
Protein Kinase C-Delta Mediates Cell Cycle Reentry and Apoptosis Induced by Amyloid-Beta Peptide in Post-Mitotic Cortical Neurons

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
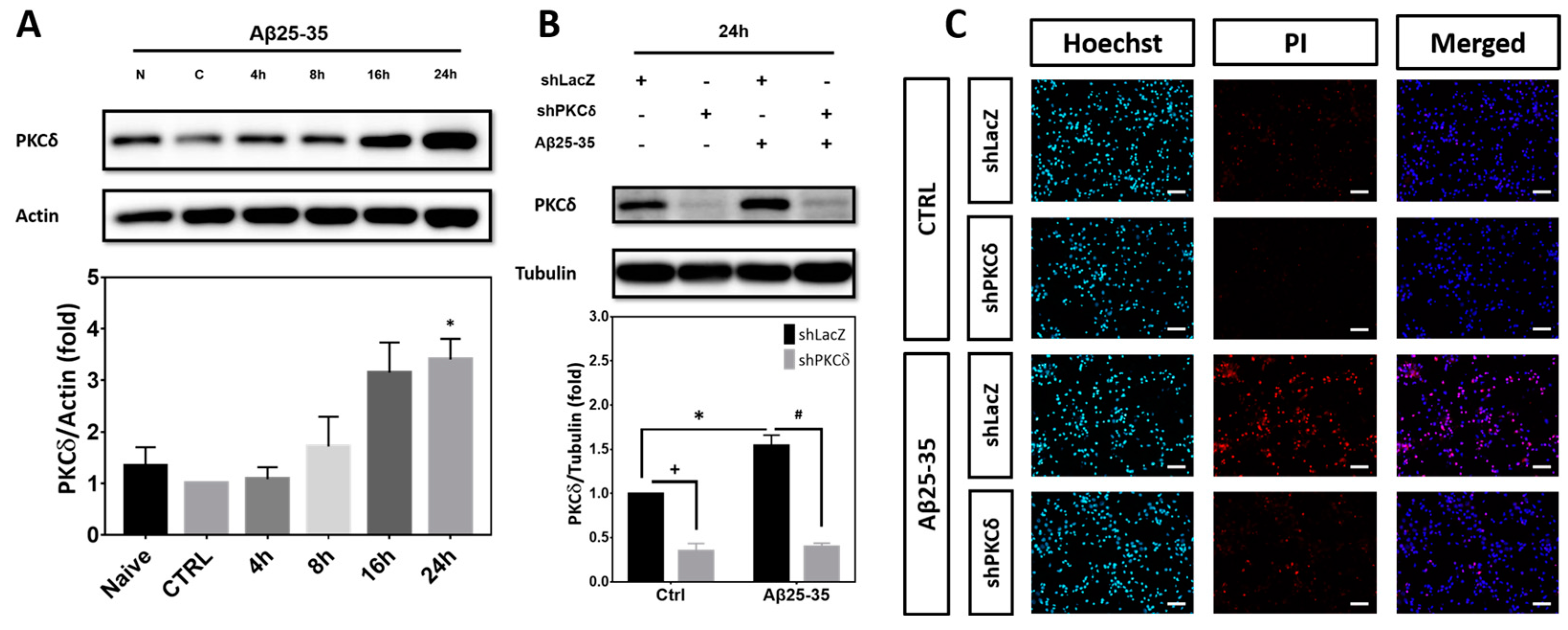
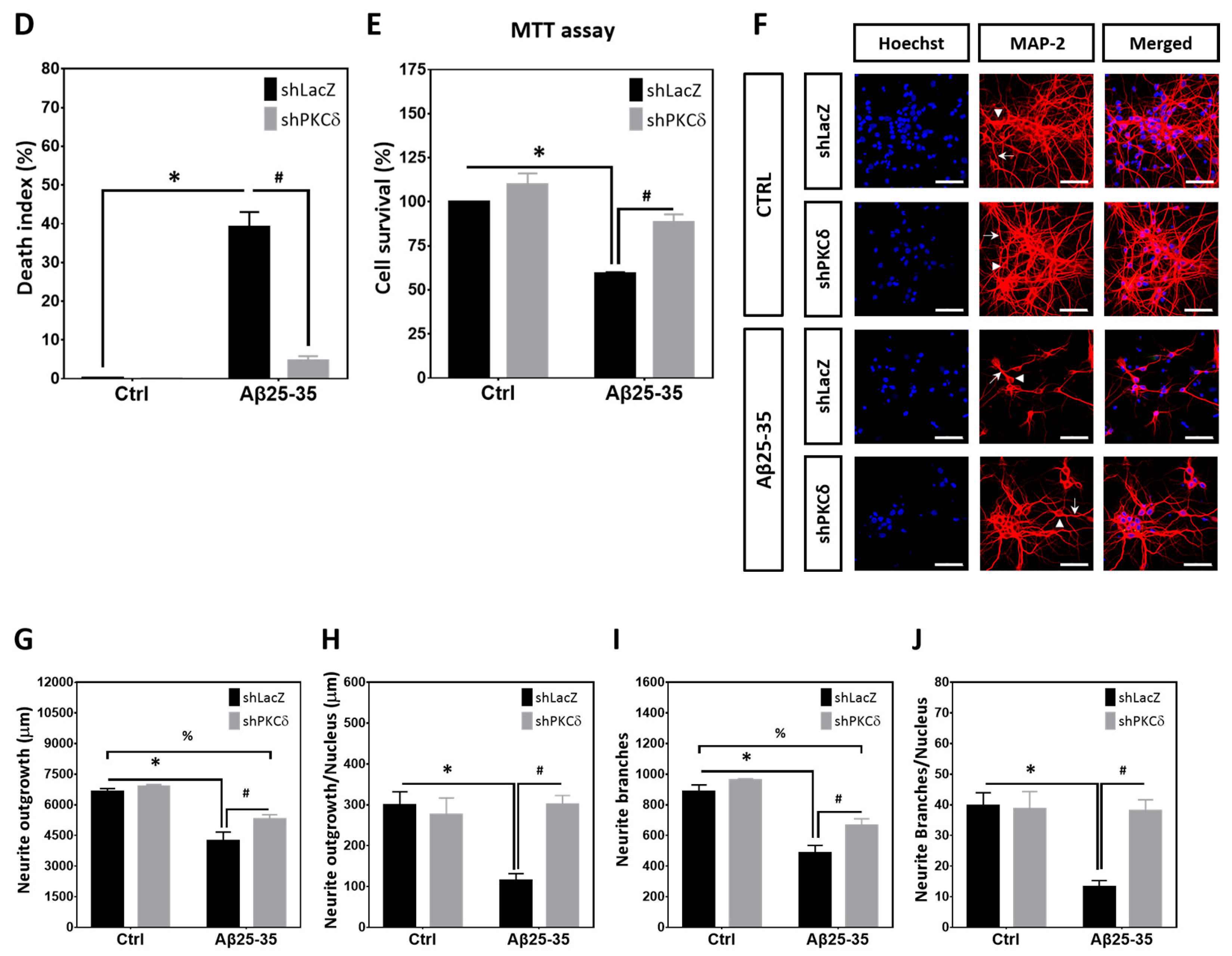
2.1. PKCδ Inhibition Exerts Neuroprotective Effects against Aβ25-35 Toxicity
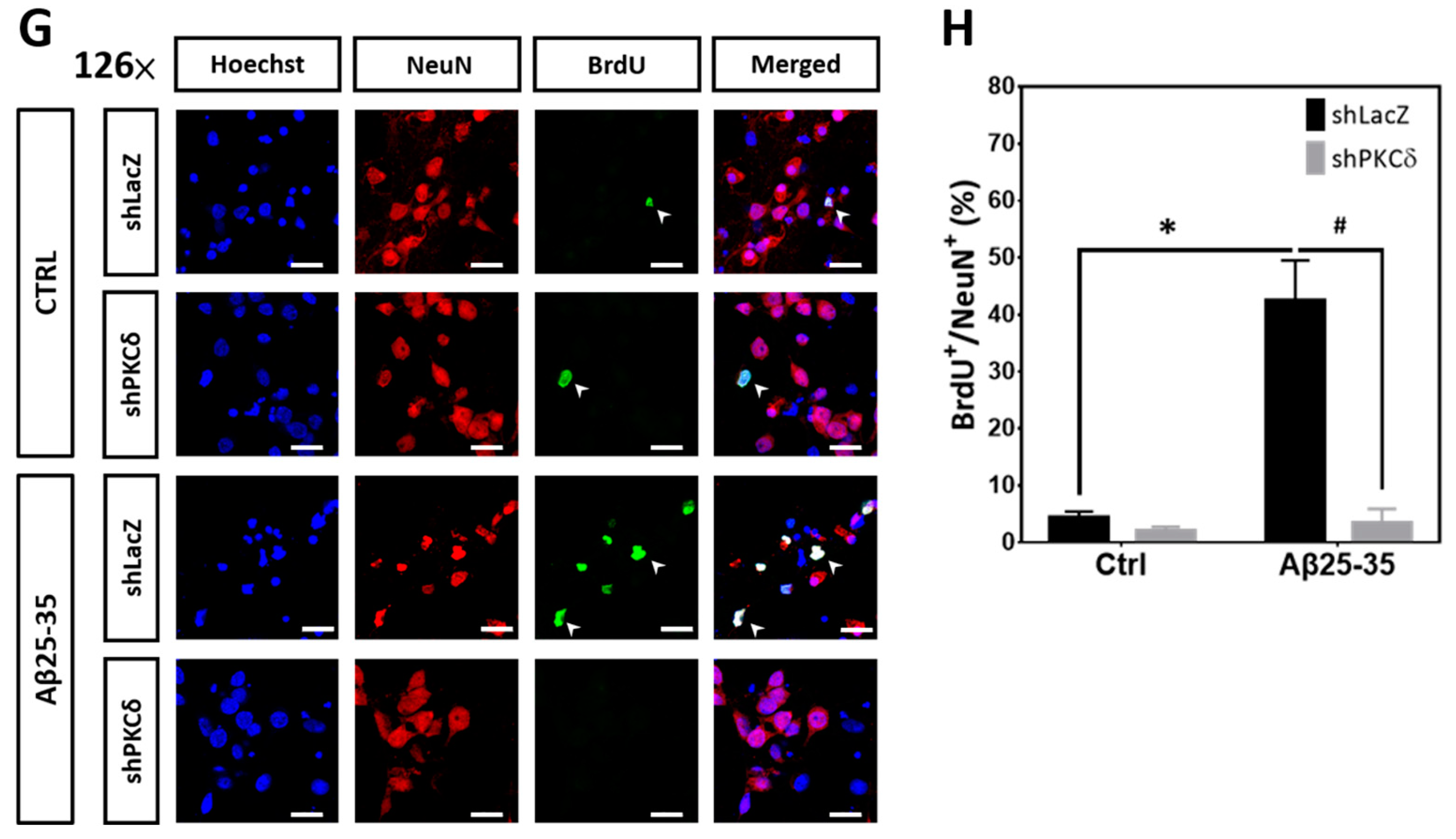
2.2. PKCδ Inhibition Blocks Aberrant CCR and Apoptosis Induced by Aβ25–35 in Differentiated Cortical Neurons
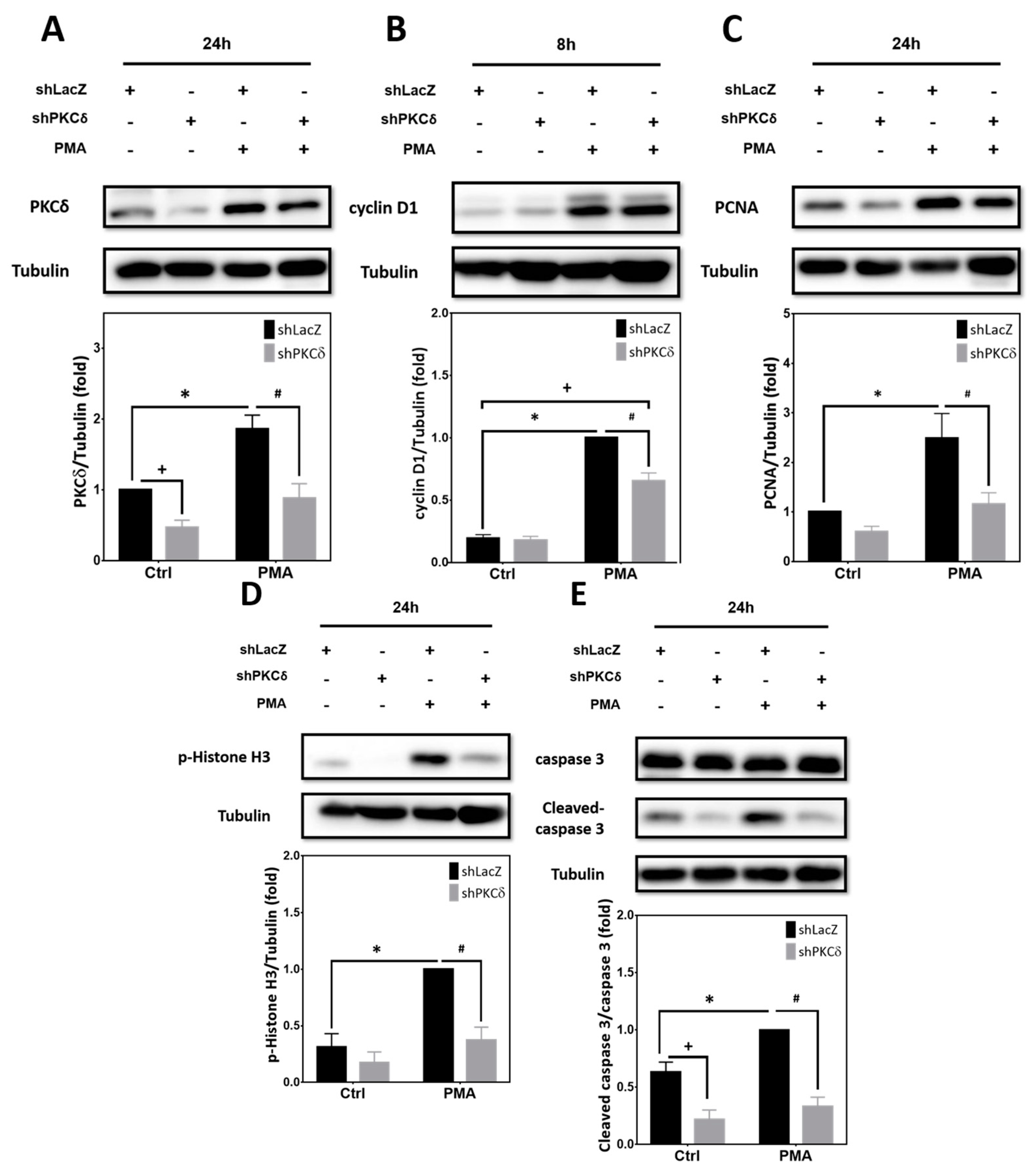
2.3. Pharmacological Activation of PKCs by PMA Is Sufficient to Upregulate Expression of Cell Cycle and Apoptotic Markers That Can Be Blocked by PKCδ Knockdown
2.4. PKCδ Inhibition Lessens Aβ25-35- and PMA-Mediated CCR by Attenuating CDK5 Hyperactivation in Primary Cortical Cultures
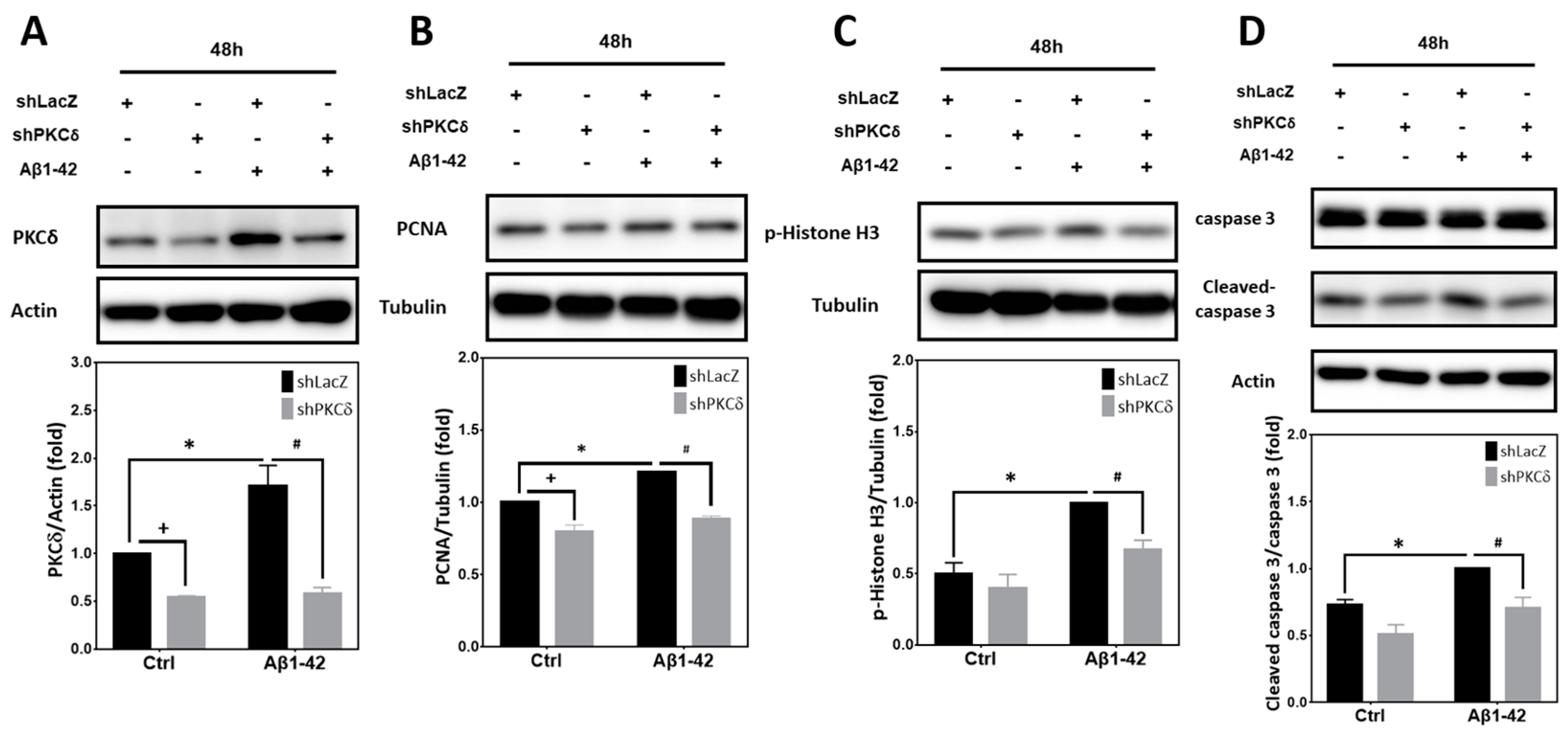
2.5. PKCδ Inhibition Blocks Neuronal CCR and Apoptosis Induced by Aβ1–42
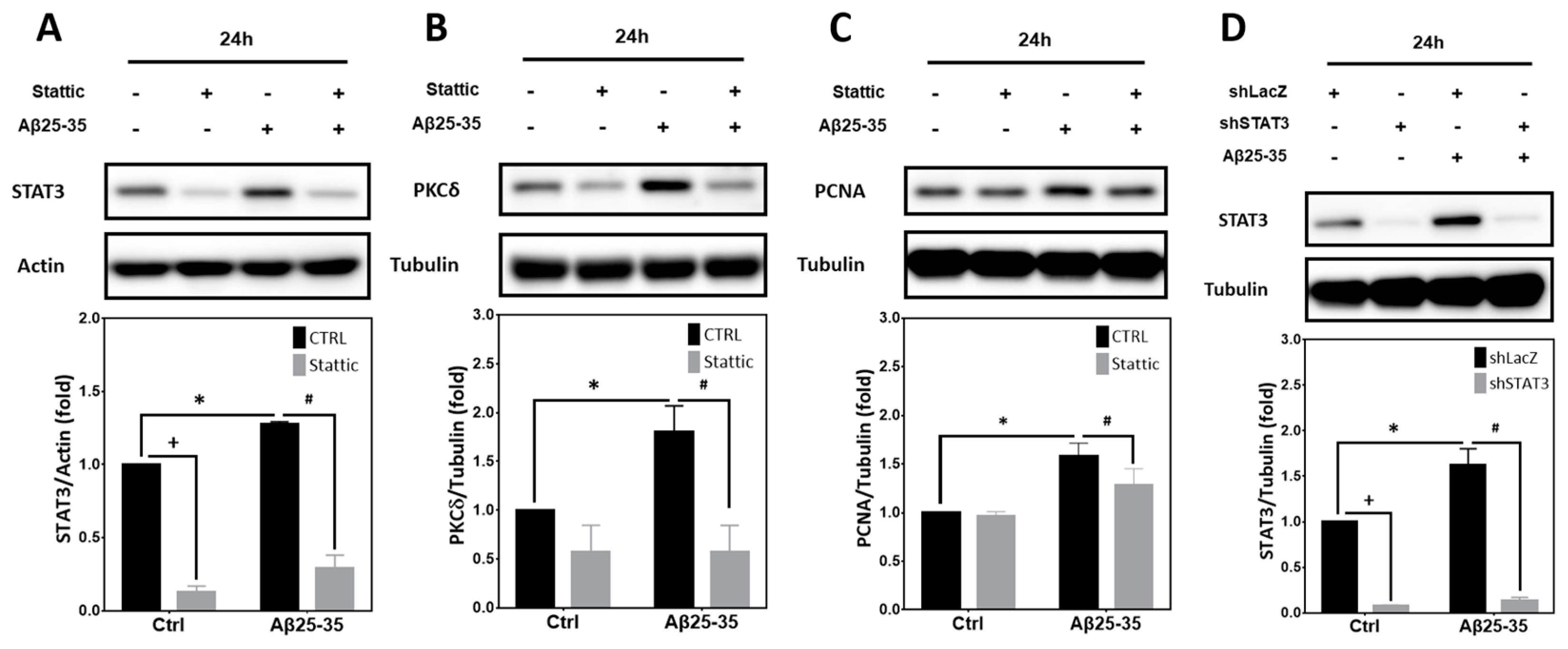
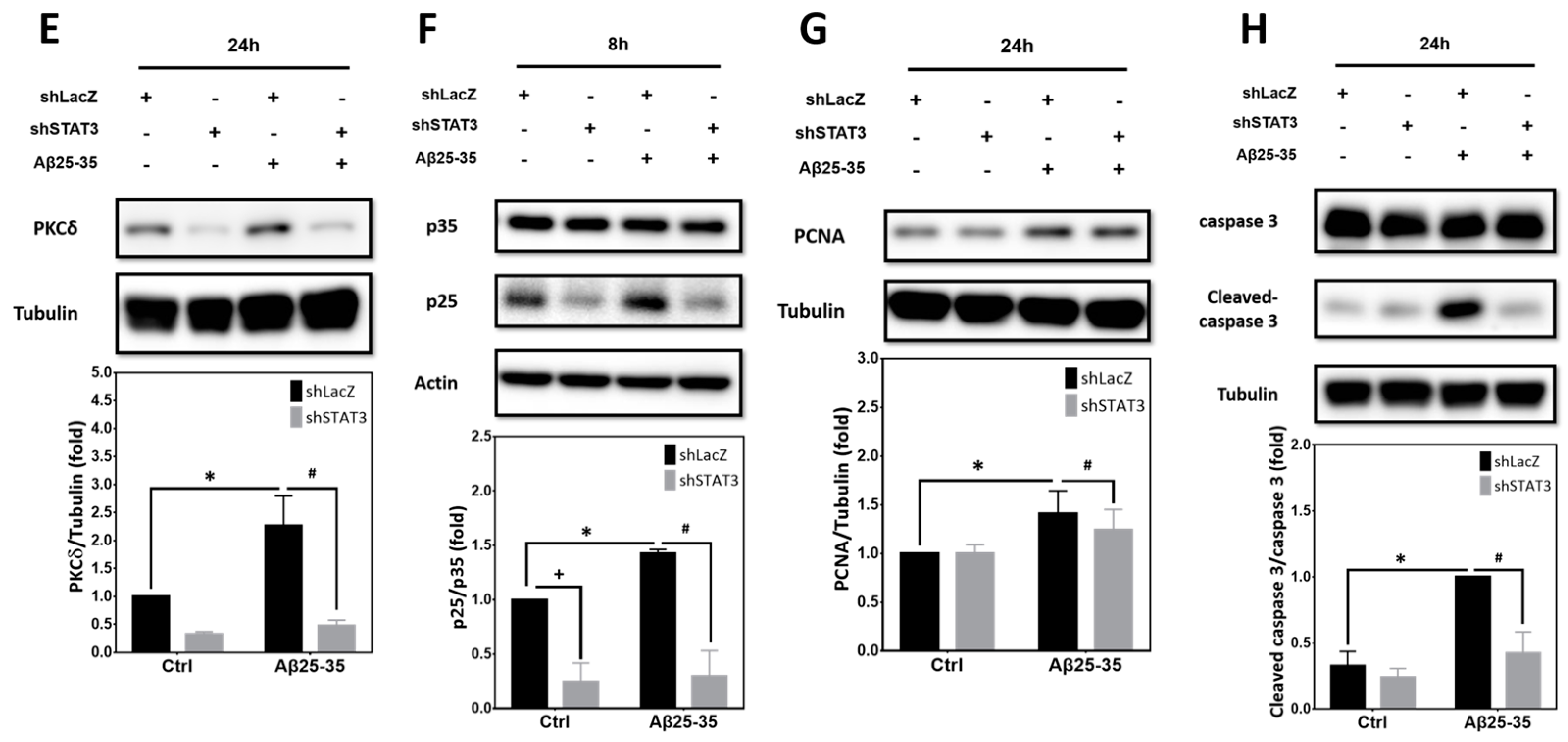
2.6. STAT3 Contributes to Aβ25-35- and PKCδ-Dependent Neuronal CCR and Apoptosis
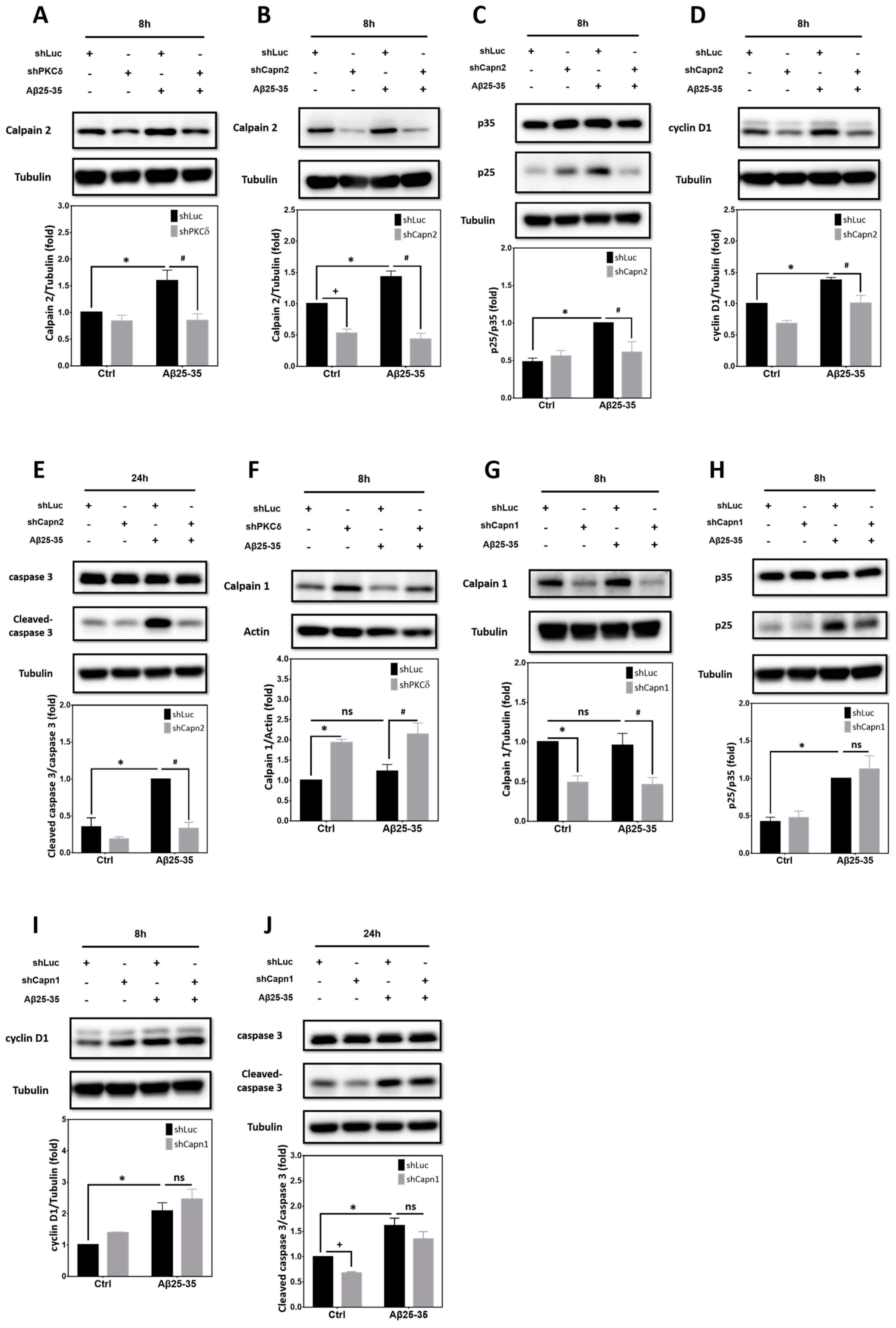
2.7. Calpain2, but Not calpain1, Contributes to PKCδ-Dependent CCR Induced by Aβ25–35 in Primary Cortical Cultures
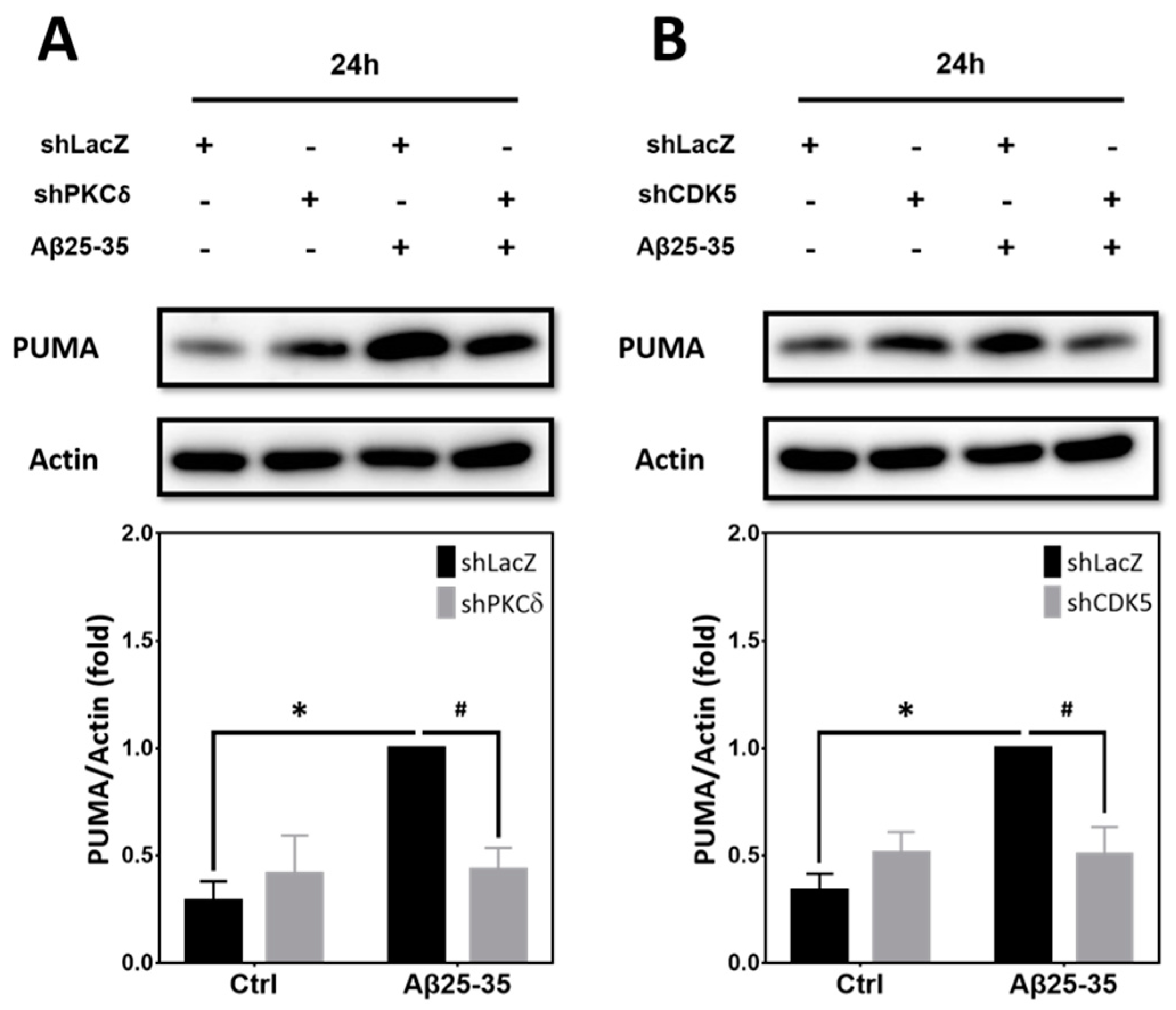
2.8. Inhibition of PKCδ and CDK5 Downregulates PUMA Expression Induced by Aβ25–35
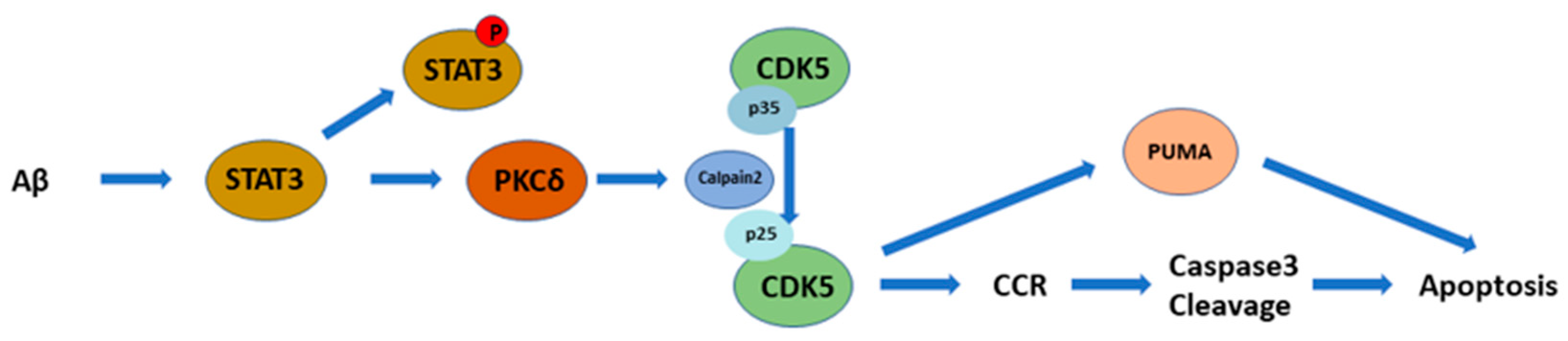
3. Discussion
4. Materials and Methods
4.1. Reagents, Preparations of Aβs, and Primary Culture of Rat Cortical Neurons
4.2. Western Blotting
4.3. Lentiviral Infection of shRNA in Primary Rat Cortical Cultures
4.4. Cell Survival Assays
4.5. Immunocytochemistry and Quantification of Neurite Lengths and Neurite Branches
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Carr, D.J.J.; Yamada, H.Y. “Amyloid-beta accumulation cycle” as a prevention and/or therapy target for Alzheimer’s disease. Aging Cell 2020, 19, e13109. [Google Scholar] [CrossRef]
