
Glial Cells as Key Regulators in Neuroinflammatory Mechanisms Associated with Multiple Sclerosis



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Glial Cell Function and Communication
2. Oligodendrocytes and Neuroinflammation
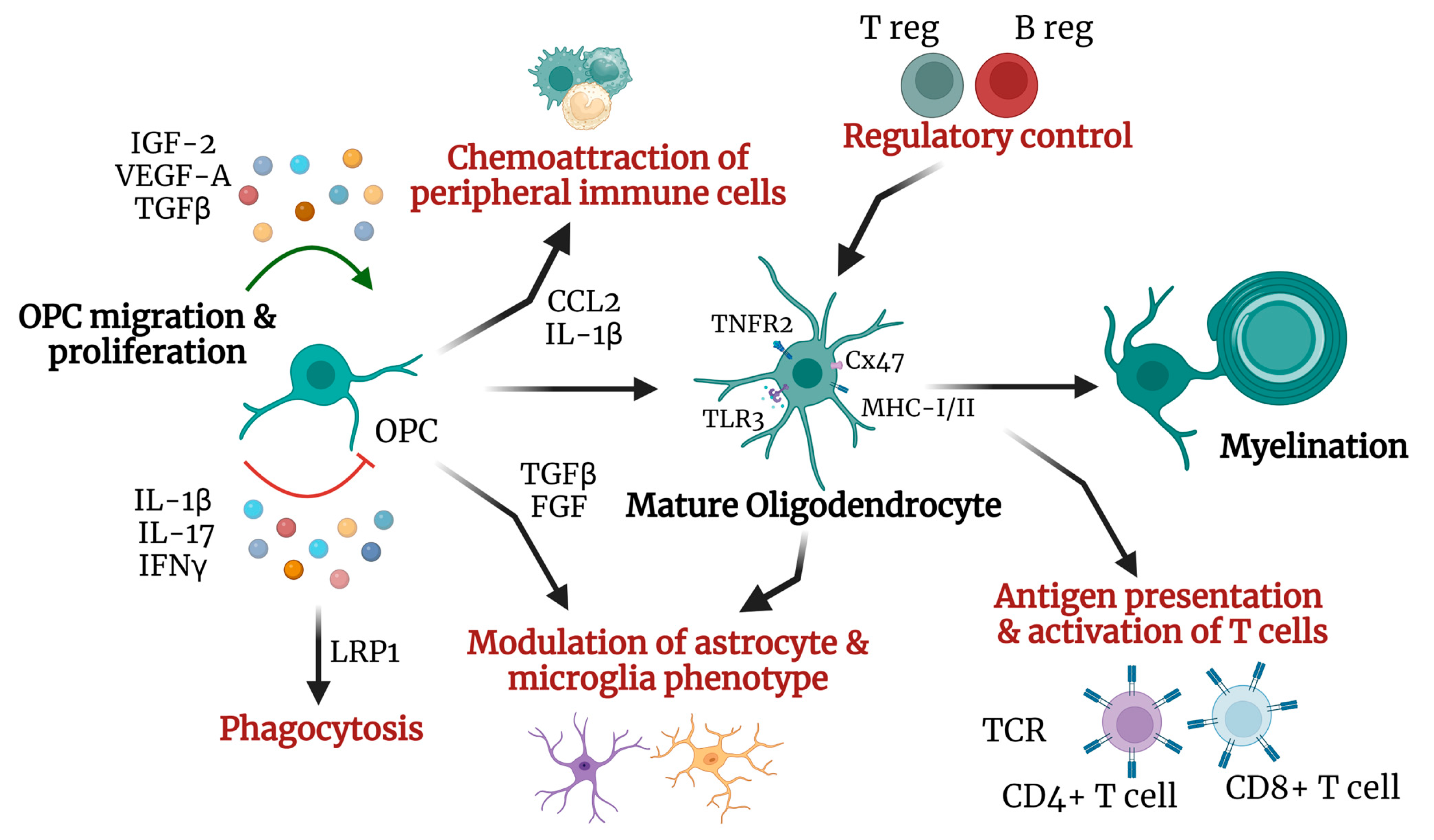
2.1. Oligodendrocytic Immunomodulatory Role
2.2. Oligodendrocyte and Remyelination
3. Astrocytes and Neuroinflammation
3.1. Astrocyte Dysregulation in MS
3.2. The Role of Astrocytes in Remyelination
4. Microglia and Neuroinflammation
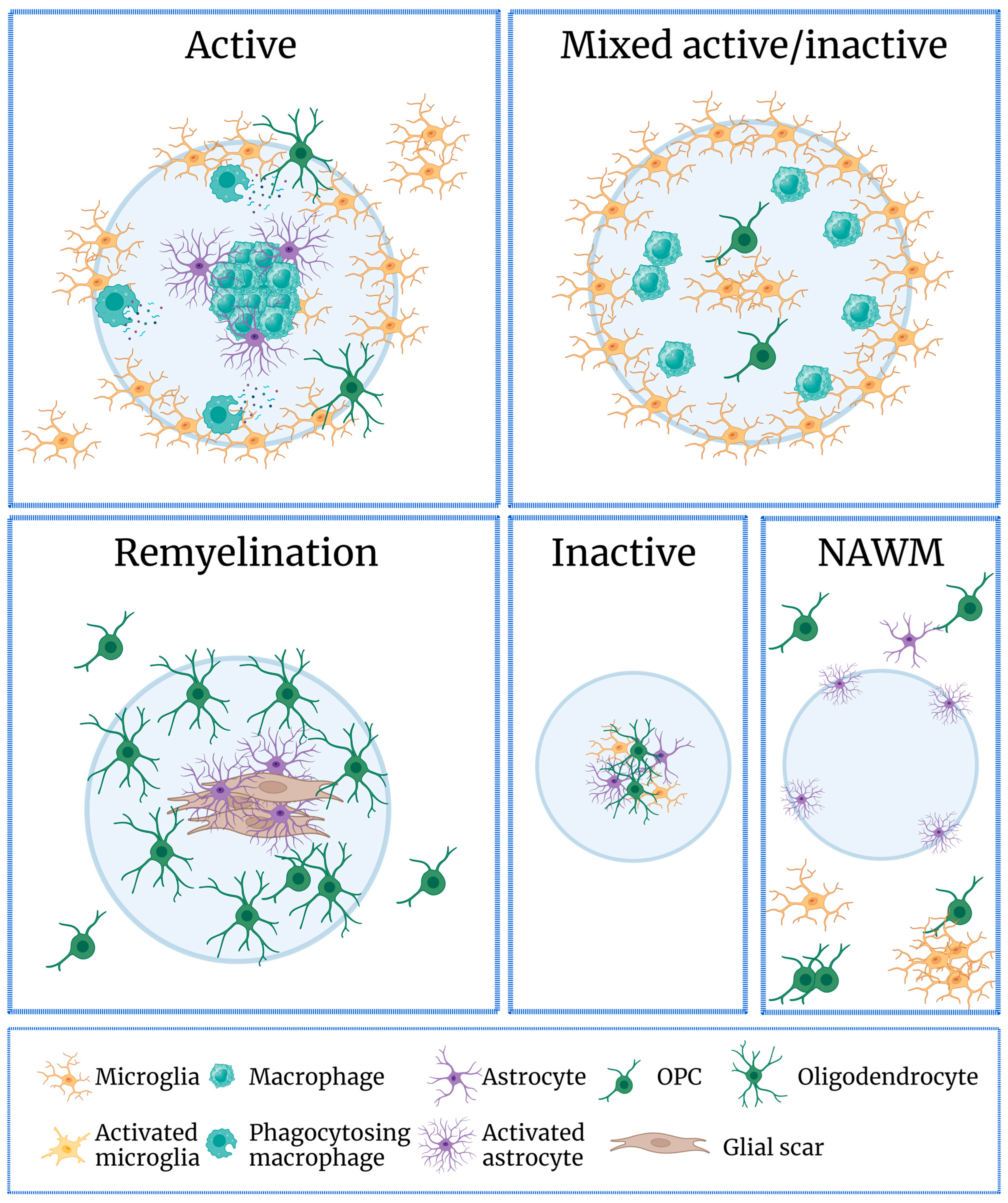
4.1. The Role of Microglia in MS
4.2. Microglia in Remyelination
5. Glial Cell Targeting in MS Treatment
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Composton, A.; Coles, A. Multiple Sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Rocca, M.A. MR imaging of multiple sclerosis. Radiology 2011, 259, 659–681. [Google Scholar] [CrossRef]
- Brosnan, C.F.; Battistini, L.; Raine, C.S.; Dickson, D.W.; Casadevall, A.; Lee, S.C. Reactive nitrogen intermediates in human neuropathology: An overview. Dev. Neurosci. 1994, 16, 152–161. [Google Scholar] [CrossRef]
- Brambilla, R.; Morton, P.D.; Ashbaugh, J.J.; Karmally, S.; Lambertsen, K.L.; Bethea, J.R. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia 2014, 62, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Szychowski, K.A.; Skora, B.; Wojtowicz, A.K. Elastin-Derived Peptides in the Central Nervous System: Friend or Foe. Cell. Mol. Neurobiol. 2022, 42, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, B.; Wei, X.; Tian, M.; Bao, X.; Zhang, Y.; Qi, H.; Zhang, Y.; Hu, M. Accumulation of extracellular elastin-derived peptides disturbed neuronal morphology and neuron-microglia crosstalk in aged brain. J. Neurochem. 2024, 168, 1460–1474. [Google Scholar] [CrossRef]
- Pisa, M.; Watson, J.L.; Spencer, J.I.; Niblett, G.; Mahjoub, Y.; Lockhart, A.; Yates, R.L.; Yee, S.A.; Hadley, G.; Ruiz, J.; et al. A role for vessel-associated extracellular matrix proteins in multiple sclerosis pathology. Brain Pathol. 2024, e13263. [Google Scholar] [CrossRef]
- Durelli, L.; Conti, L.; Clerico, M.; Boselli, D.; Contessa, G.; Ripellino, P.; Ferrero, B.; Eid, P.; Novelli, F. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann. Neurol. 2009, 65, 499–509. [Google Scholar] [CrossRef]
- Dalakas, M.C. B cells as therapeutic targets in autoimmune neurological disorders. Nat. Clin. Pract. Neurol. 2008, 4, 557–567. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130 Pt 4, 1089–1104. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef] [PubMed]
- Trobisch, T.; Zulji, A.; Stevens, N.A.; Schwarz, S.; Wischnewski, S.; Ozturk, M.; Perales-Paton, J.; Haeussler, M.; Saez-Rodriguez, J.; Velmeshev, D.; et al. Cross-regional homeostatic and reactive glial signatures in multiple sclerosis. Acta Neuropathol. 2022, 144, 987–1003. [Google Scholar] [CrossRef]
- Sen, M.K.; Mahns, D.A.; Coorssen, J.R.; Shortland, P.J. The roles of microglia and astrocytes in phagocytosis and myelination: Insights from the cuprizone model of multiple sclerosis. Glia 2022, 70, 1215–1250. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Lee, S. Microglia subtypes in acute, subacute, and chronic multiple sclerosis. J. Neuropathol. Exp. Neurol. 2023, 82, 674–694. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Ramanan, S.; Mubarak, M.; Housley, W.; Lee, S.; Sahinkaya, F.R.; Vortmeyer, A.; Raine, C.S.; Pitt, D. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain 2017, 140, 399–413. [Google Scholar] [CrossRef]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Healy, L.M.; Stratton, J.A.; Kuhlmann, T.; Antel, J. The role of glial cells in multiple sclerosis disease progression. Nat. Rev. Neurol. 2022, 18, 237–248. [Google Scholar] [CrossRef]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef]
- Sun, R.; Jiang, H. Border-associated macrophages in the central nervous system. J. Neuroinflammation 2024, 21, 67. [Google Scholar] [CrossRef]
- Chu, T.; Shields, L.B.E.; Zeng, W.; Zhang, Y.P.; Wang, Y.; Barnes, G.N.; Shields, C.B.; Cai, J. Dynamic glial response and crosstalk in demyelination-remyelination and neurodegeneration processes. Neural Regen. Res. 2021, 16, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, N.; Coles, A. Promoting remyelination in multiple sclerosis. J. Neurol. 2021, 268, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef]
- Franklin, R.J.; Gallo, V. The translational biology of remyelination: Past, present, and future. Glia 2014, 62, 1905–1915. [Google Scholar] [CrossRef]
- Maciak, K.; Dziedzic, A.; Saluk, J. Remyelination in multiple sclerosis from the miRNA perspective. Front. Mol. Neurosci. 2023, 16, 1199313. [Google Scholar] [CrossRef]
- Chari, D.M. Remyelination in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [CrossRef]
- Parrilla, G.E.; Gupta, V.; Wall, R.V.; Salkar, A.; Basavarajappa, D.; Mirzaei, M.; Chitranshi, N.; Graham, S.L.; You, Y. The role of myelin in neurodegeneration: Implications for drug targets and neuroprotection strategies. Rev. Neurosci. 2024, 35, 271–292. [Google Scholar] [CrossRef]
- Medzhitov, R. The spectrum of inflammatory responses. Science 2021, 374, 1070–1075. [Google Scholar] [CrossRef]
- Magni, G.; Riboldi, B.; Ceruti, S. Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies. Cells 2024, 13, 606. [Google Scholar] [CrossRef]
- Ahmad, S.; Srivastava, R.K.; Singh, P.; Naik, U.P.; Srivastava, A.K. Role of Extracellular Vesicles in Glia-Neuron Intercellular Communication. Front. Mol. Neurosci. 2022, 15, 844194. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef]
- Howell, O.W.; Rundle, J.L.; Garg, A.; Komada, M.; Brophy, P.J.; Reynolds, R. Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 1017–1033. [Google Scholar] [CrossRef]
- Fields, R.D.; Stevens-Graham, B. New insights into neuron-glia communication. Science 2002, 298, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Molina-Gonzalez, I.; Holloway, R.K.; Jiwaji, Z.; Dando, O.; Kent, S.A.; Emelianova, K.; Lloyd, A.F.; Forbes, L.H.; Mahmood, A.; Skripuletz, T.; et al. Astrocyte-oligodendrocyte interaction regulates central nervous system regeneration. Nat. Commun. 2023, 14, 3372. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 01416. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Kumar Jha, M.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neurosci. 2018. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef]
- Lombardi, M.; Parolisi, R.; Scaroni, F.; Bonfanti, E.; Gualerzi, A.; Gabrielli, M.; Kerlero de Rosbo, N.; Uccelli, A.; Giussani, P.; Viani, P.; et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: Astrocyte involvement in remyelination failure. Acta Neuropathol. 2019, 138, 987–1012. [Google Scholar] [CrossRef]
- Kleopa, K.A.; Orthmann-Murphy, J.; Sargiannidou, I. Gap Junction Disorders of Myelinating Cells. Rev. Neurosci. 2010, 21, 397–419. [Google Scholar] [CrossRef]
