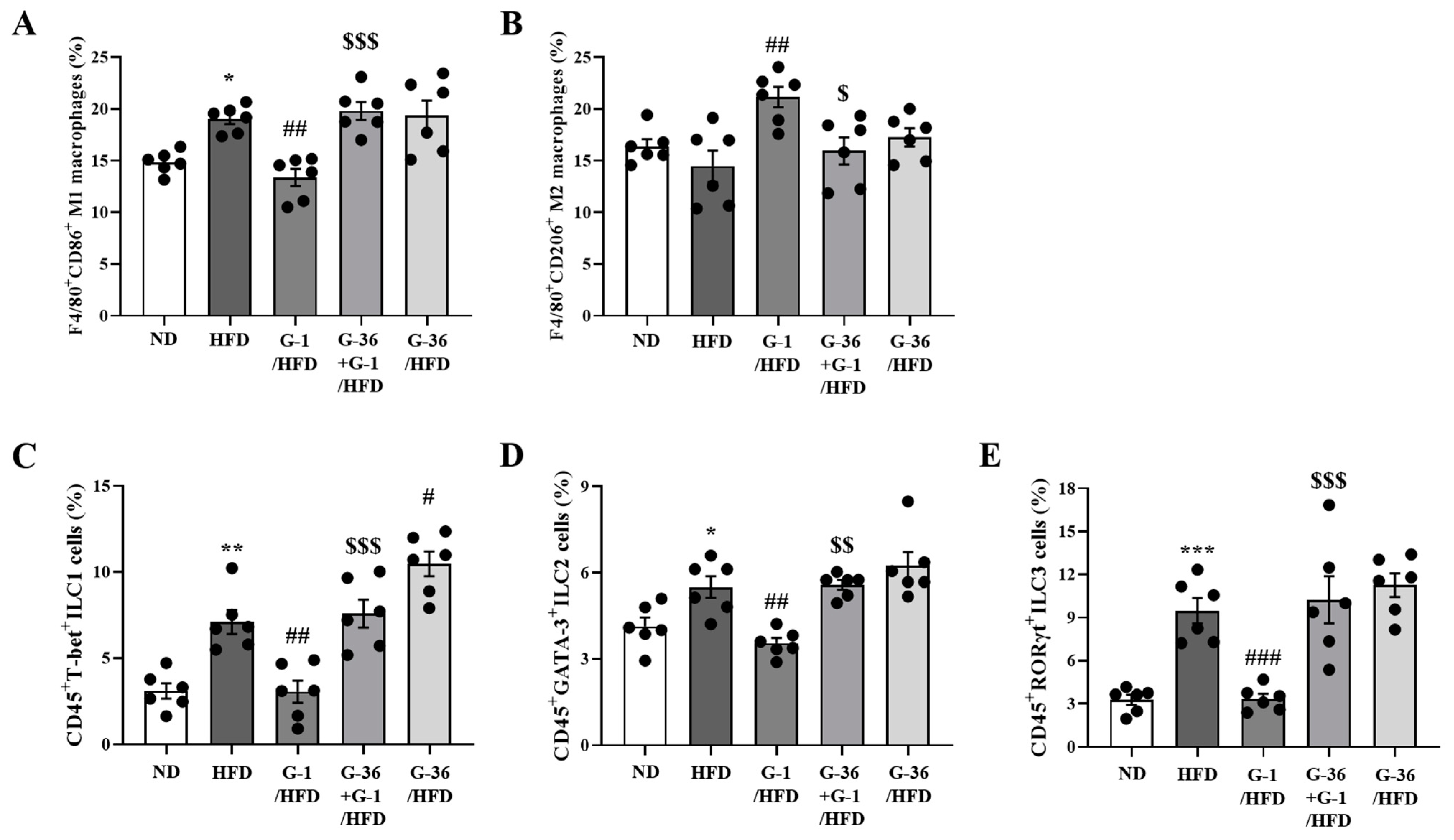
2.4. G-1 Increased Anti-Inflammatory M2 Macrophages and Suppressed Innate Lymphoid Cells (ILCs) in the Lungs of C57BL/6 Mice
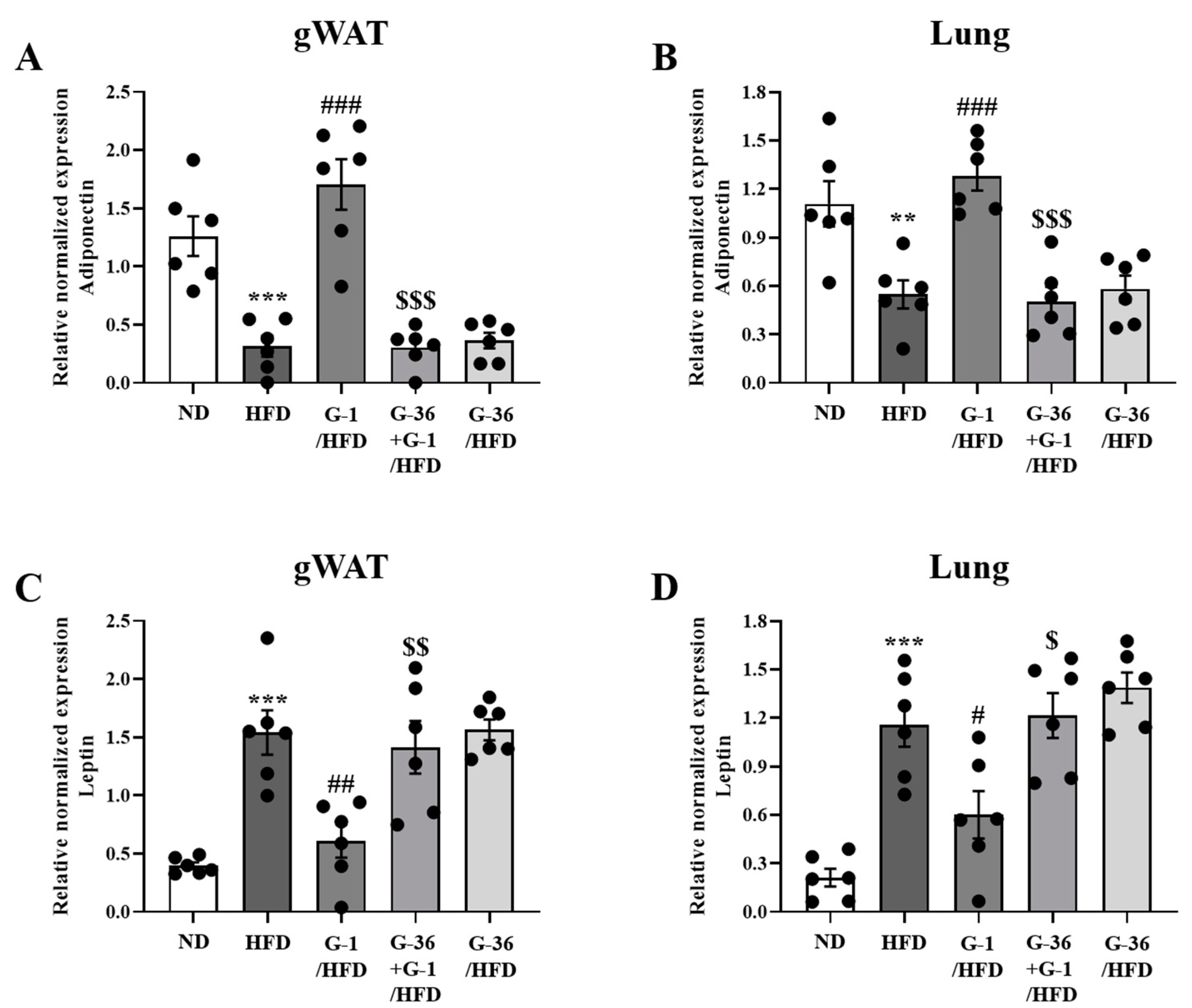
2.5. G-1 Increased Adiponectin Levels and Suppressed Leptin Levels in the Lungs and Gonadal White Adipose Tissue of C57BL/6 Mice
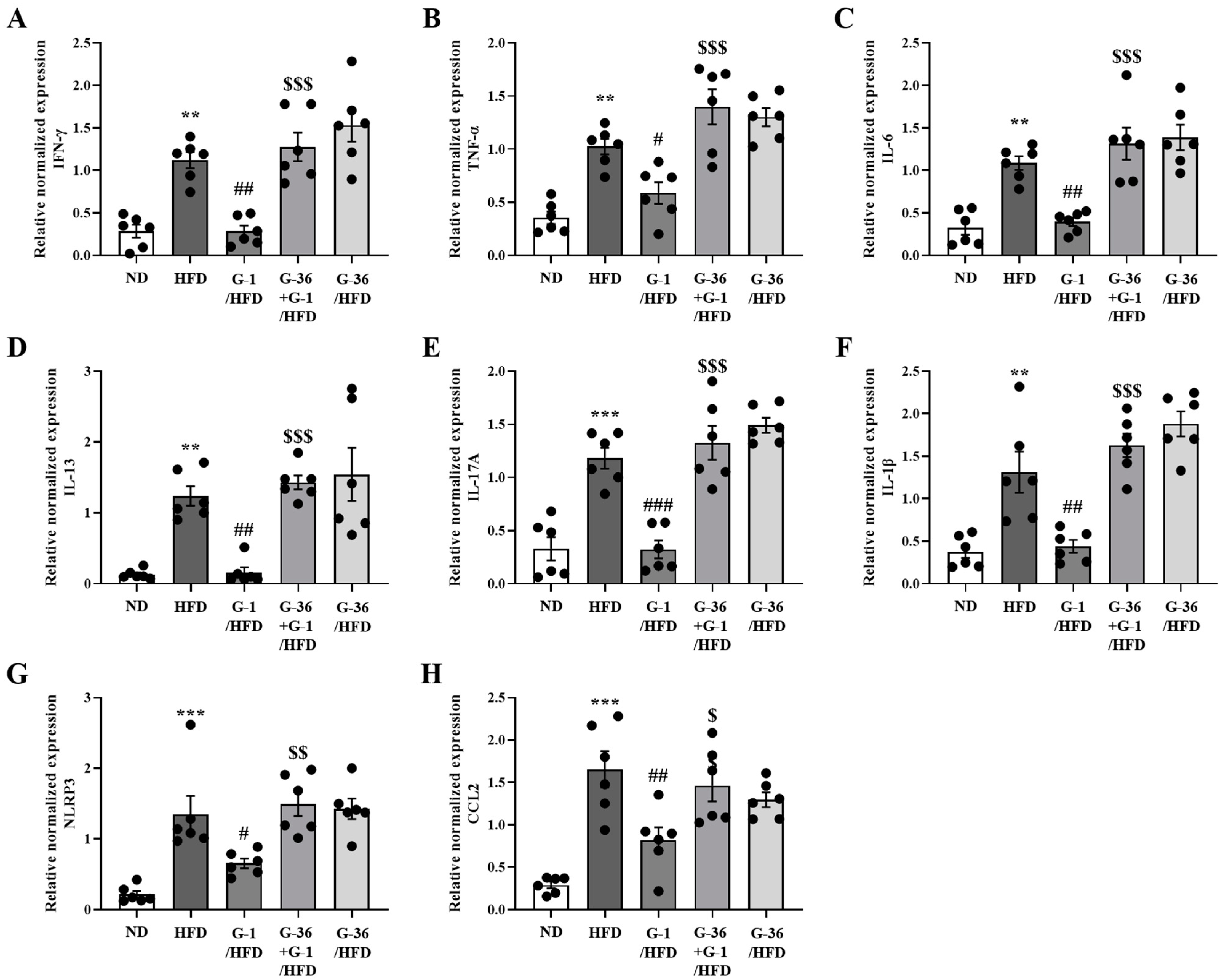
2.6. G-1 Suppressed Inflammatory Cytokine Levels in the Gonadal White Adipose Tissue of C57BL/6 Mice
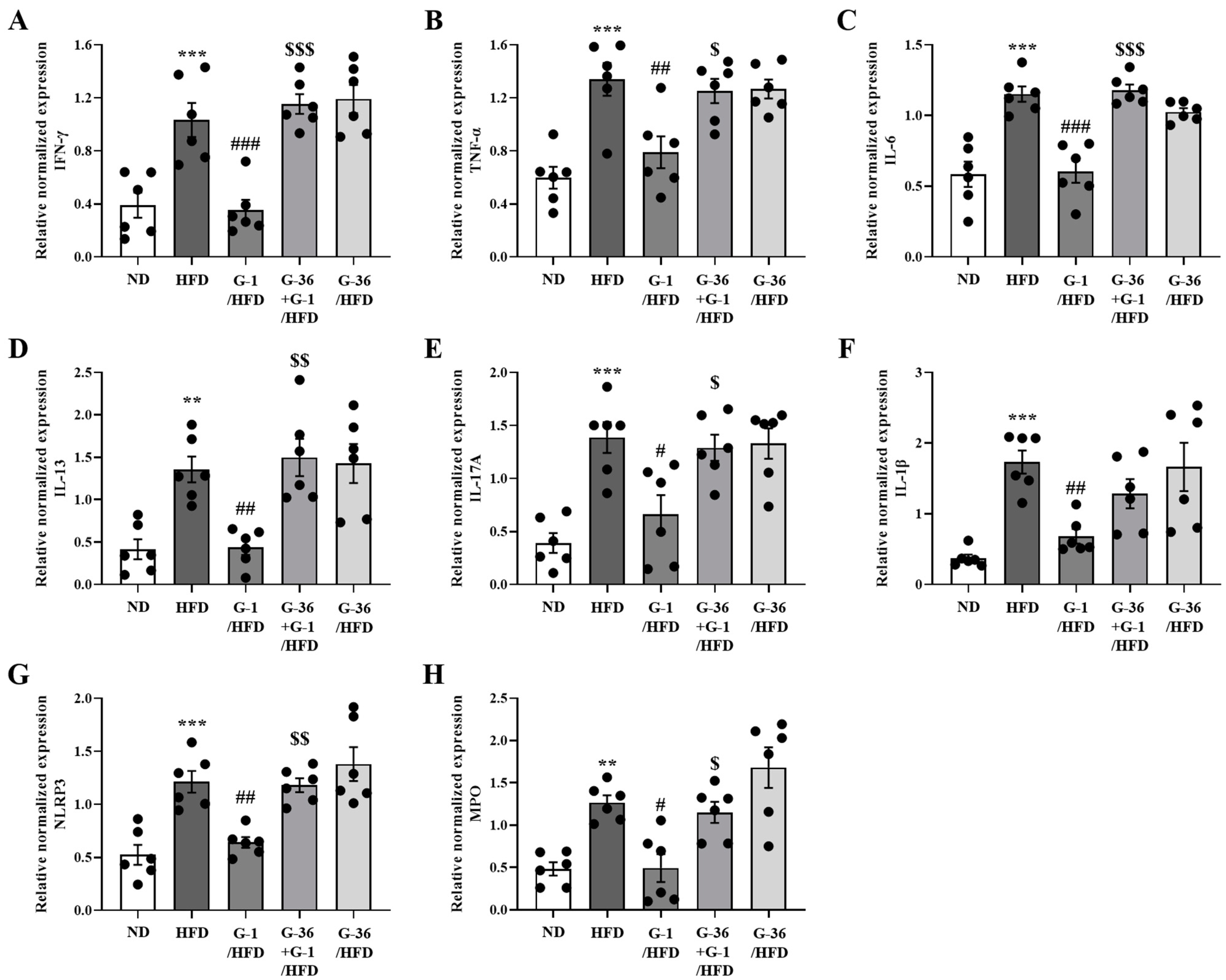
2.7. G-1 Suppressed Inflammatory Cytokine Levels in the Lungs of C57BL/6 Mice
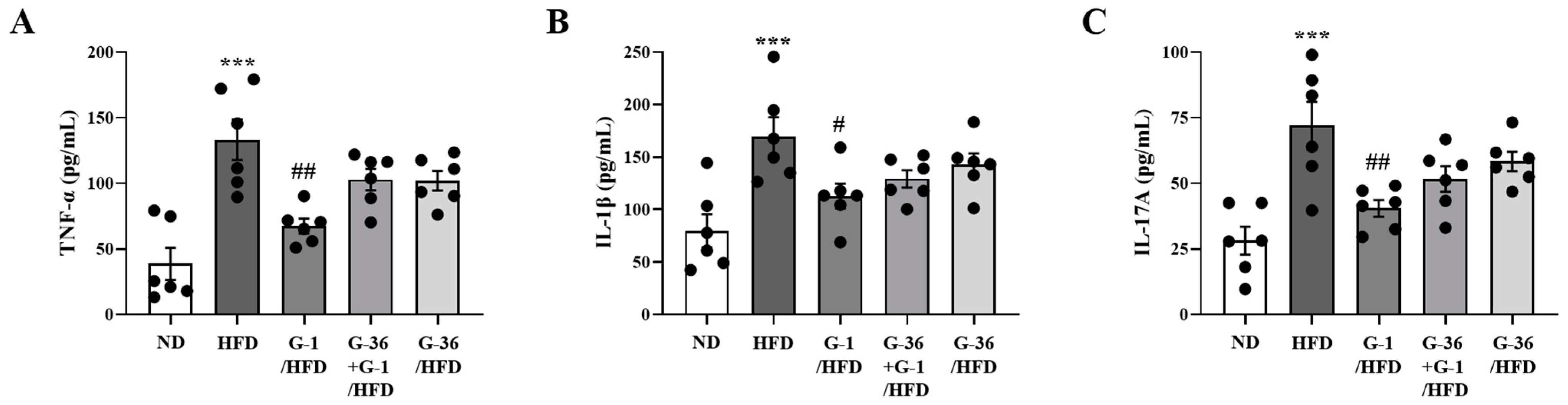
2.8. G-1 Suppressed Inflammatory Cytokine Levels in the BALF of C57BL/6 Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mouse Strain
4.3. HFD Feeding
4.4. Oral Glucose Tolerance Test (OGTT)
4.5. Airway Hyperresponsiveness (AHR) to Methacholine
4.6. Bronchial Alveolar Lavage Fluid (BALF) Cell Enumeration and Analysis
4.7. Histological Examination of Lung Tissue
4.8. Quantitative Real-Time PCR (qRT-PCR)
4.9. Flow Cytometry
4.10. Enzyme-Linked Immunosorbent Assay (ELISA)
4.11. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Albracht-Schulte, K.; Kalupahana, N.S.; Ramalingam, L.; Wang, S.; Rahman, S.M.; Robert-McComb, J.; Moustaid-Moussa, N. Omega-3 fatty acids in obesity and metabolic syndrome: A mechanistic update. J. Nutr. Biochem. 2018, 58, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodríguez, J.A. Relationship Between Obesity and Asthma. Arch. Bronconeumol. 2007, 43, 171–175. [Google Scholar] [CrossRef]