- Hradek, A.C.; Lee, H.P.; Siedlak, S.L.; Torres, S.L.; Jung, W.; Han, A.H.; Lee, H.G. Distinct chronology of neuronal cell cycle re-entry and tau pathology in the 3xTg-AD mouse model and Alzheimer’s disease patients. J. Alzheimers Dis. 2015, 43, 57–65. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, L.A.; Hoozemans, J.J. Physiological and pathophysiological functions of cell cycle proteins in post-mitotic neurons: Implications for Alzheimer’s disease. Acta Neuropathol. 2015, 129, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.; Currais, A.; Andres, A.; Towlson, C.; Pitsi, D.; Nunes, A.; Niblock, M.; Cooper, J.; Hortobagyi, T.; Soriano, S. Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the presenilin-1 Alzheimer’s disease brain. Cell Cycle 2008, 7, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bu, B.; Xie, M.; Zhang, M.; Yu, Z.; Tao, D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009, 89, 1–17. [Google Scholar] [CrossRef]
- Singh, N.; Nandy, S.K.; Jyoti, A.; Saxena, J.; Sharma, A.; Siddiqui, A.J.; Sharma, L. Protein kinase C (PKC) in neurological health: Implications for Alzheimer’s disease and chronic alcohol consumption. Brain Sci. 2024, 14, 554. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Simpkins, J.W.; Alkon, D.L.; Smith, K.E.; Chen, Y.W.; Tan, Z.; Huber, J.D.; Rosen, C.L. Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. J. Alzheimers Dis. 2015, 43, 711–724. [Google Scholar] [CrossRef]
- Conboy, L.; Foley, A.G.; O’Boyle, N.M.; Lawlor, M.; Gallagher, H.C.; Murphy, K.J.; Regan, C.M. Curcumin-induced degradation of PKC delta is associated with enhanced dentate NCAM PSA expression and spatial learning in adult and aged Wistar rats. Biochem. Pharmacol. 2009, 77, 1254–1265. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, Y.; Li, C.; Zheng, Q.; Tian, J.; Li, Z.; Huang, T.Y.; Zhang, W.; Xu, H. Inhibition of PKCdelta reduces amyloid-beta levels and reverses Alzheimer disease phenotypes. J. Exp. Med. 2018, 215, 1665–1677. [Google Scholar] [CrossRef]
