
VCD Analysis of Axial Chirality in Synthetic Stereoisomeric Biaryl-Type bis-Isochroman Heterodimers with Isolated Blocks of Central and Axial Chirality
 , , , ,
, , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
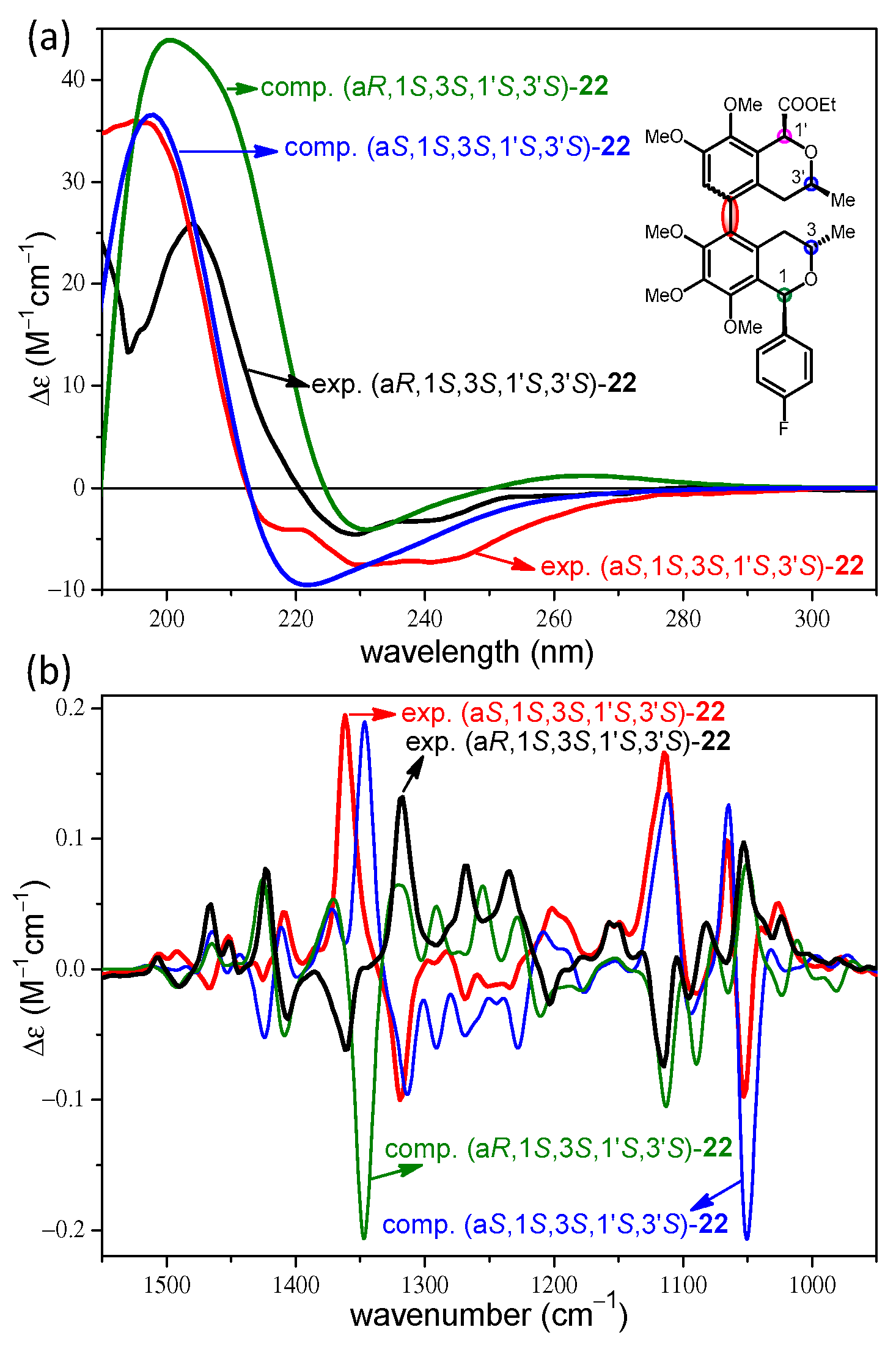
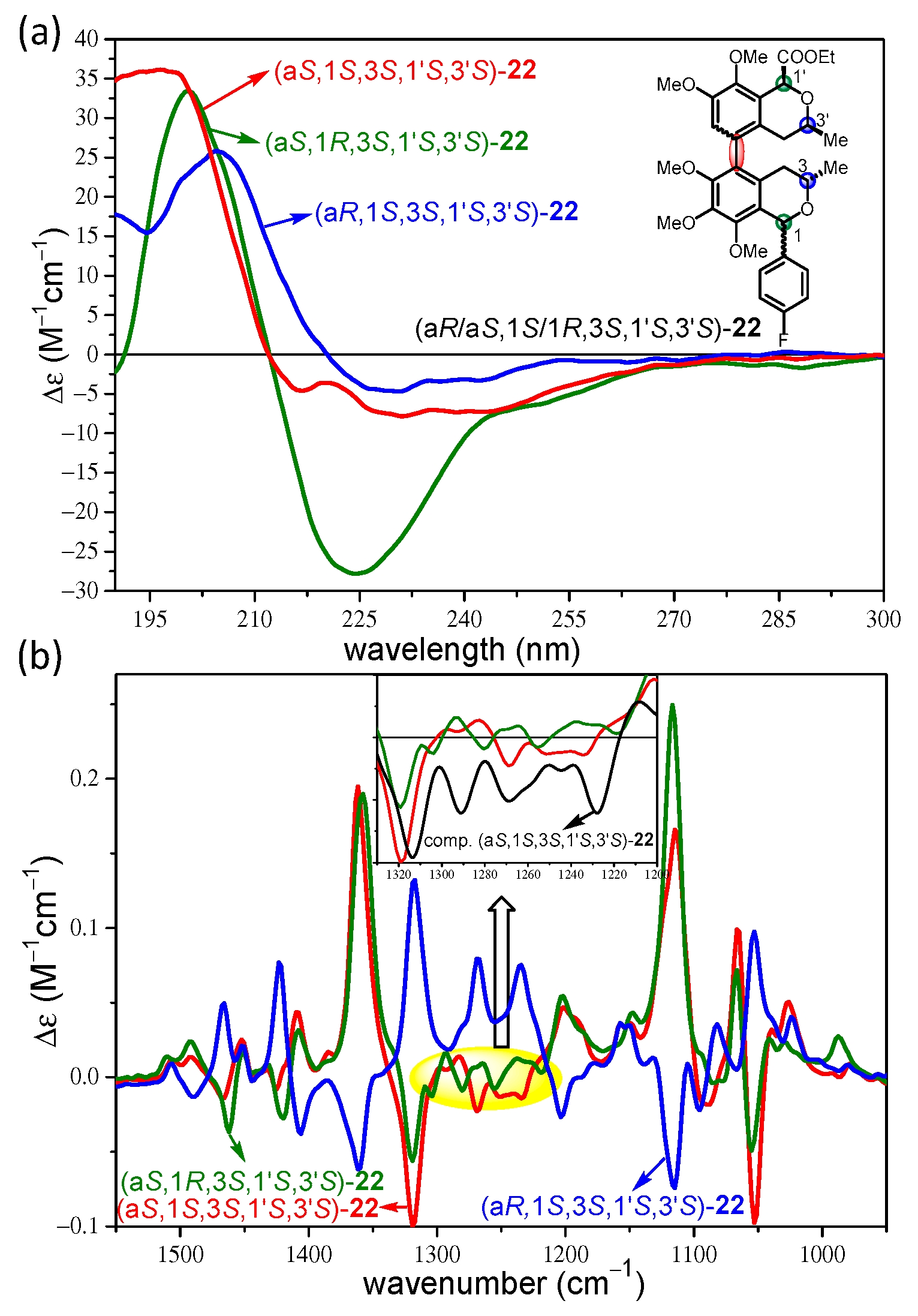
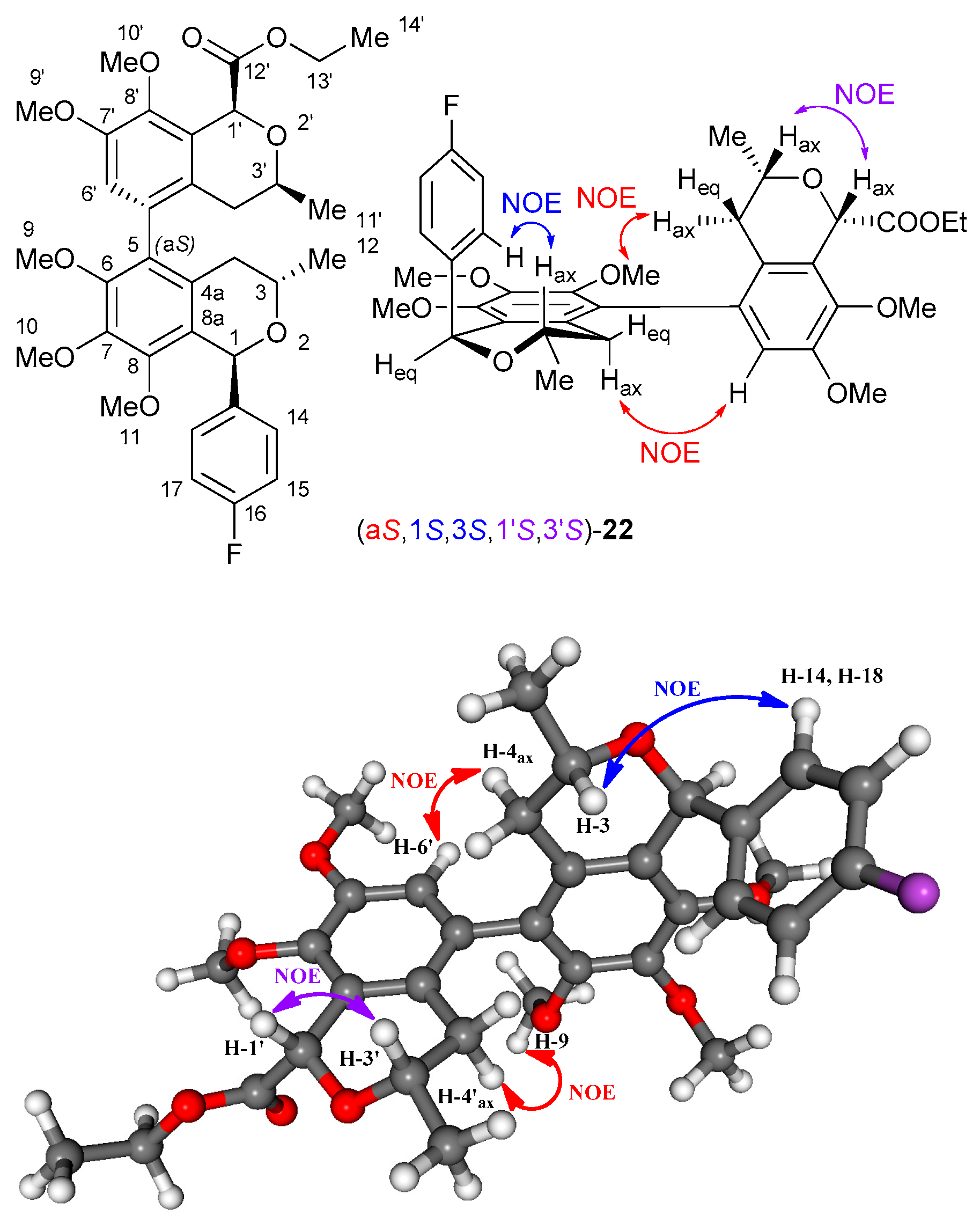
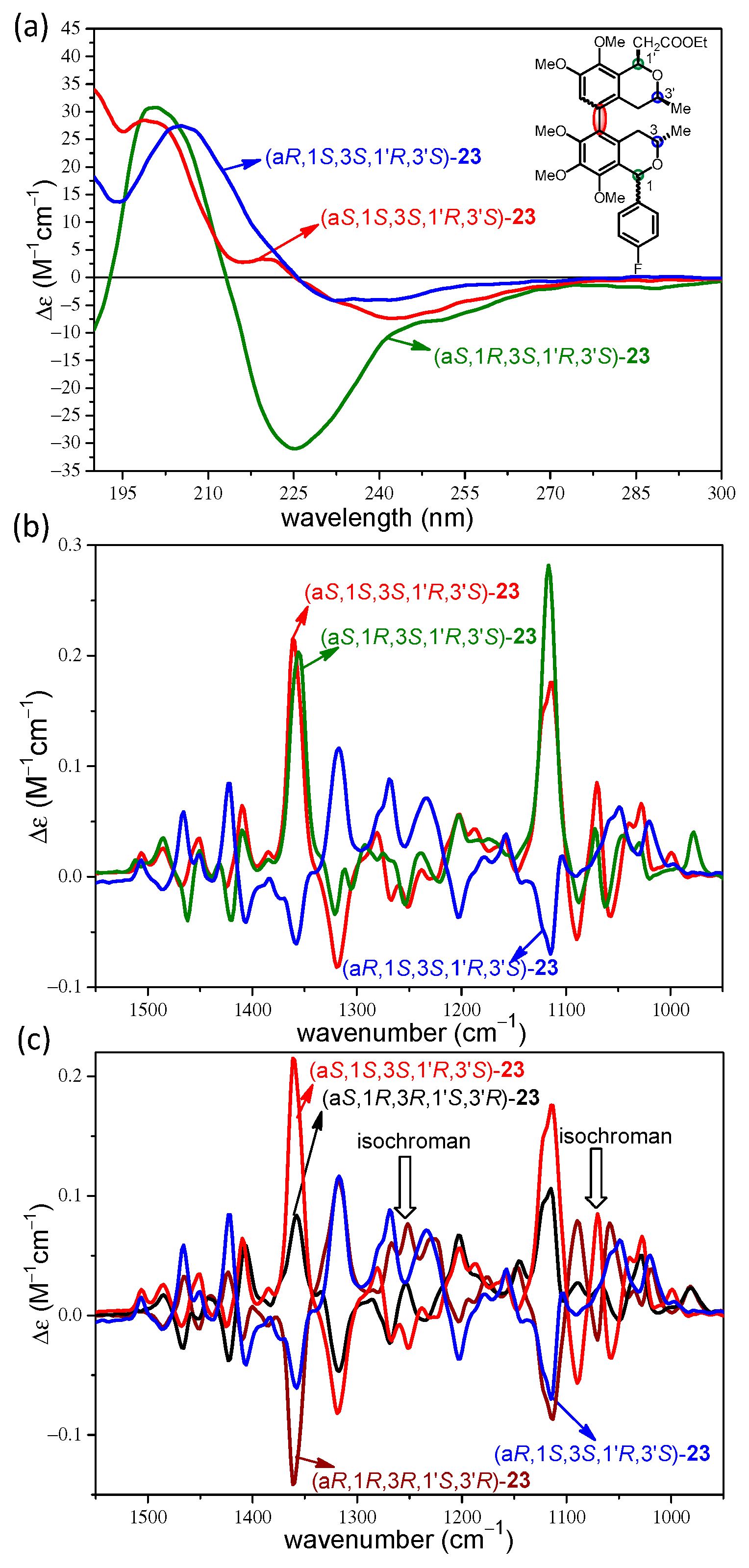
2. Results and Discussion
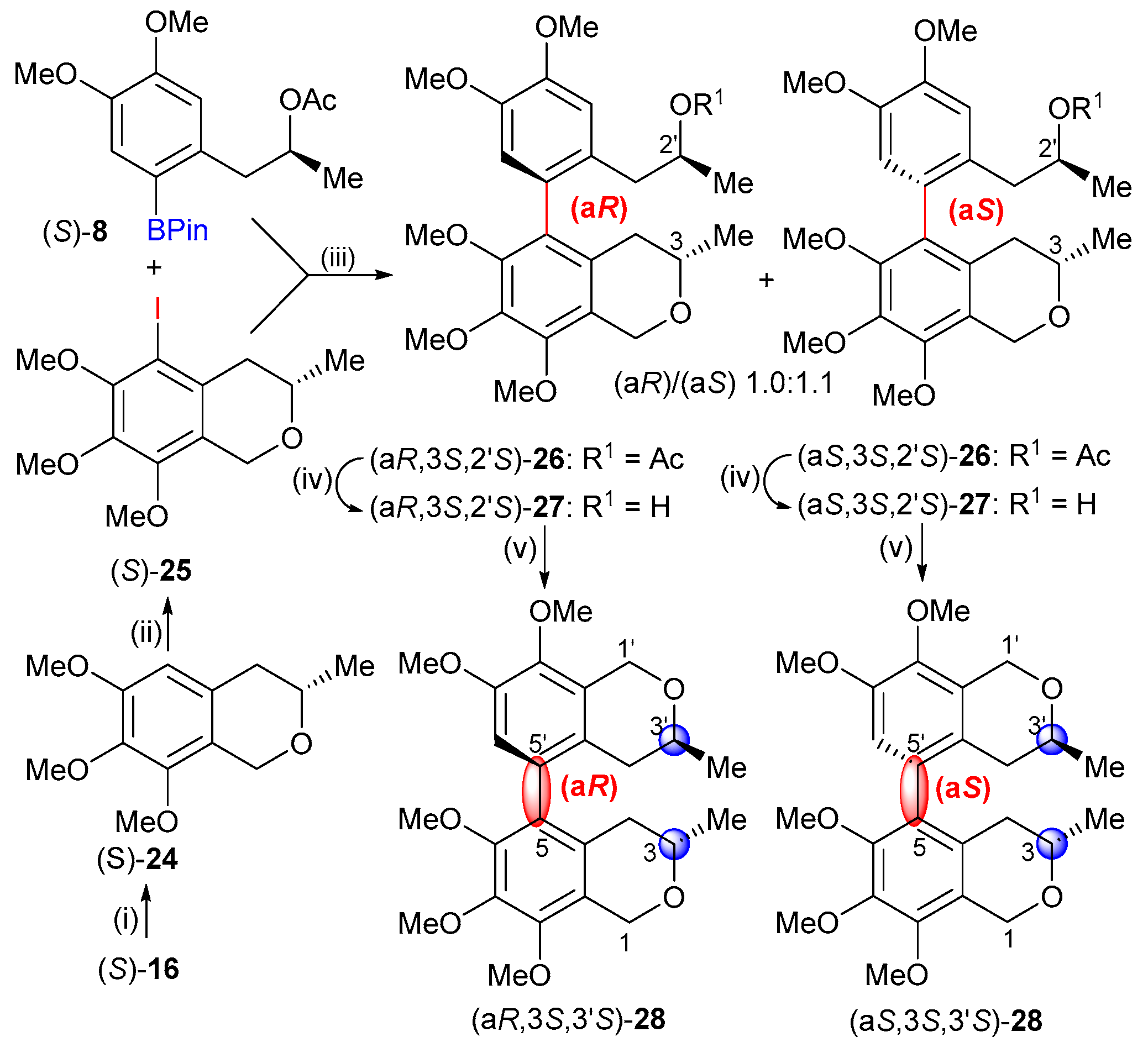
2.1. Synthesis

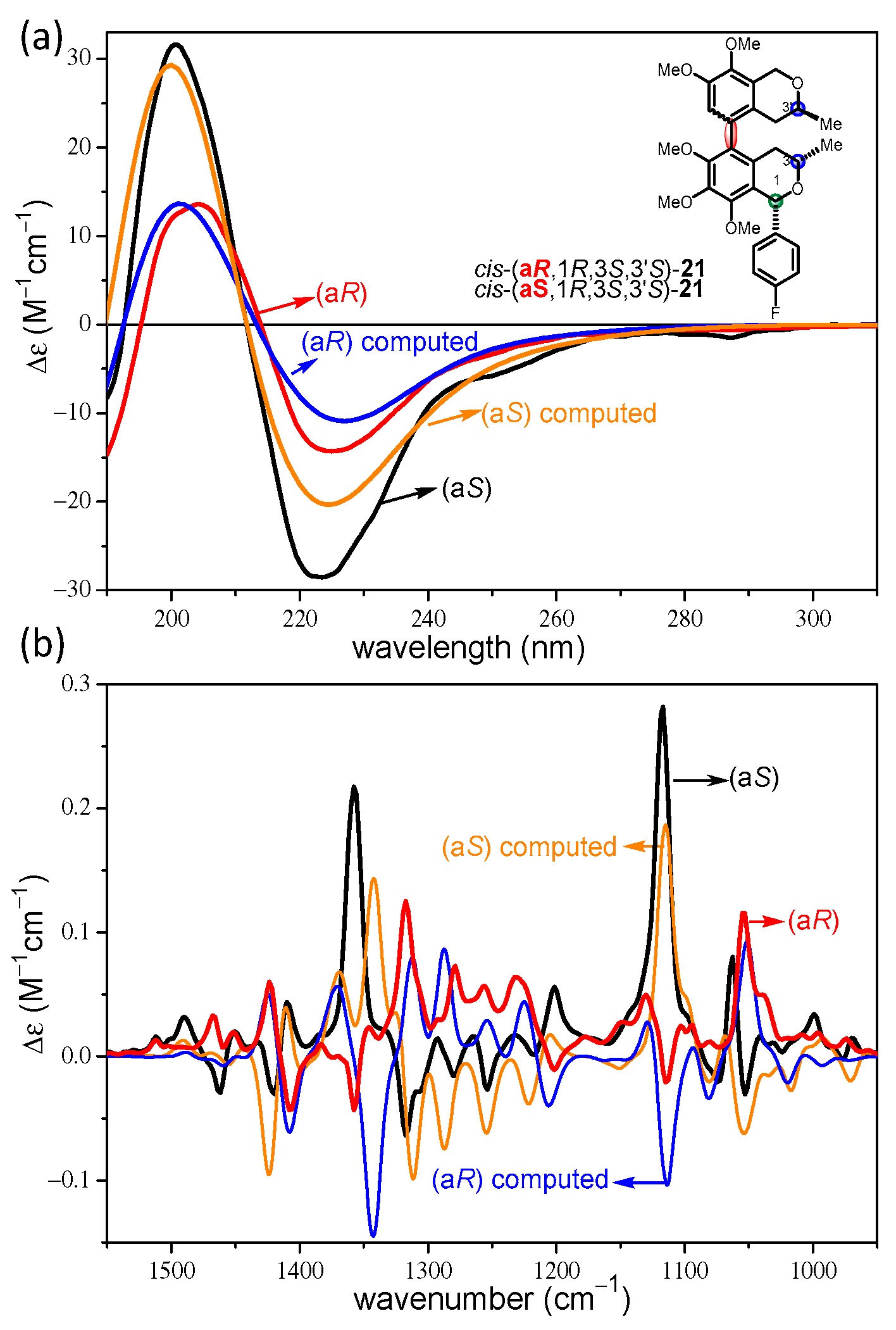
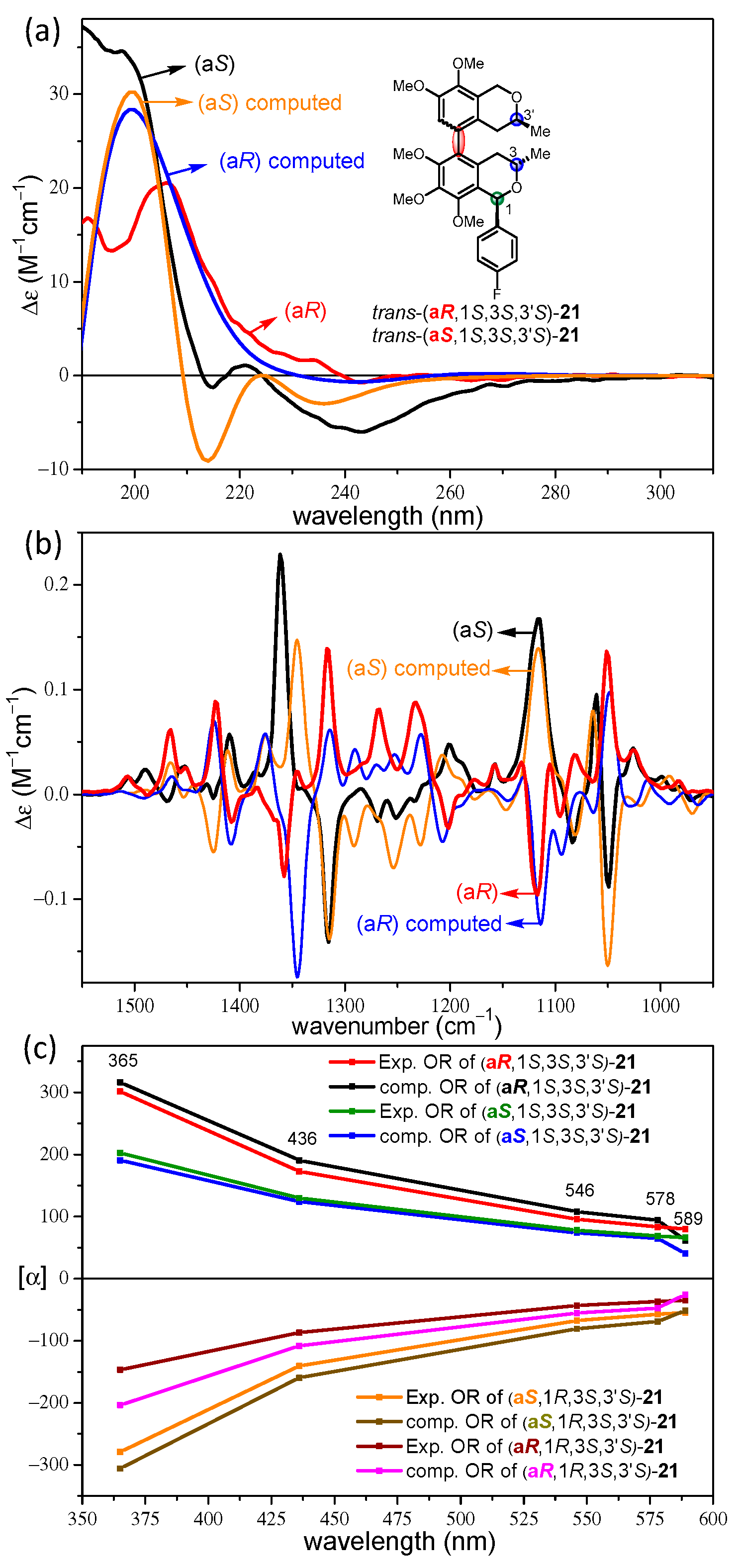
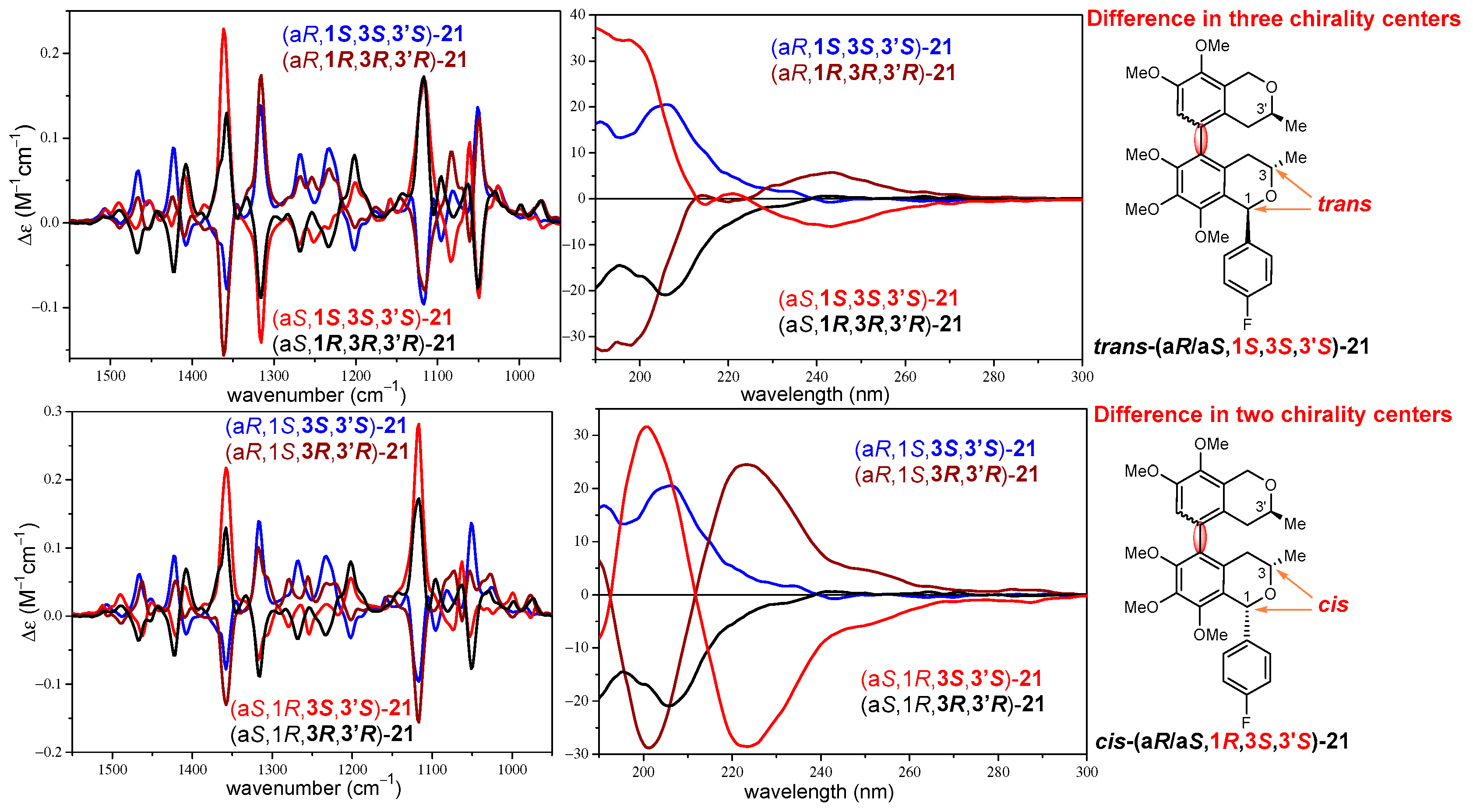
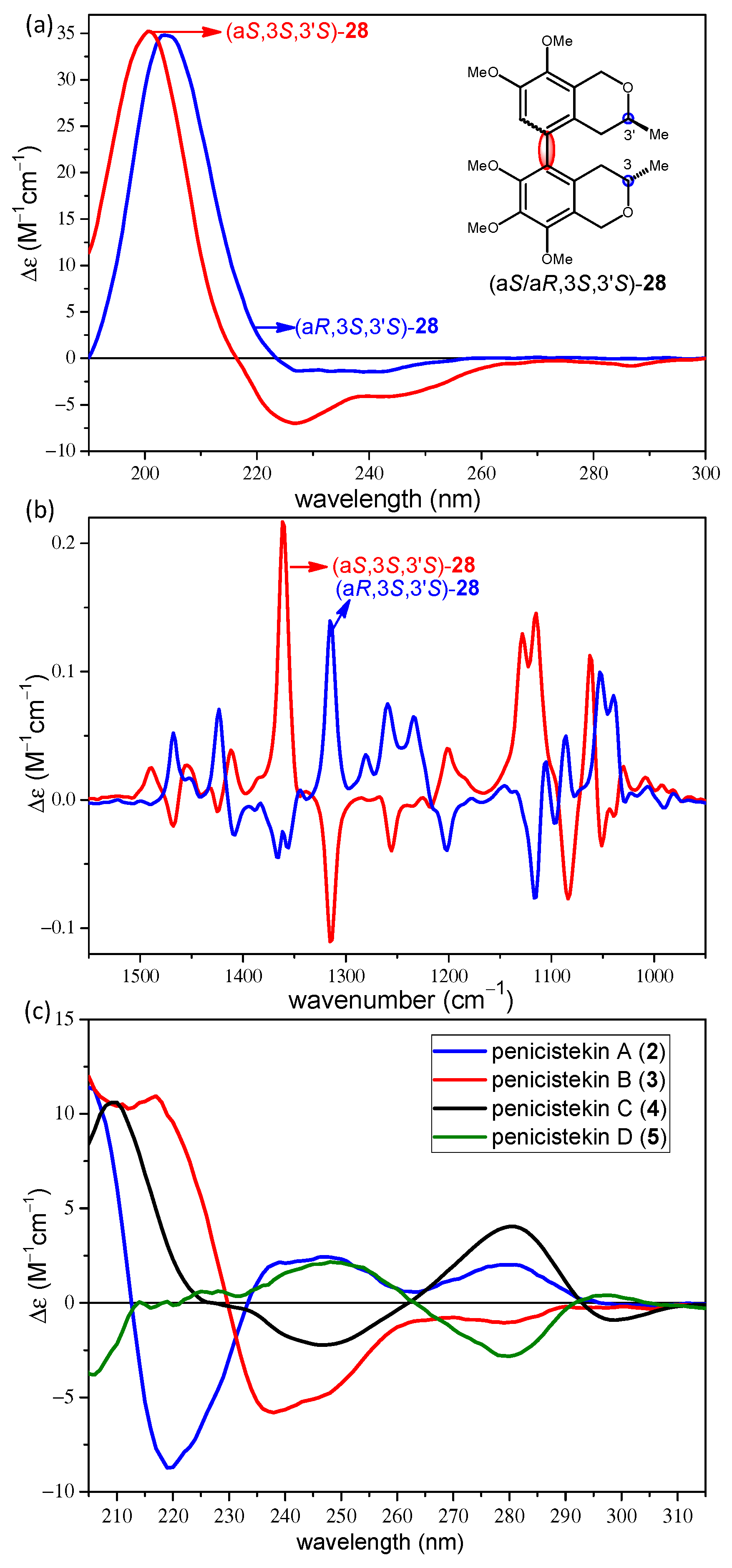
2.2. Stereochemical Analysis
3. Materials and Methods
3.1. General Information
3.2. Computational Section