- Kleopa, K.A.; Orthmann, J.L.; Enriquez, A.; Paul, D.L.; Scherer, S.S. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia 2004, 47, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia 2003, 44, 205–218. [Google Scholar] [CrossRef]
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins Are Critical for Normal Myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, B.; Wellershaus, K.; Wallraff, A.; Seifert, G.; Degen, J.; Euwens, C.; Fuss, B.; Bussow, H.; Schilling, K.; Steinjauser, C.; et al. Connexin 47 (Cx47)-Deficient Mice with Enhanced Green Fluorescent Protein Reporter Gene Reveal Predominant Oligodendrocytic Expression of Cx47 and Display Vacuolized Myelin in the CNS. J. Neurosci. 2003, 23, 4549–4559. [Google Scholar] [CrossRef]
- Basu, R.; Das Sarma, J. Connexin 43/47 channels are important for astrocyte/oligodendrocyte cross-talk in myelination and demyelination. J. Biosci. 2018, 43, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Kleopa, K.A.; Sargiannidou, I.; Markoullis, K. Connexin pathology in chronic multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Neuroimmunol. 2013, 4, 45–58. [Google Scholar] [CrossRef]
- Markoullis, K.; Sargiannidou, I.; Schiza, N.; Hadjisavvas, A.; Roncaroli, F.; Reynolds, R.; Kleopa, K.A. Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol. 2012, 123, 873–886. [Google Scholar] [CrossRef]
- Markoullis, K.; Sargiannidou, I.; Schiza, N.; Roncaroli, F.; Reynolds, R.; Kleopa, K.A. Oligodendrocyte Gap Junction Loss and Disconnection from Reactive Astrocytes in Multiple Sclerosis Gray Matter. J. Neuropathol. Exp. Neurol. 2014, 73, 865–879. [Google Scholar]
- Markoullis, K.; Sargiannidou, I.; Gardner, C.; Hadjisavvas, A.; Reynolds, R.; Kleopa, K.A. Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 2012, 60, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Papaneophytou, C.P.; Georgiou, E.; Karaiskos, C.; Sargiannidou, I.; Markoullis, K.; Freidin, M.M.; Abrams, C.K.; Kleopa, K.A. Regulatory role of oligodendrocyte gap junctions in inflammatory demyelination. Glia 2018, 66, 2589–2603. [Google Scholar] [CrossRef]
- Stavropoulos, F.; Georgiou, E.; Sargiannidou, I.; Kleopa, K.A. Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE. Pharmaceuticals 2021, 14, 621. [Google Scholar] [CrossRef]
- Li, T.; Niu, J.; Yu, G.; Ezan, P.; Yi, C.; Wang, X.; Koulakoff, A.; Gao, X.; Chen, X.; Saez, J.C.; et al. Connexin 43 deletion in astrocytes promotes CNS remyelination by modulating local inflammation. Glia 2020, 68, 1201–1212. [Google Scholar] [CrossRef]
- Zhao, Y.; Yamasaki, R.; Yamaguchi, H.; Nagata, S.; Une, H.; Cui, Y.; Masaki, K.; Nakamuta, Y.; Ionuma, K.; Watanabe, M.; et al. Oligodendroglial connexin 47 regulates neuroinflammation upon autoimmune demyelination in a novel mouse model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 2160–2169. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Di Pietro, L.; Caraci, F. Gap Junctions and Connexins in Microglia-Related Oxidative Stress and Neuroinflammation: Perspectives for Drug Discovery. Biomolecules 2023, 13, 505. [Google Scholar] [CrossRef]
- Juszczak, G.R.; Swiergiel, A.H. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: Animal and human studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 181–198. [Google Scholar] [CrossRef]
- Takase, E.O.; Yamasaki, R.; Nagata, S.; Watanabe, M.; Masaki, K.; Yamaguchi, H.; Kira, J.I.; Takeuchi, H.; Isobe, N. Astroglial connexin 43 is a novel therapeutic target for chronic multiple sclerosis model. Sci. Rep. 2024, 14, 10877. [Google Scholar] [CrossRef]
- Kister, A.; Kister, I. Overview of myelin, major myelin lipids, and myelin-associated proteins. Front. Chem. 2023, 10, 1041961. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, H.; Guan, Y. Glia Connect Inflammation and Neurodegeneration in Multiple Sclerosis. Neurosci. Bull. 2023, 39, 466–478. [Google Scholar] [CrossRef]
- Fruhbeis, C.; Kuo-Elsner, W.P.; Muller, C.; Barth, K.; Peris, L.; Tenzer, S.; Mobius, W.; Werner, H.B.; Nave, K.A.; Frohlich, D.; et al. Oligodendrocytes support axonal transport and maintenance via exosome secretion. PLoS Biol. 2020, 18, e3000621. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, E.; Bosch-Queralt, M.; Edgar, J.M.; Lehning, M.; Strauss, J.; Fleischer, N.; Kungl, T.; Wieghofer, P.; Berghoff, S.A.; Reinert, T.; et al. Myelin insulation as a risk factor for axonal degeneration in autoimmune demyelinating disease. Nat. Neurosci. 2023, 26, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.R.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Niu, J.; Tsai, H.H.; Hoi, K.K.; Huang, N.; Yu, G.; Kim, K.; Baranzini, S.E.; Xiao, L.; Chan, J.R.; Fancy, S.P.J. Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat. Neurosci. 2019, 22, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 2016, 351, 379–384. [Google Scholar] [CrossRef]
- Falcao, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jakel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; Ffrench-Constant, C.; et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef]
- Jakel, S.; Agirre, E.; Mendanha Falcao, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef]
- Valihrach, L.; Matusova, Z.; Zucha, D.; Klassen, R.; Benesova, S.; Abaffy, P.; Kubista, M.; Anderova, M. Recent advances in deciphering oligodendrocyte heterogeneity with single-cell transcriptomics. Front. Cell. Neurosci. 2022, 16, 1025012. [Google Scholar] [CrossRef]
- Chapman, T.W.; Kamen, Y.; Piedra, E.T.; Hill, R.A. Oligodendrocyte Maturation Alters the Cell Death Mechanisms That Cause Demyelination. J. Neurosci. Off. J. Soc. Neurosci. 2024, 44, e1794232024. [Google Scholar] [CrossRef]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef]
- Zeis, T.; Enz, L.; Schaeren-Wiemers, N. The immunomodulatory oligodendrocyte. Brain Res. 2016, 1641, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.B.; Takada, N.; Latimer, A.J.; Shin, J.; Carney, T.J.; Kelsh, R.N.; Appel, B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci. 2006, 9, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.M.; Hage, Z.; Tsirka, S.E. Beyond Myelination: Possible Roles of the Immune Proteasome in Oligodendroglial Homeostasis and Dysfunction. Front. Neurosci. 2022, 16, 867357. [Google Scholar] [CrossRef]
- Boccazzi, M.; Raffaele, S.; Fumagalli, M. Not only myelination: The immuneinflammatory functions of oligodendrocytes. Neural Regen. Res. 2022, 17, 2661–2663. [Google Scholar]
- Nguyen, D.; Stangel, M. Expression of the chemokine receptors CXCR1 and CXCR2 in rat oligodendroglial cells. Dev. Brain Res. 2001, 128, 77–81. [Google Scholar] [CrossRef]
- Omari, K.M.; John, G.R.; Sealfon, S.C.; Raine, C.S. CXC chemokine receptors on human oligodendrocytes: Implications for multiple sclerosis. Brain 2005, 128, 1003–1015. [Google Scholar] [CrossRef]