- Shupp, A.; Casimiro, M.C.; Pestell, R.G. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget 2017, 8, 17373–17382. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Li, W.; Allen, M.E.; Rui, Y.; Ku, L.; Liu, G.; Bankston, A.N.; Zheng, J.Q.; Feng, Y. p39 is responsible for increasing Cdk5 activity during postnatal neuron differentiation and governs neuronal network formation and epileptic responses. J. Neurosci. 2016, 36, 11283–11294. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.Y.; Shang, L.; Hu, Y.Y.; Jiang, L.P.; Wan, Y.Y.; Zhou, Q.Q.; Zhang, K.; Liao, H.F.; Yi, J.L.; Han, X.J. The role of Cdk5-mediated Drp1 phosphorylation in Abeta(1-42) induced mitochondrial fission and neuronal apoptosis. J. Cell. Biochem. 2018, 119, 4815–4825. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Anwar, F.; Al-Abbasi, F.A.; Zamzami, M.A.; Al-Talhi, H.A.; Kamal, M.A. Neuroprotective mechanisms mediated by CDK5 inhibition. Curr. Pharm. Des. 2016, 22, 527–534. [Google Scholar] [CrossRef]
- Bhounsule, A.S.; Bhatt, L.K.; Prabhavalkar, K.S.; Oza, M. Cyclin dependent kinase 5: A novel avenue for Alzheimer’s disease. Brain Res. Bull. 2017, 132, 28–38. [Google Scholar] [CrossRef]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Takahashi, S.; Saito, T.; Hisanaga, S.; Pant, H.C.; Kulkarni, A.B. Tau phosphorylation by cyclin-dependent kinase 5/p39 during brain development reduces its affinity for microtubules. J. Biol. Chem. 2003, 278, 10506–10515. [Google Scholar] [CrossRef]
- Lapresa, R.; Agulla, J.; Sanchez-Moran, I.; Zamarreno, R.; Prieto, E.; Bolanos, J.P.; Almeida, A. Amyloid-β promotes neurotoxicity by Cdk5-induced p53 stabilization. Neuropharmacology 2019, 146, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Meng, C.; Xing, D. Abeta induces PUMA activation: A new mechanism for Abeta-mediated neuronal apoptosis. Neurobiol. Aging 2015, 36, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, X.; Ma, S.; Zhang, N.; Yang, D.; Wang, L.; Ye, S.; Zhang, Q.; Ruan, J.; Ma, J.; et al. ChK1 activation induces reactive astrogliosis through CIP2A/PP2A/STAT3 pathway in Alzheimer’s disease. FASEB J. 2022, 36, e22209. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Patino-Lopez, G.; Gomez-Lira, G.; Gutierrez, R.; Becerril-Perez, F.; Rosales-Cordova, A.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Leon-Rivera, I.; Soto-Cruz, I.; et al. Activation of STAT3 regulates reactive astrogliosis and neuronal death induced by AbetaO neurotoxicity. Int. J. Mol. Sci. 2020, 21, 7458. [Google Scholar] [CrossRef]