- Visser, M.; Bouter, L.M.; McQuillan, G.M.; Wener, M.H.; Harris, T.B. Low-grade systemic inflammation in overweight children. Pediatrics 2001, 107, E13. [Google Scholar] [CrossRef]
- Miethe, S.; Karsonova, A.; Karaulov, A.; Renz, H. Obesity and asthma. J. Allergy Clin. Immunol. 2020, 146, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.A.; Terry, R.D.; Flynt, L.; Xu, A.; Hug, C. Adiponectin attenuates allergen-induced airway inflammation and hyperresponsiveness in mice. J. Allergy Clin. Immunol. 2006, 118, 389–395. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, X.; Chong, L.; Kong, L.; Wen, S.; Zhang, H.; Zhang, W.; Li, C. Adiponectin alleviates exacerbation of airway inflammation and oxidative stress in obesity-related asthma mice partly through AMPK signaling pathway. Int. Immunopharmacol. 2019, 67, 396–407. [Google Scholar] [CrossRef]
- Matarese, G.; La Cava, A.; Sanna, V.; Lord, G.M.; Lechler, R.I.; Fontana, S.; Zappacosta, S. Balancing susceptibility to infection and autoimmunity: A role for leptin? Trends Immunol. 2002, 23, 182–187. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Holguin, F. Sex Hormones and Asthma. Am. J. Respir. Crit. Care Med. 2020, 201, 127–128. [Google Scholar] [CrossRef]
- Vink, N.M.; Postma, D.S.; Schouten, J.P.; Rosmalen, J.G.; Boezen, H.M. Gender differences in asthma development and remission during transition through puberty: The TRacking Adolescents’ Individual Lives Survey (TRAILS) study. J. Allergy Clin. Immunol. 2010, 126, 498–504.e6. [Google Scholar] [CrossRef]