- Tsai, H.; Frost, E.; To, V.; Robinson, S.; Ffrench-Constant, C.; Geertman, R.; Ransohoff, R.M.; Miller, R.H. The Chemokine Receptor CXCR2 Controls Positioning of Oligodendrocyte Precursors in Developing Spinal Cord by Arresting Their Migration. Cell 2002, 110, 373–383. [Google Scholar] [CrossRef]
- Hosking, M.P.; Tirotta, E.; Ransohoff, R.M.; Lane, T.E. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS ONE 2010, 5, e11340. [Google Scholar] [CrossRef]
- Liu, L.; Belkadi, A.; Darnall, L.; Hu, T.; Drescher, C.; Cotleur, A.C.; Padovani-Claudio, D.; He, T.; Choi, K.; Lane, T.E.; et al. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: Relevance to multiple sclerosis. Nat. Neurosci. 2010, 13, 319–326. [Google Scholar] [CrossRef]
- Patel, J.R.; McCandless, E.E.; Dorsey, D.; Klein, R.S. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc. Natl. Acad. Sci. USA 2010, 107, 11062–11067. [Google Scholar] [CrossRef]
- Tian, Y.; Yin, H.; Deng, X.; Tang, B.; Ren, X.; Jiang, T. CXCL12 induces migration of oligodendrocyte precursor cells through the CXCR4-activated MEK/ERK and PI3K/AKT pathways. Mol. Med. Rep. 2018, 18, 4374–4380. [Google Scholar] [CrossRef]
- Suo, N.; Guo, Y.E.; He, B.; Gu, H.; Xie, X. Inhibition of MAPK/ERK pathway promotes oligodendrocytes generation and recovery of demyelinating diseases. Glia 2019, 67, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.; et al. Demyelination Causes Adult CNS Progenitors to Revert to an Immature State and Express Immune Cues That Support Their Migration. J. Neurosci. 2015, 34, 4–20. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef]
- Zveik, O.; Rechtman, A.; Brill, L.; Vaknin-Dembinsky, A. Anti- and pro-inflammatory milieu differentially regulate differentiation and immune functions of oligodendrocyte progenitor cells. Immunology 2024, 171, 618–633. [Google Scholar] [CrossRef]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 3887. [Google Scholar] [CrossRef]
- Furlan, R.; Brambilla, E.; Ruffini, F.; Poliani, P.L.; Bergami, A.; Marconi, P.C.; Franciotta, D.M.; Penna, G.; Comi, G.; Adorini, L.; et al. Intrathecal delivery of IFN-gamma protects C57BL/6 mice from chronic-progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system-infiltrating lymphocytes. J. Immunol. 2001, 167, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alvarado, M.N.; Aprato, J.; Baumeister, M.; Lippert, M.; Ekici, A.B.; Kirchner, P.; Welz, T.; Hoffmann, A.; Winkler, J.; Wegner, M.; et al. Oligodendrocytes regulate the adhesion molecule ICAM-1 in neuroinflammation. Glia 2022, 70, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Vautier, F.; Belachew, S.; Chittajallu, R.; Gallo, V. Shaker-type potassium channel subunits differentially control oligodendrocyte progenitor proliferation. Glia 2004, 48, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alvarado, M.N.; Rotger, C.; Berger, L.; London, B.; Haase, S.; Kuhbandner, K.; Lee, D.H.; Linker, R.A. Functional role of endogenous Kv1.4 in experimental demyelination. J. Neuroimmunol. 2020, 343, 577227. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131 Pt 7, 1749–1758. [Google Scholar] [CrossRef]
- Furusho, M.; Roulois, A.J.; Franklin, R.J.; Bansal, R. Fibroblast growth factor signaling in oligodendrocyte-lineage cells facilitates recovery of chronically demyelinated lesions but is redundant in acute lesions. Glia 2015, 63, 1714–1728. [Google Scholar] [CrossRef]
- Madsen, P.M.; Desu, H.L.; de Rivero Vaccari, J.P.; Florimon, Y.; Ellman, D.G.; Keane, R.W.; Clausen, B.H.; Lambertsen, K.L.; Brambilla, R. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav. Immun. 2020, 84, 132–146. [Google Scholar] [CrossRef]
- Cudrici, C.; Niculescu, T.; Niculescu, F.; Shin, M.L.; Rus, H. Oligodendrocyte cell death in pathogenesis of multiple sclerosis: Protection of oligodendrocytes from apoptosis by complement. J. Rehabil. Res. Dev. 2006, 43, 123–132. [Google Scholar] [CrossRef]
- Ingram, G.; Loveless, S.; Howell, O.W.; Hakobyan, S.; Dancey, B.; Harris, C.L.; Robertson, N.P.; Neal, J.W.; Morgan, B.P. Complement activation in multiple sclerosis plaques: An immunohistochemical analysis. Acta Neuropathol. Commun. 2014, 2, 1–15. [Google Scholar] [CrossRef]
- Liu, Y.; Given, K.S.; Harlow, D.E.; Matschulat, A.M.; Macklin, W.B.; Bennett, J.L.; Owens, G.P. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol. Commun. 2017, 5, 25. [Google Scholar] [CrossRef]
- Soane, L.; Cho, H.J.; Niculescu, F.; Rus, H.; Shin, M.L. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J. Immunol. 2001, 167, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Cudrici, C.; Niculescu, F.; Jensen, T.; Zafranskaia, E.; Fosbrink, M.; Rus, V.; Shin, M.L.; Rus, H. C5b-9 terminal complex protects oligodendrocytes from apoptotic cell death by inhibiting caspase-8 processing and up-regulating FLIP. J. Immunol. 2006, 176, 3173–3180. [Google Scholar] [CrossRef]
- Ghorbani, S.; Jelinek, E.; Jain, R.; Buehner, B.; Li, C.; Lozinski, B.M.; Sarkar, S.; Kaushik, D.K.; Dong, Y.; Wight, T.N.; et al. Versican promotes T helper 17 cytotoxic inflammation and impedes oligodendrocyte precursor cell remyelination. Nat. Commun. 2022, 13, 2445. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, C.; Wasser, B.; Jamann, H.; Loffel, J.T.; Cui, Q.L.; Tastet, O.; Schillner, M.; Luchtman, D.; Birkenstock, J.; Stroh, A.; et al. Pro-inflammatory T helper 17 directly harms oligodendrocytes in neuroinflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2025813118. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Blakemore, W.F.; McDonald, W.I. Central remyelination restores secure conduction. Nature 1979, 280, 395–396. [Google Scholar] [CrossRef]
- Mallucci, G.; Peruzzotti-Jametti, L.; Bernstock, J.D.; Pluchino, S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog. Neurobiol. 2015, 127–128, 1–22. [Google Scholar] [CrossRef]
- Hess, K.; Starost, L.; Kieran, N.W.; Thomas, C.; Vincenten, M.C.J.; Antel, J.; Martino, G.; Huitinga, I.; Healy, L.; Kuhlmann, T. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol. 2020, 140, 359–375. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Bruck, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Moll, N.M.; Hong, E.; Fauveau, M.; Naruse, M.; Kerninon, C.; Tepavcevic, V.; Klopstein, A.; Seilhean, D.; Chew, L.J.; Gallo, V.; et al. SOX17 is expressed in regenerating oligodendrocytes in experimental models of demyelination and in multiple sclerosis. Glia 2013, 61, 1659–1672. [Google Scholar] [CrossRef]
- Neely, S.A.; Williamson, J.M.; Klingseisen, A.; Zoupi, L.; Early, J.J.; Williams, A.; Lyons, D.A. New oligodendrocytes exhibit more abundant and accurate myelin regeneration than those that survive demyelination. Nat. Neurosci. 2022, 25, 415–420. [Google Scholar] [CrossRef]