- Choi, M.; Kim, H.; Yang, E.J.; Kim, H.S. Inhibition of STAT3 phosphorylation attenuates impairments in learning and memory in 5XFAD mice, an animal model of Alzheimer’s disease. J. Pharmacol. Sci. 2020, 143, 290–299. [Google Scholar] [CrossRef]
- Mehla, J.; Singh, I.; Diwan, D.; Nelson, J.W.; Lawrence, M.; Lee, E.; Bauer, A.Q.; Holtzman, D.M.; Zipfel, G.J. STAT3 inhibitor mitigates cerebral amyloid angiopathy and parenchymal amyloid plaques while improving cognitive functions and brain networks. Acta Neuropathol. Commun. 2021, 9, 193. [Google Scholar] [CrossRef]
- Yang, G.; Tong, Y.; Wang, X.; Zhao, C.; Ba, Z.; Ahelijiang, R.; Liu, X.; Gao, W.; Zhao, Y.; Gu, Y.; et al. Guizhi Fuling capsule relieves memory deficits by inhibition of microglial neuroinflammation through blocking JAK2/STAT3 pathway in presenilin1/2 conditional double knockout mice. Front. Immunol. 2023, 14, 1185570. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Q.; Wang, S.; Liao, Z.; Jin, H.; Huang, S.; Hong, X.; Liu, Y.; Pang, J.; Shen, Q.; et al. The food additive beta-caryophyllene exerts its neuroprotective effects through the JAK2-STAT3-BACE1 pathway. Front. Aging Neurosci. 2022, 14, 814432. [Google Scholar]
- Wan, H.L.; Hong, X.Y.; Zhao, Z.H.; Li, T.; Zhang, B.G.; Liu, Q.; Wang, Q.; Zhao, S.; Wang, J.Z.; Shen, X.F.; et al. STAT3 ameliorates cognitive deficits via regulation of NMDAR expression in an Alzheimer’s disease animal model. Theranostics 2021, 11, 5511–5524. [Google Scholar] [CrossRef]
- Chao, A.C.; Chen, C.H.; Wu, M.H.; Hou, B.Y.; Yang, D.I. Roles of Id1/HIF-1 and CDK5/HIF-1 in cell cycle reentry induced by amyloid-beta peptide in post-mitotic cortical neuron. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118628. [Google Scholar] [CrossRef]
- Prigent, C.; Dimitrov, S. Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 2003, 116, 3677–3685. [Google Scholar] [CrossRef]