- Matheson, M.C.; Burgess, J.A.; Lau, M.Y.; Lowe, A.J.; Gurrin, L.C.; Hopper, J.L.; Giles, G.G.; Johns, D.P.; Walters, E.H.; Abramson, M.J.; et al. Hormonal contraception increases risk of asthma among obese but decreases it among nonobese subjects: A prospective, population-based cohort study. ERJ Open Res. 2015, 1, 00026–02015. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.Y.; Forno, E.; Celedón, J.C. Sex Steroid Hormones and Asthma in a Nationwide Study of U.S. Adults. Am. J. Respir. Crit. Care Med. 2020, 201, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Teixeira, D.; Calhau, C. Estrogen signaling in metabolic inflammation. Mediat. Inflamm. 2014, 2014, 615917. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.A.; Al-Kabariti, A.Y.; Sutton, C. Comprehensive understanding of the role of GPER in estrogen receptor-alpha negative breast cancer. J. Steroid Biochem. Mol. Biol. 2024, 241, 106523. [Google Scholar] [CrossRef] [PubMed]
- Żabińska, M.; Wiśniewska, K.; Węgrzyn, G.; Pierzynowska, K. Exploring the physiological role of the G protein-coupled estrogen receptor (GPER) and its associations with human diseases. Psychoneuroendocrinology 2024, 166, 107070. [Google Scholar] [CrossRef]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of human cDNA encoding a novel heptahelix receptor expressed in Burkitt’s lymphoma and widely distributed in brain and peripheral tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- Feng, Y.; Gregor, P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem. Biophys. Res. Commun. 1997, 231, 651–654. [Google Scholar] [CrossRef]