3.3. X-ray Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.; Lee, C.; Bang, S.H.; Ma, J.Y.; Kim, S.; Koh, Y.-S.; Shim, S.H. Isochromans and Related Constituents from the Endophytic Fungus Annulohypoxylon truncatum of Zizania caduciflora and Their Anti-Inflammatory Effects. J. Nat. Prod. 2017, 80, 205–209. [Google Scholar] [CrossRef]
- He, G.; Matsuura, H.; Takushi, T.; Kawano, S.; Yoshihara, T. A New Antifungal Metabolite from Penicillium expansum. J. Nat. Prod. 2004, 67, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Kock, I.; Draeger, S.; Schulz, B.; Elsässer, B.; Kurtán, T.; Kenéz, Á.; Antus, S.; Pescitelli, G.; Salvadori, P.; Speakman, J.-B.; et al. Pseudoanguillosporin A and B: Two New Isochromans Isolated from the Endophytic Fungus Pseudoanguillospora sp. Eur. J. Org. Chem. 2009, 2009, 1427–1434. [Google Scholar] [CrossRef]
- Bianco, A.; Coccioli, F.; Guiso, M.; Marra, C. The occurrence in olive oil of a new class of phenolic compounds: Hydroxy-isochromans. Food Chem. 2002, 77, 405–411. [Google Scholar] [CrossRef]
- Lorenz, P.; Zeh, M.; Martens-Lobenhoffer, J.; Schmidt, H.; Wolf, G.; Horn, T.F.W. Natural and newly synthesized hydroxy-1-aryl-isochromans: A class of potential antioxidants and radical scavengers. Free Radical Res. 2005, 39, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Togna, A.R.; Latina, V.; Trefiletti, G.; Guiso, M.; Moschini, S.; Togna, G.I. 1-Phenil-6,7-dihydroxy-isochroman inhibits inflammatory activation of microglia. Brain Res. Bull. 2013, 95, 33–39. [Google Scholar] [CrossRef]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A twist of nature—The significance of atropisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef]