- Chao, A.C.; Chen, C.H.; Chang, S.H.; Huang, C.T.; Hwang, W.C.; Yang, D.I. Id1 and sonic hedgehog mediate cell cycle reentry and apoptosis induced by amyloid beta-peptide in post-mitotic cortical neurons. Mol. Neurobiol. 2019, 56, 465–489. [Google Scholar] [CrossRef]
- Wen, H.C.; Huo, Y.N.; Chou, C.M.; Lee, W.S. PMA inhibits endothelial cell migration through activating the PKC-delta/Syk/NF-kappaB-mediated up-regulation of Thy-1. Sci. Rep. 2018, 8, 16247. [Google Scholar] [CrossRef]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef] [PubMed]
- Vosler, P.S.; Brennan, C.S.; Chen, J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef] [PubMed]
- Pao, P.C.; Tsai, L.H. Three decades of Cdk5. J. Biomed. Sci. 2021, 28, 79. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, T.; Hao, Y.; Li, C.; Tang, L.; Li, X.; Zhang, X.; Li, L.; Yao, D.; Xu, X.; et al. PKCdelta serves as a potential biomarker and therapeutic target for microglia-mediated neuroinflammation in Alzheimer’s disease. Alzheimers Dement. 2024, 20, 5511–5527. [Google Scholar] [CrossRef]
- Lu, W.; Tang, S.; Li, A.; Huang, Q.; Dou, M.; Zhang, Y.; Hu, X.; Chang, R.C.C.; Wong, G.T.C.; Huang, C. The role of PKC/PKR in aging, Alzheimer’s disease, and perioperative neurocognitive disorders. Front. Aging Neurosci. 2022, 14, 973068. [Google Scholar] [CrossRef]
- Lee, B.K.; Jee, H.J.; Jung, Y.S. Abeta(1-40)-induced platelet adhesion is ameliorated by rosmarinic acid through inhibition of NADPH oxidase/PKC-delta/integrin alpha(IIb)beta(3) signaling. Antioxidants (Basel) 2021, 10, 1671. [Google Scholar] [CrossRef]
- Gschwendt, M.; Muller, H.J.; Kielbassa, K.; Zang, R.; Kittstein, W.; Rincke, G.; Marks, F. Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 1994, 199, 93–98. [Google Scholar] [CrossRef]
- Bhavanasi, D.; Kostyak, J.C.; Swindle, J.; Kilpatrick, L.E.; Kunapuli, S.P. CGX1037 is a novel PKC isoform delta selective inhibitor in platelets. Platelets 2015, 26, 2–9. [Google Scholar] [CrossRef]
- Hou, B.Y.; Wu, M.H.; Hsu, H.Y.; Lin, Y.C.; Yang, D.I. Polysaccharides from Basella alba protect post-mitotic neurons against cell cycle re-entry and apoptosis induced by the amyloid-beta peptide by blocking sonic hedgehog expression. Int. J. Mol. Sci. 2024, 25, 7316. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Kanthasamy, A.; Kitazawa, M.; Anantharam, V.; Kanthasamy, A.G. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: Relevance to oxidative stress in dopaminergic degeneration. Eur. J. Neurosci. 2003, 18, 1387–1401. [Google Scholar] [CrossRef]