- Macchi, M.; Magalon, K.; Zimmer, C.; Peeva, E.; El Waly, B.; Brousse, B.; Jaekel, S.; Grobe, K.; Kiefer, F.; Williams, A.; et al. Mature oligodendrocytes bordering lesions limit demyelination and favor myelin repair via heparan sulfate production. eLife 2020, 9, 51735. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Kirby, L.; Castelo-Branco, G. Crossing boundaries: Interplay between the immune system and oligodendrocyte lineage cells. Semin. Cell Dev. Biol. 2021, 116, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Simons, M. CNS remyelination and inflammation: From basic mechanisms to therapeutic opportunities. Neuron 2022, 110, 3549–3565. [Google Scholar] [CrossRef]
- Marangon, D.; Castro, E.S.J.H.; Cerrato, V.; Boda, E.; Lecca, D. Oligodendrocyte Progenitors in Glial Scar: A Bet on Remyelination. Cells 2024, 13, 1024. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, K.; Frost, E.E.; Pillai, P.P. Role of PDGF-A-Activated ERK Signaling Mediated FAK-Paxillin Interaction in Oligodendrocyte Progenitor Cell Migration. J. Mol. Neurosci. 2019, 67, 564–573. [Google Scholar] [CrossRef]
- Ghorbani, S.; Yong, V.W. The extracellular matrix as modifier of neuroinflammation and remyelination in multiple sclerosis. Brain 2021, 144, 1958–1973. [Google Scholar] [CrossRef]
- Fancy, S.P.; Baranzini, S.E.; Zhao, C.; Yuk, D.I.; Irvine, K.A.; Kaing, S.; Sanai, N.; Franklin, R.J.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes. Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef]
- Wang, J.; Saraswat, D.; Sinha, A.K.; Polanco, J.; Dietz, K.; O’Bara, M.A.; Pol, S.U.; Shayya, H.J.; Sim, F.J. Paired Related Homeobox Protein 1 Regulates Quiescence in Human Oligodendrocyte Progenitors. Cell Rep. 2018, 25, 3435–3450.e6. [Google Scholar] [CrossRef]
- Tepavcevic, V.; Blakemore, W.F. Glial grafting for demyelinating disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1775–1795. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Bieri, G.; Gontier, G.; Muller, S.; Smith, L.K.; Snethlage, C.E.; White, C.W., 3rd; Maybury-Lewis, S.Y.; Villeda, S.A. MHC class I H2-Kb negatively regulates neural progenitor cell proliferation by inhibiting FGFR signaling. PLoS Biol. 2021, 19, e3001311. [Google Scholar] [CrossRef]
- Kaya, T.; Mattugini, N.; Liu, L.; Ji, H.; Cantuti-Castelvetri, L.; Wu, J.; Schifferer, M.; Groh, J.; Martini, R.; Besson-Girard, S.; et al. CD8+ T cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci. 2022, 25, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Tepavcevic, V.; Lubetzki, C. Oligodendrocyte progenitor cell recruitment and remyelination in multiple sclerosis: The more, the merrier? Brain 2022, 145, 4178–4192. [Google Scholar] [CrossRef] [PubMed]
- Baxi, E.G.; DeBruin, J.; Tosi, D.M.; Grishkan, I.V.; Smith, M.D.; Kirby, L.A.; Strasburger, H.J.; Fairchild, A.N.; Calabresi, P.A.; Gocke, A.R. Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 8626–8639. [Google Scholar] [CrossRef]
- Choi, E.H.; Xu, Y.; Medynets, M.; Monaco, M.C.G.; Major, E.O.; Nath, A.; Wang, T. Activated T cells induce proliferation of oligodendrocyte progenitor cells via release of vascular endothelial cell growth factor-A. Glia 2018, 66, 2503–2513. [Google Scholar] [CrossRef]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef]
- Li, W.; Berlinicke, C.; Huang, Y.; Giera, S.; McGrath, A.G.; Fang, W.; Chen, C.; Takaesu, F.; Chang, X.; Duan, Y.; et al. High-throughput screening for myelination promoting compounds using human stem cell-derived oligodendrocyte progenitor cells. iScience 2023, 26, 106156. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Robel, S.; Mishra, A. Astrocyte Endfeet in Brain Function and Pathology: Open Questions. Annu. Rev. Neurosci. 2023, 46, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 28. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Deneen, B. Astrocyte development: A Guide for the Perplexed. Glia 2015, 63, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Cajal, S.R.Y.; Azoulay, L. Histologie du Système Nerveux de L’homme et des Vertébrés; Maloine: Paris, France, 1909. [Google Scholar]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Das Neves, S.P.; Sousa, J.C.; Magalhaes, R.; Gao, F.; Coppola, G.; Meriaux, S.; Boumezbeur, F.; Sousa, N.; Cerqueira, J.J.; Marques, F. Astrocytes Undergo Metabolic Reprogramming in the Multiple Sclerosis Animal Model. Cells 2023, 12, 2484. [Google Scholar] [CrossRef]
- Brambilla, R.; Bracchi-Ricard, V.; Hu, W.H.; Frydel, B.; Bramwell, A.; Karmally, S.; Green, E.J.; Bethea, J.R. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 2005, 202, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Ceyzeriat, K.; Abjean, L.; Carrillo-de Sauvage, M.A.; Ben Haim, L.; Escartin, C. The complex STATes of astrocyte reactivity: How are they controlled by the JAK-STAT3 pathway? Neuroscience 2016, 330, 205–218. [Google Scholar] [CrossRef]
- Choudhury, R.G.; Ryou, M.G.; Poteet, E.; Wen, Y.; He, R.; Sun, F.; Yuan, F.; Jin, K.; Yang, S.H. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res. 2014, 1551, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Blank, T.; Prinz, M. NF-kappaB signaling regulates myelination in the CNS. Front. Mol. Neurosci. 2014, 7, 47. [Google Scholar] [CrossRef]
- Brambilla, R.; Hurtado, A.; Persaud, T.; Esham, K.; Pearse, D.D.; Oudega, M.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J. Neurochem. 2009, 110, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Liu, L.; Spangler, R.; Spear, C.; Wang, C.; Gulen, M.F.; Veenstra, M.; Ouyang, W.; Ransohoff, R.M.; Li, X. IL-17-induced Act1-mediated signaling is critical for cuprizone-induced demyelination. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 8284–8292. [Google Scholar] [CrossRef]
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; DiCorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat. Neurosci. 2013, 16, 1401–1408. [Google Scholar] [CrossRef]
- Guerrero-García, J.J. The role of astrocytes in multiple sclerosis pathogenesis. Neurol. Engl. Ed. 2020, 35, 400–408. [Google Scholar] [CrossRef]
- Boccazzi, M.; Van Steenwinckel, J.; Schang, A.L.; Faivre, V.; Le Charpentier, T.; Bokobza, C.; Csaba, Z.; Verderio, C.; Fumagalli, M.; Mani, S.; et al. The immune-inflammatory response of oligodendrocytes in a murine model of preterm white matter injury: The role of TLR3 activation. Cell Death Dis. 2021, 12, 166. [Google Scholar] [CrossRef]
- Gambuzza, M.; Licata, N.; Palella, E.; Celi, D.; Foti Cuzzola, V.; Italiano, D.; Marino, S.; Bramanti, P. Targeting Toll-like receptors: Emerging therapeutics for multiple sclerosis management. J. Neuroimmunol. 2011, 239, 1–12. [Google Scholar] [CrossRef]
- Bsibsi, M.; Persoon-Deen, C.; Verwer, R.W.; Meeuwsen, S.; Ravid, R.; Van Noort, J.M. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006, 53, 688–695. [Google Scholar] [CrossRef]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef] [PubMed]