- Rönsberg, D.; Debbab, A.; Mándi, A.; Vasylyeva, V.; Böhler, P.; Stork, B.; Engelke, L.; Hamacher, A.; Sawadogo, R.; Diederich, M.; et al. Pro-Apoptotic and Immunostimulatory Tetrahydroxanthone Dimers from the Endophytic Fungus Phomopsis longicolla. J. Org. Chem. 2013, 78, 12409–12425. [Google Scholar] [CrossRef]
- Yang, Y.D.; Yang, B.B.; Li, L.A.-O. A nonneglectable stereochemical factor in drug development: Atropisomerism. Chirality 2022, 34, 1355–1370. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J.; Moran, W.J.; Edwards, P.J.; LaPlante, S.R. The Challenge of Atropisomerism in Drug Discovery. Angew. Chem. Int. Ed. 2009, 48, 6398–6401. [Google Scholar] [CrossRef]
- Sayed, A.M.; Ibrahim, A.H.; Tajuddeen, N.; Seibel, J.; Bodem, J.; Geiger, N.; Striffler, K.; Bringmann, G.; Abdelmohsen, U.R. Korupensamine A, but not its atropisomer, korupensamine B, inhibits SARS-CoV-2 in vitro by targeting its main protease (Mpro). Eur. J. Med. Chem. 2023, 251, 115226. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Sandha, K.; Waingankar, A.; Jain, P.; Abhyankar, A. Atropisomerism transforming anti-cancer drug discovery. Chem. Biol. Drug Des. 2023, 101, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Perreault, S.; Chandrasekhar, J.; Patel, L. Atropisomerism in Drug Discovery: A Medicinal Chemistry Perspective Inspired by Atropisomeric Class I PI3K Inhibitors. Acc. Chem. Res. 2022, 55, 2581–2593. [Google Scholar] [CrossRef]