- Kaul, S.; Anantharam, V.; Yang, Y.; Choi, C.J.; Kanthasamy, A.; Kanthasamy, A.G. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J. Biol. Chem. 2005, 280, 28721–28730. [Google Scholar] [CrossRef] [PubMed]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Lucas, C.D.; MacKellar, A.; Li, F.; Hiersemenzel, K.; Haslett, C.; Davidson, D.J.; Rossi, A.G. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J. Inflamm. (Lond) 2013, 10, 12. [Google Scholar] [CrossRef]
- Zhao, C.T.; Li, K.; Li, J.T.; Zheng, W.; Liang, X.J.; Geng, A.Q.; Li, N.; Yuan, X.B. PKCdelta regulates cortical radial migration by stabilizing the Cdk5 activator p35. Proc. Natl. Acad. Sci. U S A 2009, 106, 21353–21358. [Google Scholar] [CrossRef]
- Sahin, B.; Hawasli, A.H.; Greene, R.W.; Molkentin, J.D.; Bibb, J.A. Negative regulation of cyclin-dependent kinase 5 targets by protein kinase C. Eur. J. Pharmacol. 2008, 581, 270–275. [Google Scholar] [CrossRef]
- Zhang, H.; Chang, L.; Zhang, H.; Nie, J.; Zhang, Z.; Yang, X.; Vuong, A.M.; Wang, Z.; Chen, A.; Niu, Q. Calpain-2/p35-p25/Cdk5 pathway is involved in the neuronal apoptosis induced by polybrominated diphenyl ether-153. Toxicol. Lett. 2017, 277, 41–53. [Google Scholar] [CrossRef]
- Requejo-Aguilar, R. Cdk5 and aberrant cell cycle activation at the core of neurodegeneration. Neural Regen. Res. 2023, 18, 1186–1190. [Google Scholar] [CrossRef]
- Tian, B.; Yang, Q.; Mao, Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat. Cell Biol. 2009, 11, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.N.; Kim, D.W.; Kim, I.H.; Park, J.H.; Ahn, J.H.; Kang, I.J.; Lee, Y.L.; Lee, C.H.; Hwang, I.K.; Kim, Y.M.; et al. Down-regulation of cyclin-dependent kinase 5 attenuates p53-dependent apoptosis of hippocampal CA1 pyramidal neurons following transient cerebral ischemia. Sci. Rep. 2019, 9, 13032. [Google Scholar] [CrossRef] [PubMed]