- Kramann, N.; Menken, L.; Pfortner, R.; Schmid, S.N.; Stadelmann, C.; Wegner, C.; Bruck, W. Glial fibrillary acidic protein expression alters astrocytic chemokine release and protects mice from cuprizone-induced demyelination. Glia 2019, 67, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Schiro, G.; Sorbello, G.; Di Liegro, I. Involvement of Astrocytes in the Formation, Maintenance, and Function of the Blood-Brain Barrier. Cells 2024, 13, 150. [Google Scholar] [CrossRef]
- Sharma, R.; Fischer, M.T.; Bauer, J.; Felts, P.A.; Smith, K.J.; Misu, T.; Fujihara, K.; Bradl, M.; Lassmann, H. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010, 120, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Parratt, J.D.; Prineas, J.W. Neuromyelitis optica: A demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult. Scler. 2010, 16, 1156–1172. [Google Scholar] [CrossRef]
- Gimenez, M.A.; Sim, J.E.; Russell, J.H. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J. Neuroimmunol. 2004, 151, 116–125. [Google Scholar] [CrossRef]
- Popescu, B.; Guo, Y.; Jentoft, M.E.; Parisi, J.E.; Lennon, V.A.; Pittock, S.J.; Weinshenker, B.G.; Wingerchuk, D.M.; Giannini, C.; Metz, I.; et al. Diagnostic utility of aquaporin-4 in the analysis of active demyelinating lesions. Neurology 2015, 84, 148–158. [Google Scholar] [CrossRef]
- Miyamoto, K.; Nagaosa, N.; Motoyama, M.; Kataoka, K.; Kusunoki, S. Upregulation of water channel aquaporin-4 in experimental autoimmune encephalomyeritis. J. Neurol. Sci. 2009, 276, 103–107. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Verkman, A.S. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009, 10, 94. [Google Scholar] [CrossRef]
- Kim, R.Y.; Hoffman, A.S.; Itoh, N.; Ao, Y.; Spence, R.; Sofroniew, M.V.; Voskuhl, R.R. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 274, 53–61. [Google Scholar] [CrossRef]
- Kunkl, M.; Amormino, C.; Tedeschi, V.; Fiorillo, M.T.; Tuosto, L. Astrocytes and Inflammatory T Helper Cells: A Dangerous Liaison in Multiple Sclerosis. Front. Immunol. 2022, 13, 824411. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Damsker, J.M.; Hansen, A.M.; Caspi, R.R. Th1 and Th17 cells: Adversaries and collaborators. Ann. N. Y. Acad. Sci. 2010, 1183, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Kronisch, J.; Khorooshi, R.; Knier, B.; Toft-Hansen, H.; Gudi, V.; Floess, S.; Huehn, J.; Owens, T.; Korn, T.; et al. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflammation 2017, 14, 204. [Google Scholar] [CrossRef]
- McWilliams, I.L.; Rajbhandari, R.; Nozell, S.; Benveniste, E.; Harrington, L.E. STAT4 controls GM-CSF production by both Th1 and Th17 cells during EAE. J. Neuroinflammation 2015, 12, 128. [Google Scholar] [CrossRef]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I. Granulocyte-macrophage colony-stimulating factor as a mediator of autoimmunity in multiple sclerosis. J. Neuroimmunol. 2018, 323, 1–9. [Google Scholar] [CrossRef]
- Watanabe, M.; Masaki, K.; Yamasaki, R.; Kawanokuchi, J.; Takeuchi, H.; Matsushita, T.; Suzumura, A.; Kira, J.I. Th1 cells downregulate connexin 43 gap junctions in astrocytes via microglial activation. Sci. Rep. 2016, 6, 38387. [Google Scholar] [CrossRef]
- Senecal, V.; Deblois, G.; Beauseigle, D.; Schneider, R.; Brandenburg, J.; Newcombe, J.; Moore, C.S.; Prat, A.; Antel, J.; Arbour, N. Production of IL-27 in multiple sclerosis lesions by astrocytes and myeloid cells: Modulation of local immune responses. Glia 2016, 64, 553–569. [Google Scholar] [CrossRef]
- Lemaitre, F.; Farzam-Kia, N.; Carmena Moratalla, A.; Carpentier Solorio, Y.; Clenet, M.L.; Tastet, O.; Cleret-Buhot, A.; Guimond, J.V.; Haddad, E.; Duquette, P.; et al. IL-27 shapes the immune properties of human astrocytes and their impact on encountered human T lymphocytes. J. Neuroinflammation 2022, 19, 212. [Google Scholar] [CrossRef]
- Clarner, T.; Janssen, K.; Nellessen, L.; Stangel, M.; Skripuletz, T.; Krauspe, B.; Hess, F.M.; Denecke, B.; Beutner, C.; Linnartz-Gerlach, B.; et al. CXCL10 triggers early microglial activation in the cuprizone model. J. Immunol. 2015, 194, 3400–3413. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 10, 1147–1156. [Google Scholar] [CrossRef]
- Tenner, A.J.; Stevens, B.; Woodruff, T.M. New tricks for an ancient system: Physiological and pathological roles of complement in the CNS. Mol. Immunol. 2018, 102, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P.; Singhrao, S.K.; Neal, J.W.; Gotze, O.; Morgan, B.P. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am. J. Pathol. 1997, 150, 31–41. [Google Scholar]
- Crane, J.W.; Baiquni, G.P.; Sullivan, R.K.; Lee, J.D.; Sah, P.; Taylor, S.M.; Noakes, P.G.; Woodruff, T.M. The C5a anaphylatoxin receptor CD88 is expressed in presynaptic terminals of hippocampal mossy fibres. J. Neuroinflammation 2009, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Loveless, S.; Neal, J.W.; Howell, O.W.; Harding, K.E.; Sarkies, P.; Evans, R.; Bevan, R.J.; Hakobyan, S.; Harris, C.L.; Robertson, N.P.; et al. Tissue microarray methodology identifies complement pathway activation and dysregulation in progressive multiple sclerosis. Brain Pathol. 2018, 28, 507–520. [Google Scholar] [CrossRef]
- Olivero, G.; Taddeucci, A.; Vallarino, G.; Trebesova, H.; Roggeri, A.; Gagliani, M.C.; Cortese, K.; Grilli, M.; Pittaluga, A. Complement tunes glutamate release and supports synaptic impairments in an animal model of multiple sclerosis. Br. J. Pharmacol. 2024, 181, 1812–1828. [Google Scholar] [CrossRef]
- Schroder, L.J.; Mulenge, F.; Pavlou, A.; Skripuletz, T.; Stangel, M.; Gudi, V.; Kalinke, U. Dynamics of reactive astrocytes fosters tissue regeneration after cuprizone-induced demyelination. Glia 2023, 71, 2573–2590. [Google Scholar] [CrossRef]
- Rawji, K.S.; Gonzalez Martinez, G.A.; Sharma, A.; Franklin, R.J.M. The Role of Astrocytes in Remyelination. Trends Neurosci. 2020, 43, 596–607. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef]
- Hammond, T.R.; McEllin, B.; Morton, P.D.; Raymond, M.; Dupree, J.; Gallo, V. Endothelin-B Receptor Activation in Astrocytes Regulates the Rate of Oligodendrocyte Regeneration during Remyelination. Cell Rep. 2015, 13, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Watzlawik, J.O.; Warrington, A.E.; Rodriguez, M. PDGF is required for remyelination-promoting IgM stimulation of oligodendrocyte progenitor cell proliferation. PLoS ONE 2013, 8, e55149. [Google Scholar] [CrossRef] [PubMed]
- Thummler, K.; Rom, E.; Zeis, T.; Lindner, M.; Brunner, S.; Cole, J.J.; Arseni, D.; Mucklisch, S.; Edgar, J.M.; Schaeren-Wiemers, N.; et al. Polarizing receptor activation dissociates fibroblast growth factor 2 mediated inhibition of myelination from its neuroprotective potential. Acta Neuropathol. Commun. 2019, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Skripuletz, T.; Hackstette, D.; Bauer, K.; Gudi, V.; Pul, R.; Voss, E.; Berger, K.; Kipp, M.; Baumgartner, W.; Stangel, M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 2013, 136 Pt 1, 147–167. [Google Scholar] [CrossRef]