- Wu, Z.-J.; Ouyang, M.-A.; Tan, Q.-W. New asperxanthone and asperbiphenyl from the marine fungus Aspergillus sp. Pest Manag. Sci. 2009, 65, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Z.; Huang, W.-J.; Liu, W.; Mándi, A.; Zhang, Q.; Zhang, L.; Zhang, W.; Kurtán, T.; Yuan, C.-S.; Zhang, C. Penicisteckins A–F, Isochroman-Derived Atropisomeric Dimers from Penicillium steckii HNNU-5B18. J. Nat. Prod. 2021, 84, 2953–2960. [Google Scholar] [CrossRef]
- Zhang, Q.; Mándi, A.; Li, S.; Chen, Y.; Zhang, W.; Tian, X.; Zhang, H.; Li, H.; Zhang, W.; Zhang, S.; et al. N–N-Coupled Indolo-sesquiterpene Atropo-Diastereomers from a Marine-Derived Actinomycete. Eur. J. Org. Chem. 2012, 2012, 5256–5262. [Google Scholar] [CrossRef]
- Wu, G.; Yu, G.; Kurtán, T.; Mándi, A.; Peng, J.; Mo, X.; Liu, M.; Li, H.; Sun, X.; Li, J.; et al. Versixanthones A–F, Cytotoxic Xanthone–Chromanone Dimers from the Marine-Derived Fungus Aspergillus versicolor HDN1009. J. Nat. Prod. 2015, 78, 2691–2698. [Google Scholar] [CrossRef]