- Olde, B.; Leeb-Lundberg, L.M. GPR30/GPER1: Searching for a role in estrogen physiology. Trends Endocrinol. Metab. 2009, 20, 409–416. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.E.; Carstens, E.J.; Irani, B.G.; Gent, L.M.; Hahner, L.M.; Clegg, D.J. Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm. Behav. 2014, 66, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Mauvais-Jarvis, F.; Prossnitz, E.R. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J. Steroid Biochem. Mol. Biol. 2018, 176, 31–37. [Google Scholar] [CrossRef]
- Lin, X.; Wang, L.; Lu, X.; Zhang, Y.; Zheng, R.; Chen, R.; Zhang, W. Targeting of G-protein coupled receptor 40 alleviates airway hyperresponsiveness through RhoA/ROCK1 signaling pathway in obese asthmatic mice. Respir. Res. 2023, 24, 56. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.J.; Chang, Y.J.; Pichavant, M.; Shore, S.A.; Fitzgerald, K.A.; Iwakura, Y.; Israel, E.; Bolger, K.; Faul, J.; et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med. 2014, 20, 54–61. [Google Scholar] [CrossRef]
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Hsu, A.T.; Gottschalk, T.A.; Tsantikos, E.; Hibbs, M.L. The Role of Innate Lymphoid Cells in Chronic Respiratory Diseases. Front. Immunol. 2021, 12, 733324. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef]
- Capllonch-Amer, G.; Lladó, I.; Proenza, A.M.; García-Palmer, F.J.; Gianotti, M. Opposite effects of 17-β estradiol and testosterone on mitochondrial biogenesis and adiponectin synthesis in white adipocytes. J. Mol. Endocrinol. 2014, 52, 203–214. [Google Scholar] [CrossRef]