- Moore, C.S.; Abdullah, S.L.; Brown, A.; Arulpragasam, A.; Crocker, S.J. How factors secreted from astrocytes impact myelin repair. J. Neurosci. Res. 2011, 89, 13–21. [Google Scholar] [CrossRef]
- Cheng, N.; Xiong, Y.; Zhang, W.; Wu, X.; Sun, Z.; Zhang, L.; Wu, H.; Tang, Y.; Peng, Y. Astrocytes promote the proliferation of oligodendrocyte precursor cells through connexin 47-mediated LAMB2 secretion in exosomes. Mol. Biol. Rep. 2022, 49, 7263–7273. [Google Scholar] [CrossRef]
- Xu, D.; Liu, Z.; Wang, S.; Peng, Y.; Sun, X. Astrocytes regulate the expression of Sp1R3 on oligodendrocyte progenitor cells through Cx47 and promote their proliferation. Biochem. Biophys. Res. Commun. 2017, 490, 670–675. [Google Scholar] [CrossRef]
- Silva Oliveira Junior, M.; Reiche, L.; Daniele, E.; Kortebi, I.; Faiz, M.; Kury, P. Star power: Harnessing the reactive astrocyte response to promote remyelination in multiple sclerosis. Neural Regen. Res. 2024, 19, 578–582. [Google Scholar] [CrossRef]
- Yang, S.; Qin, C.; Hu, Z.W.; Zhou, L.Q.; Yu, H.H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.J.; Tian, D.S. Microglia reprogram metabolic profiles for phenotype and function changes in central nervous system. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef]
- McNamara, N.B.; Munro, D.A.D.; Bestard-Cuche, N.; Uyeda, A.; Bogie, J.F.J.; Hoffmann, A.; Holloway, R.K.; Molina-Gonzalez, I.; Askew, K.E.; Mitchell, S.; et al. Microglia regulate central nervous system myelin growth and integrity. Nature 2023, 613, 120–129. [Google Scholar] [CrossRef]
- Olcum, M.; Tastan, B.; Kiser, C.; Genc, S.; Genc, K. Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv. Protein Chem. Struct. Biol. 2020, 119, 247–308. [Google Scholar] [CrossRef] [PubMed]
- Andoh, M.; Koyama, R. Microglia regulate synaptic development and plasticity. Dev. Neurobiol. 2021, 81, 568–590. [Google Scholar] [CrossRef]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705–716. [Google Scholar] [CrossRef]
- Zhou, L.J.; Peng, J.; Xu, Y.N.; Zeng, W.J.; Zhang, J.; Wei, X.; Mai, C.L.; Lin, Z.J.; Liu, Y.; Murugan, M.; et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019, 27, 3844–3859.e6. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; Carrier, M.; Tremblay, M.E. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019, 2034, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, F.; Sun, M.; Wu, N.; Liu, B.; Yi, X.; Ge, R.; Fan, X. Microglia in the context of multiple sclerosis. Front. Neurol. 2023, 14, 1157287. [Google Scholar] [CrossRef]
- Hou, K.; Li, G.; Yu, J.; Xu, K.; Wu, W. Receptors, Channel Proteins, and Enzymes Involved in Microglia-mediated Neuroinflammation and Treatments by Targeting Microglia in Ischemic Stroke. Neuroscience 2021, 460, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef]
- Zia, S.; Rawji, K.S.; Michaels, N.J.; Burr, M.; Kerr, B.J.; Healy, L.M.; Plemel, J.R. Microglia Diversity in Health and Multiple Sclerosis. Front. Immunol. 2020, 11, 588021. [Google Scholar] [CrossRef]
- Van den Bosch, A.M.R.; van der Poel, M.; Fransen, N.L.; Vincenten, M.C.J.; Bobeldijk, A.M.; Jongejan, A.; Engelenburg, H.J.; Moerland, P.D.; Smolders, J.; Huitinga, I.; et al. Profiling of microglia nodules in multiple sclerosis reveals propensity for lesion formation. Nat. Commun. 2024, 15, 1667. [Google Scholar] [CrossRef] [PubMed]
- Marzan, D.E.; Brugger-Verdon, V.; West, B.L.; Liddelow, S.; Samanta, J.; Salzer, J.L. Activated microglia drive demyelination via CSF1R signaling. Glia 2021, 69, 1583–1604. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Willis, C.M.; Krzak, G.; Hamel, R.; Pirvan, L.; Ionescu, R.B.; Reisz, J.A.; Prag, H.A.; Garcia-Segura, M.E.; Wu, V.; et al. Mitochondrial complex I activity in microglia sustains neuroinflammation. Nature 2024, 628, 195–203. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Vogel, D.Y.; Vereyken, E.J.; Glim, J.E.; Heijnen, P.D.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflammation 2013, 10, 809. [Google Scholar] [CrossRef]
- O’Loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial Phenotypes and Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef]
- Nowacki, P.; Koziarska, D.; Masztalewicz, M. Microglia and astroglia proliferation within the normal appearing white matter in histologically active and inactive multiple sclerosis. Folia Neuropathol. 2019, 57, 249–257. [Google Scholar] [CrossRef]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Mado, H.; Adamczyk-Sowa, M.; Sowa, P. Role of Microglial Cells in the Pathophysiology MS: Synergistic or Antagonistic? Int. J. Mol. Sci. 2023, 24, 1861. [Google Scholar] [CrossRef]
- Seo, J.E.; Hasan, M.; Han, J.S.; Kang, M.J.; Jung, B.H.; Kwok, S.K.; Kim, H.Y.; Kwon, O.S. Experimental autoimmune encephalomyelitis and age-related correlations of NADPH oxidase, MMP-9, and cell adhesion molecules: The increased disease severity and blood-brain barrier permeability in middle-aged mice. J. Neuroimmunol. 2015, 287, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, M.S.; Joly-Amado, A.; Bickford, P.C.; Nash, K.R. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacol. Ther. 2022, 231, 107989. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Sanchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.E.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Wasser, B.; Luchtman, D.; Loffel, J.; Robohm, K.; Birkner, K.; Stroh, A.; Vogelaar, C.F.; Zipp, F.; Bittner, S. CNS-localized myeloid cells capture living invading T cells during neuroinflammation. J. Exp. Med. 2020, 217, e20190812. [Google Scholar] [CrossRef]
- Moser, T.; Akgun, K.; Proschmann, U.; Sellner, J.; Ziemssen, T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun. Rev. 2020, 19, 102647. [Google Scholar] [CrossRef]
- Van Langelaar, J.; van der Vuurst de Vries, R.M.; Janssen, M.; Wierenga-Wolf, A.F.; Spilt, I.M.; Siepman, T.A.; Dankers, W.; Verjans, G.; de Vries, H.E.; Lubberts, E.; et al. T helper 17.1 cells associate with multiple sclerosis disease activity: Perspectives for early intervention. Brain 2018, 141, 1334–1349. [Google Scholar] [CrossRef]
- Strachan-Whaley, M.; Rivest, S.; Yong, V.W. Interactions between microglia and T cells in multiple sclerosis pathobiology. J. Interferon Cytokine Res. 2014, 34, 615–622. [Google Scholar] [CrossRef]
- Dong, Y.; Yong, V.W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 704–717. [Google Scholar] [CrossRef]