- Samidurai, M.; Tarale, P.; Janarthanam, C.; Estrada, C.G.; Gordon, R.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances activation of STAT3/NLRC4 inflammasome signaling axis through PKCdelta in astrocytes: Implications for Parkinson’s disease. Cells 2020, 9, 1831. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef]
- Zhao, Y.; Ding, M.; Yan, F.; Yin, J.; Shi, W.; Yang, N.; Zhao, H.; Fang, Y.; Huang, Y.; Zheng, Y.; et al. Inhibition of the JAK2/STAT3 pathway and cell cycle re-entry contribute to the protective effect of remote ischemic pre-conditioning of rat hindlimbs on cerebral ischemia/reperfusion injury. CNS Neurosci. Ther. 2023, 29, 866–877. [Google Scholar] [CrossRef]
- Olah, Z.; Kalman, J.; Toth, M.E.; Zvara, A.; Santha, M.; Ivitz, E.; Janka, Z.; Pakaski, M. Proteomic analysis of cerebrospinal fluid in Alzheimer’s disease: Wanted dead or alive. J. Alzheimers Dis. 2015, 44, 1303–1312. [Google Scholar] [CrossRef]
- Zhuang, J.; Cai, P.; Chen, Z.; Yang, Q.; Chen, X.; Wang, X.; Zhuang, X. Long noncoding RNA MALAT1 and its target microRNA-125b are potential biomarkers for Alzheimer’s disease management via interactions with FOXQ1, PTGS2 and CDK5. Am. J. Transl. Res. 2020, 12, 5940–5954. [Google Scholar]
- Liu, J.; Jiao, L.; Zhong, X.; Yao, W.; Du, K.; Lu, S.; Wu, Y.; Ma, T.; Tong, J.; Xu, M.; et al. Platelet activating factor receptor exaggerates microglia-mediated microenvironment by IL10-STAT3 signaling: A novel potential biomarker and target for diagnosis and treatment of Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 856628. [Google Scholar] [CrossRef]
- Arias-Vasquez, A.; Aulchenko, Y.S.; Isaacs, A.; van Oosterhout, A.; Sleegers, K.; Hofman, A.; van Broeckhoven, C.; Oostra, B.A.; Breteler, M.; van Duijn, C.M. Cyclin-dependent kinase 5 is associated with risk for Alzheimer’s disease in a Dutch population-based study. J. Neurol. 2008, 255, 655–662. [Google Scholar] [CrossRef]
- Vazquez-Higuera, J.L.; Mateo, I.; Sanchez-Juan, P.; Rodriguez-Rodriguez, E.; Infante, J.; Berciano, J.; Combarros, O. No association of CDK5 genetic variants with Alzheimer’s disease risk. BMC Med. Genet. 2009, 10, 68. [Google Scholar] [CrossRef]