- Tanaka, M.; Nakaya, S.; Kumai, T.; Watanabe, M.; Tateishi, T.; Shimizu, H.; Kobayashi, S. Effects of estrogen on serum leptin levels and leptin mRNA expression in adipose tissue in rats. Horm. Res. 2001, 56, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.A.; Johnston, R.A. Obesity and asthma. Pharmacol. Ther. 2006, 110, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.A. Obesity, airway hyperresponsiveness, and inflammation. J. Appl. Physiol. 2010, 108, 735–743. [Google Scholar] [CrossRef]
- Kwon, O.; Kang, E.S.; Kim, I.; Shin, S.; Kim, M.; Kwon, S.; Oh, S.R.; Ahn, Y.S.; Kim, C.H. GPR30 mediates anorectic estrogen-induced STAT3 signaling in the hypothalamus. Metabolism 2014, 63, 1455–1461. [Google Scholar] [CrossRef]
- Zhu, P.; Yuen, J.M.; Sham, K.W.; Cheng, C.H. GPER mediates the inhibitory actions of estrogen on adipogenesis in 3T3-L1 cells through perturbation of mitotic clonal expansion. Gen. Comp. Endocrinol. 2013, 193, 19–26. [Google Scholar] [CrossRef]
- Tiano, J.P.; Delghingaro-Augusto, V.; Le May, C.; Liu, S.; Kaw, M.K.; Khuder, S.S.; Latour, M.G.; Bhatt, S.A.; Korach, K.S.; Najjar, S.M.; et al. Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents β cell failure in rodent models of type 2 diabetes. J. Clin. Investig. 2011, 121, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Prossnitz, E.R. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic beta-cells. Endocrinology 2011, 152, 3030–3039. [Google Scholar] [CrossRef]