- Kalafatakis, I.; Karagogeos, D. Oligodendrocytes and Microglia: Key Players in Myelin Development, Damage and Repair. Biomolecules 2021, 11, 1058. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Lohr, K.; Floess, S.; Zimmermann, J.; Ulrich, R.; Gudi, V.; Beineke, A.; Baumgartner, W.; Muller, M.; Huehn, J.; et al. Effector molecules released by Th1 but not Th17 cells drive an M1 response in microglia. Brain Behav. Immun. 2014, 37, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Frascolla, S.; Amormino, C.; Volpe, E.; Tuosto, L. T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells 2020, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Sloane, J.A.; Batt, C.; Ma, Y.; Harris, Z.M.; Trapp, B.; Vartanian, T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc. Natl. Acad. Sci. USA 2010, 107, 11555–11560. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Tran, D.Q.; Pesu, M.; Davidson, T.S.; Ramsey, H.; O’Shea, J.J.; Shevach, E.M. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J. Exp. Med. 2008, 205, 1975–1981. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Garcia-Rodriguez, C.; Villalobos, C.; Nunez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef]
- Miranda-Hernandez, S.; Baxter, A.G. Role of toll-like receptors in multiple sclerosis. Am. J. Clin. Exp. Immunol. 2013, 2, 75–93. [Google Scholar]
- Fornari Laurindo, L.; Aparecido Dias, J.; Cressoni Araujo, A.; Torres Pomini, K.; Machado Galhardi, C.; Rucco Penteado Detregiachi, C.; Santos de Argollo Haber, L.; Donizeti Roque, D.; Dib Bechara, M.; Vialogo Marques de Castro, M.; et al. Immunological dimensions of neuroinflammation and microglial activation: Exploring innovative immunomodulatory approaches to mitigate neuroinflammatory progression. Front. Immunol. 2023, 14, 1305933. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Lee, M.J.; Bing, S.J.; Choi, J.; Jang, M.; Lee, G.; Lee, H.; Chang, B.S.; Jee, Y.; Lee, S.J.; Cho, I.H. IKKbeta-mediated inflammatory myeloid cell activation exacerbates experimental autoimmune encephalomyelitis by potentiating Th1/Th17 cell activation and compromising blood brain barrier. Mol. Neurodegener. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.9. [Google Scholar] [CrossRef]
- Hagan, N.; Kane, J.L.; Grover, D.; Woodworth, L.; Madore, C.; Saleh, J.; Sancho, J.; Liu, J.; Li, Y.; Proto, J.; et al. CSF1R signaling is a regulator of pathogenesis in progressive MS. Cell Death Dis. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, C.; Fernandez-Zapata, C.; Schlickeiser, S.; Kunkel, D.; Schulz, A.R.; Mei, H.E.; Weidinger, C.; Giess, R.M.; Asseyer, S.; Siegmund, B.; et al. Multi-parameter immune profiling of peripheral blood mononuclear cells by multiplexed single-cell mass cytometry in patients with early multiple sclerosis. Sci. Rep. 2019, 9, 19471. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Serafini, B.; Adorini, L. Glia-T cell dialogue. J. Neuroimmunol. 2000, 107, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-Examining the Role of TNF in MS Pathogenesis and Therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef]
- Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. [Google Scholar] [CrossRef]
- Guadalupi, L.; Vanni, V.; Balletta, S.; Caioli, S.; De Vito, F.; Fresegna, D.; Sanna, K.; Nencini, M.; Donninelli, G.; Volpe, E.; et al. Interleukin-9 protects from microglia- and TNF-mediated synaptotoxicity in experimental multiple sclerosis. J. Neuroinflammation 2024, 21, 128. [Google Scholar] [CrossRef]
- Batoulis, H.; Recks, M.S.; Holland, F.O.; Thomalla, F.; Williams, R.O.; Kuerten, S. Blockade of tumour necrosis factor-alpha in experimental autoimmune encephalomyelitis reveals differential effects on the antigen-specific immune response and central nervous system histopathology. Clin. Exp. Immunol. 2014, 175, 41–48. [Google Scholar] [CrossRef]
- Deczkowska, A.; Baruch, K.; Schwartz, M. Type I/II Interferon Balance in the Regulation of Brain Physiology and Pathology. Trends Immunol. 2016, 37, 181–192. [Google Scholar] [CrossRef]
- Takeuchi, H.; Wang, J.; Kawanokuchi, J.; Mitsuma, N.; Mizuno, T.; Suzumura, A. Interferon-gamma induces microglial-activation-induced cell death: A hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiol. Dis. 2006, 22, 33–39. [Google Scholar] [CrossRef]
- Papageorgiou, I.E.; Lewen, A.; Galow, L.V.; Cesetti, T.; Scheffel, J.; Regen, T.; Hanisch, U.K.; Kann, O. TLR4-activated microglia require IFN-gamma to induce severe neuronal dysfunction and death in situ. Proc. Natl. Acad. Sci. USA 2016, 113, 212–217. [Google Scholar] [CrossRef]
- Tichauer, J.E.; Arellano, G.; Acuna, E.; Gonzalez, L.F.; Kannaiyan, N.R.; Murgas, P.; Panadero-Medianero, C.; Ibanez-Vega, J.; Burgos, P.I.; Loda, E.; et al. Interferon-gamma ameliorates experimental autoimmune encephalomyelitis by inducing homeostatic adaptation of microglia. Front. Immunol. 2023, 14, 1191838. [Google Scholar] [CrossRef] [PubMed]
- Wies Mancini, V.S.B.; Mattera, V.S.; Pasquini, J.M.; Pasquini, L.A.; Correale, J.D. Microglia-derived extracellular vesicles in homeostasis and demyelination/remyelination processes. J. Neurochem. 2024, 168, 3–25. [Google Scholar] [CrossRef]
- Mahmood, A.; Miron, V.E. Microglia as therapeutic targets for central nervous system remyelination. Curr. Opin. Pharmacol. 2022, 63, 102188. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Zarini, D.; Pasbakhsh, P.; Mojaverrostami, S.; Amirizadeh, S.; Hashemi, M.; Shabani, M.; Noshadian, M.; Kashani, I.R. Microglia/macrophage polarization regulates spontaneous remyelination in intermittent cuprizone model of demyelination. Biochem. Biophys. Rep. 2024, 37, 101630. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef]
- Plemel, J.R.; Manesh, S.B.; Sparling, J.S.; Tetzlaff, W. Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression. Glia 2013, 61, 1471–1487. [Google Scholar] [CrossRef]
- Kotter, M.R.; Setzu, A.; Sim, F.J.; Van Rooijen, N.; Franklin, R.J. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 2001, 35, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, R.J. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Duking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2021, 24, 47–60. [Google Scholar] [CrossRef]
- Laflamme, N.; Cisbani, G.; Prefontaine, P.; Srour, Y.; Bernier, J.; St-Pierre, M.K.; Tremblay, M.E.; Rivest, S. mCSF-Induced Microglial Activation Prevents Myelin Loss and Promotes Its Repair in a Mouse Model of Multiple Sclerosis. Front. Cell. Neurosci. 2018, 12, 178. [Google Scholar] [CrossRef]
- Church, J.S.; Kigerl, K.A.; Lerch, J.K.; Popovich, P.G.; McTigue, D.M. TLR4 Deficiency Impairs Oligodendrocyte Formation in the Injured Spinal Cord. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 6352–6364. [Google Scholar] [CrossRef] [PubMed]
- Wasko, N.J.; Kulak, M.H.; Paul, D.; Nicaise, A.M.; Yeung, S.T.; Nichols, F.C.; Khanna, K.M.; Crocker, S.; Pachter, J.S.; Clark, R.B. Systemic TLR2 tolerance enhances central nervous system remyelination. J. Neuroinflammation 2019, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Piaton, G.; Aigrot, M.S.; Belhadi, A.; Theaudin, M.; Petermann, F.; Thomas, J.L.; Zalc, B.; Lubetzki, C. Semaphorin 3A and 3F: Key players in myelin repair in multiple sclerosis? Brain 2007, 130, 2554–2565. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Hlavica, M.; Delparente, A.; Good, A.; Good, N.; Plattner, P.S.; Seyedsadr, M.S.; Schwab, M.E.; Figlewicz, D.P.; Ineichen, B.V. Intrathecal insulin-like growth factor 1 but not insulin enhances myelin repair in young and aged rats. Neurosci. Lett. 2017, 648, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.L.; Suzuki, K.; Chaplin, D.D.; Matsushima, G.K. Interleukin-1beta promotes repair of the CNS. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 7046–7052. [Google Scholar] [CrossRef]