- Sjolander, A.; Andersson, M.E.; Zetterberg, H.; Minthon, L.; Bogdanovic, N.; Blennow, K. Alzheimer’s disease: No effect of the CDK5 gene on CSF biomarkers, neuropathology or disease risk. Mol Med Rep 2009, 2, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.C.; Yang, Y.T.; Yang, D.I. Protective effects of S-nitrosoglutathione against neurotoxicity of 3-nitropropionic acid in rat. Neurosci. Lett. 2004, 362, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Hwang, C.S.; Yang, D.I. Protective effects of brain-derived neurotrophic factor against neurotoxicity of 3-nitropropionic acid in rat cortical neurons. Neurotoxicology 2009, 30, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.C.; Chen, S.D.; Liu, C.C.; Yang, D.I. Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic. Biol. Med. 2005, 38, 938–949. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-H.; Chao, A.-C.; Hsieh, Y.-H.; Lien, Y.; Lin, Y.-C.; Yang, D.-I. Protein Kinase C-Delta Mediates Cell Cycle Reentry and Apoptosis Induced by Amyloid-Beta Peptide in Post-Mitotic Cortical Neurons. Int. J. Mol. Sci. 2024, 25, 9626. https://doi.org/10.3390/ijms25179626
Wu M-H, Chao A-C, Hsieh Y-H, Lien Y, Lin Y-C, Yang D-I. Protein Kinase C-Delta Mediates Cell Cycle Reentry and Apoptosis Induced by Amyloid-Beta Peptide in Post-Mitotic Cortical Neurons. International Journal of Molecular Sciences. 2024; 25(17):9626. https://doi.org/10.3390/ijms25179626
Chicago/Turabian StyleWu, Ming-Hsuan, A-Ching Chao, Yi-Heng Hsieh, You Lien, Yi-Chun Lin, and Ding-I Yang. 2024. "Protein Kinase C-Delta Mediates Cell Cycle Reentry and Apoptosis Induced by Amyloid-Beta Peptide in Post-Mitotic Cortical Neurons" International Journal of Molecular Sciences 25, no. 17: 9626. https://doi.org/10.3390/ijms25179626
APA StyleWu, M.-H., Chao, A.-C., Hsieh, Y.-H., Lien, Y., Lin, Y.-C., & Yang, D.-I. (2024). Protein Kinase C-Delta Mediates Cell Cycle Reentry and Apoptosis Induced by Amyloid-Beta Peptide in Post-Mitotic Cortical Neurons. International Journal of Molecular Sciences, 25(17), 9626. https://doi.org/10.3390/ijms25179626