- Salem, M.L.; Matsuzaki, G.; Kishihara, K.; Madkour, G.A.; Nomoto, K. β-estradiol suppresses T cell-mediated delayed-type hypersensitivity through suppression of antigen-presenting cell function and Th1 induction. Int. Arch. Allergy Immunol. 2000, 121, 161–169. [Google Scholar] [CrossRef]
- Brunsing, R.L.; Prossnitz, E.R. Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled estrogen receptor (GPER) agonist G-1. Immunology 2011, 134, 93–106. [Google Scholar] [CrossRef]
- Brunsing, R.L.; Owens, K.S.; Prossnitz, E.R. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing conditions. J. Immunother. 2013, 36, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-W.; Odegaard, J.I.; Mukundan, L.; Qiu, Y.; Molofsky, A.B.; Nussbaum, J.C.; Yun, K.; Locksley, R.M.; Chawla, A. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 2015, 160, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, H.; Yang, Y.; Jiang, Z.; Ma, H. Dehydroepiandrosterone activates the GPER-mediated AMPK signaling pathway to alleviate the oxidative stress and inflammatory response in laying hens fed with high-energy and low-protein diets. Life Sci. 2022, 308, 120926. [Google Scholar] [CrossRef]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef]
- Wculek, S.K.; Heras-Murillo, I.; Mastrangelo, A.; Mañanes, D.; Galán, M.; Miguel, V.; Curtabbi, A.; Barbas, C.; Chandel, N.S.; Enríquez, J.A.; et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity 2023, 56, 516–530.e9. [Google Scholar] [CrossRef]
- Blasko, E.; Haskell, C.A.; Leung, S.; Gualtieri, G.; Halks-Miller, M.; Mahmoudi, M.; Dennis, M.K.; Prossnitz, E.R.; Karpus, W.J.; Horuk, R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 2009, 214, 67–77. [Google Scholar] [CrossRef]