- Baaklini, C.S.; Ho, M.F.S.; Lange, T.; Hammond, B.P.; Panda, S.P.; Zirngibl, M.; Zia, S.; Himmelsbach, K.; Rana, H.; Phillips, B.; et al. Microglia promote remyelination independent of their role in clearing myelin debris. Cell Rep. 2023, 42, 113574. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Gudesblatt, M.; Bumstead, B.; Buhse, M.; Zarif, M.; Morrow, S.A.; Nicholas, J.A.; Hancock, L.M.; Wilken, J.; Weller, J.; Scott, N.; et al. De-escalation of Disease-Modifying Therapy for People with Multiple Sclerosis due to Safety Considerations: Characterizing 1-Year Outcomes in 25 People Who Switched from Ocrelizumab to Diroximel Fumarate. Adv. Ther. 2024, 41, 3059–3075. [Google Scholar] [CrossRef]
- Scolding, N.J.; Pasquini, M.; Reingold, S.C.; Cohen, J.A.; International Conference on Cell-Based Therapies for Multiple Sclerosis. Cell-based therapeutic strategies for multiple sclerosis. Brain 2017, 140, 2776–2796. [Google Scholar] [CrossRef]
- Mullin, A.P.; Cui, C.; Wang, Y.; Wang, J.; Troy, E.; Caggiano, A.O.; Parry, T.J.; Colburn, R.W.; Pavlopoulos, E. rHIgM22 enhances remyelination in the brain of the cuprizone mouse model of demyelination. Neurobiol. Dis. 2017, 105, 142–155. [Google Scholar] [CrossRef]
- Nastasijevic, B.; Wright, B.R.; Smestad, J.; Warrington, A.E.; Rodriguez, M.; Maher, J.L., III. Remyelination Induced by a DNA Aptamer in a Mouse Model of Multiple Sclerosis. PLoS ONE 2012, 7, e39595. [Google Scholar] [CrossRef]
- Healy, L.M.; Antel, J.P. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Mullershausen, F.; Craveiro, L.M.; Shin, Y.; Cortes-Cros, M.; Bassilana, F.; Osinde, M.; Wishart, W.L.; Guerini, D.; Thallmair, M.; Schwab, M.E.; et al. Phosphorylated FTY720 promotes astrocyte migration through sphingosine-1-phosphate receptors. J. Neurochem. 2007, 102, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Osinde, M.; Mullershausen, F.; Dev, K.K. Phosphorylated FTY720 stimulates ERK phosphorylation in astrocytes via S1P receptors. Neuropharmacology 2007, 52, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef]
- Ratchford, J.N.; Endres, C.J.; Hammoud, D.A.; Pomper, M.G.; Shiee, N.; McGready, J.; Pham, D.L.; Calabresi, P.A. Decreased microglial activation in MS patients treated with glatiramer acetate. J. Neurol. 2012, 259, 1199–1205. [Google Scholar] [CrossRef]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of Macrophages in Multiple Sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef]
- Kenison, J.E.; Jhaveri, A.; Li, Z.; Khadse, N.; Tjon, E.; Tezza, S.; Nowakowska, D.; Plasencia, A.; Stanton, V.P., Jr.; Sherr, D.H.; et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 32017–32028. [Google Scholar] [CrossRef]
- Yeste, A.; Nadeau, M.; Burns, E.J.; Weiner, H.L.; Quintana, F.J. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 11270–11275. [Google Scholar] [CrossRef]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Ozturk, O.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Berek, K.; Bauer, A.; Rudzki, D.; Auer, M.; Barket, R.; Zinganell, A.; Lerch, M.; Hofer, L.; Grams, A.; Poskaite, P.; et al. Immune profiling in multiple sclerosis: A single-center study of 65 cytokines, chemokines, and related molecules in cerebrospinal fluid and serum. Front. Immunol. 2023, 14, 1200146. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Markowitz, C. Deciding on the Best Multiple Sclerosis Therapy: Tough Choices. JAMA Neurol. 2018, 75, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Fissolo, N.; Benkert, P.; Sastre-Garriga, J.; Mongay-Ochoa, N.; Vilaseca-Jolonch, A.; Llufriu, S.; Blanco, Y.; Hegen, H.; Berek, K.; Perez-Miralles, F.; et al. Serum biomarker levels predict disability progression in patients with primary progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2024, 95, 410–418. [Google Scholar] [CrossRef]
- Canto, E.; Reverter, F.; Morcillo-Suarez, C.; Matesanz, F.; Fernandez, O.; Izquierdo, G.; Vandenbroeck, K.; Rodriguez-Antiguedad, A.; Urcelay, E.; Arroyo, R.; et al. Chitinase 3-like 1 plasma levels are increased in patients with progressive forms of multiple sclerosis. Mult. Scler. 2012, 18, 983–990. [Google Scholar] [CrossRef]
- Perez-Miralles, F.; Prefasi, D.; Garcia-Merino, A.; Gascon-Gimenez, F.; Medrano, N.; Castillo-Villalba, J.; Cubas, L.; Alcala, C.; Gil-Perotin, S.; Gomez-Ballesteros, R.; et al. CSF chitinase 3-like-1 association with disability of primary progressive MS. Neurol. Neuroimmunol. Neuroinflammation 2020, 7. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, M.; Jiang, L. Potential Roles and Future Perspectives of Chitinase 3-like 1 in Macrophage Polarization and the Development of Diseases. Int. J. Mol. Sci. 2023, 24, 16149. [Google Scholar] [CrossRef]
- Cubas-Nunez, L.; Gil-Perotin, S.; Castillo-Villalba, J.; Lopez, V.; Solis Tarazona, L.; Gasque-Rubio, R.; Carratala-Bosca, S.; Alcala-Vicente, C.; Perez-Miralles, F.; Lassmann, H.; et al. Potential Role of CHI3L1+ Astrocytes in Progression in MS. Neurol. Neuroimmunol. Neuroinflammation 2021, 8. [Google Scholar] [CrossRef]
- Song, Y.; Jiang, W.; Afridi, S.K.; Wang, T.; Zhu, F.; Xu, H.; Nazir, F.H.; Liu, C.; Wang, Y.; Long, Y.; et al. Astrocyte-derived CHI3L1 signaling impairs neurogenesis and cognition in the demyelinated hippocampus. Cell Rep. 2024, 43, 114226. [Google Scholar] [CrossRef]
- Zrzavy, T.; Rieder, K.; Wuketich, V.; Thalhammer, R.; Haslacher, H.; Altmann, P.; Kornek, B.; Krajnc, N.; Monschein, T.; Schmied, C.; et al. Immunophenotyping in routine clinical practice for predicting treatment response and adverse events in patients with MS. Front. Neurol. 2024, 15, 1388941. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theophanous, S.; Sargiannidou, I.; Kleopa, K.A. Glial Cells as Key Regulators in Neuroinflammatory Mechanisms Associated with Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 9588. https://doi.org/10.3390/ijms25179588
Theophanous S, Sargiannidou I, Kleopa KA. Glial Cells as Key Regulators in Neuroinflammatory Mechanisms Associated with Multiple Sclerosis. International Journal of Molecular Sciences. 2024; 25(17):9588. https://doi.org/10.3390/ijms25179588
Chicago/Turabian StyleTheophanous, Styliani, Irene Sargiannidou, and Kleopas A. Kleopa. 2024. "Glial Cells as Key Regulators in Neuroinflammatory Mechanisms Associated with Multiple Sclerosis" International Journal of Molecular Sciences 25, no. 17: 9588. https://doi.org/10.3390/ijms25179588