- Wang, C.; Dehghani, B.; Li, Y.; Kaler, L.J.; Proctor, T.; Vandenbark, A.A.; Offner, H. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J. Immunol. 2009, 182, 3294–3303. [Google Scholar] [CrossRef]
- Meyer, M.R.; Fredette, N.C.; Howard, T.A.; Hu, C.; Ramesh, C.; Daniel, C.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. G protein-coupled estrogen receptor protects from atherosclerosis. Sci. Rep. 2014, 4, 7564. [Google Scholar] [CrossRef]
- Periyalil, H.A.; Wood, L.G.; Wright, T.A.; Karihaloo, C.; Starkey, M.R.; Miu, A.S.; Baines, K.J.; Hansbro, P.M.; Gibson, P.G. Obese asthmatics are characterized by altered adipose tissue macrophage activation. Clin. Exp. Allergy 2018, 48, 641–649. [Google Scholar] [CrossRef]
- Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. [Google Scholar] [CrossRef] [PubMed]
- Dib, L.H.; Ortega, M.T.; Fleming, S.D.; Chapes, S.K.; Melgarejo, T. Bone marrow leptin signaling mediates obesity-associated adipose tissue inflammation in male mice. Endocrinology 2014, 155, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Im, D.-S. Efficacy Comparison of LPA2 Antagonist H2L5186303 and Agonist GRI977143 on Ovalbumin-Induced Allergic Asthma in BALB/c Mice. Int. J. Mol. Sci. 2022, 23, 9745. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Gene | Forward Sequence (5′ to 3′) | Reverse Sequence (5′ to 3′) |
|---|---|---|
| Il-4 | CCTCACAGCAACGAAGAACA | CTGCAGCTCCATGAGAACAC |
| Il-5 | CACCAGCTATGCATTGGAGA | CTCTTTTTGGCGGTCAATGT |
| Il-6 | CTGATGCTGGTGACAACCAC | TCCACGATTACCCAGAGAAC |
| Il-13 | CAGCATGGTATGGAGTGTGG | AGGCCATGCAATATCCTCTG |
| Il-17a | AGCTGGACCACCACATGAAT | AGCATCTTCTCGACCCTGAA |
| Ifn-γ | CACGGCACAGTCATTGAAAG | GTCACCATCCTTTTGCCAGT |
| Il-22 | CCGAGGAGTCAGTGCTAAGG | CATGTAGGGCTGGAACCTGT |
| Il-23 | GACCCACAAGGACTCAAGGA | CATGGGGCTATCAGGGAGTA |
| Il-1β | CAGGCAGGCAGTATCACTCA | TGTCCTCATCCTGGAAGGTC |
| Tnf-α | ACGGCATGGATCTCAAAGAC | AGATAGCAAATCGGCTGACG |
| Mpo | CACTGGACACTGCAACAACA | CCATTGCGATTGACTCCAGG |
| Nlrp3 | ATGCTGCTTCGACATCTCCT | GTTTCTGGAGGTTGCAGAGC |
| Ccl2 | TGAATGTGAAGTTGACCCGT | ACAGAAGTGCTTGAGGTGGT |
| Leptin | TTCCTGTGGCTTTGGTCCTA | CGACTGCGTGTGTGAAATGT |
| Adipoq | TACTGCAACATTCCGGGACT | GTAGGTGAAGAGAACGGCCT |
| Gapdh | AACTTTGGCATTGTGGAAGG | GGATGCAGGGATGATGTTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, S.-E.; Im, D.-S. Activation of G Protein-Coupled Estrogen Receptor 1 (GPER) Attenuates Obesity-Induced Asthma by Switching M1 Macrophages to M2 Macrophages. Int. J. Mol. Sci. 2024, 25, 9532. https://doi.org/10.3390/ijms25179532
Son S-E, Im D-S. Activation of G Protein-Coupled Estrogen Receptor 1 (GPER) Attenuates Obesity-Induced Asthma by Switching M1 Macrophages to M2 Macrophages. International Journal of Molecular Sciences. 2024; 25(17):9532. https://doi.org/10.3390/ijms25179532
Chicago/Turabian StyleSon, So-Eun, and Dong-Soon Im. 2024. "Activation of G Protein-Coupled Estrogen Receptor 1 (GPER) Attenuates Obesity-Induced Asthma by Switching M1 Macrophages to M2 Macrophages" International Journal of Molecular Sciences 25, no. 17: 9532. https://doi.org/10.3390/ijms25179532