- Ola, A.R.B.; Debbab, A.; Aly, A.H.; Mandi, A.; Zerfass, I.; Hamacher, A.; Kassack, M.U.; Brötz-Oesterhelt, H.; Kurtan, T.; Proksch, P. Absolute configuration and antibiotic activity of neosartorin from the endophytic fungus Aspergillus fumigatiaffinis. Tetrahedron Lett. 2014, 55, 1020–1023. [Google Scholar] [CrossRef]
- Zhu, Q.; Tang, C.; Mándi, A.A.-O.; Kurtán, T.A.-O.; Ye, Y.A.-O. Trigonostemons G and H, dinorditerpenoid dimers with axially chiral biaryl linkage from Trigonostemon chinensis. Chirality 2020, 32, 265–272. [Google Scholar] [CrossRef]
- Bara, R.; Zerfass, I.; Aly, A.H.; Goldbach-Gecke, H.; Raghavan, V.; Sass, P.; Mándi, A.; Wray, V.; Polavarapu, P.L.; Pretsch, A.; et al. Atropisomeric Dihydroanthracenones as Inhibitors of Multiresistant Staphylococcus aureus. J. Med. Chem. 2013, 56, 3257–3272. [Google Scholar] [CrossRef]
- Li, X.L.; Kurtán, T.; Hu, J.C.; Mándi, A.; Li, J.; Li, X.W.; Guo, Y.A.-O. Structural and Stereochemical Studies of Laurokamurols A-C, Uncommon Bis-sesquiterpenoids from the Chinese Red Alga Laurencia okamurai Yamada. J. Agric. Food Chem. 2017, 65, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, P.L.; Jeirath, N.; Kurtán, T.; Pescitelli, G.; Krohn, K. Determination of the absolute configurations at stereogenic centers in the presence of axial chirality. Chirality 2009, 21, E202–E207. [Google Scholar] [CrossRef]
- Csupor, D.; Kurtán, T.; Vollár, M.; Kúsz, N.; Kövér, K.E.; Mándi, A.; Szűcs, P.; Marschall, M.; Senobar Tahaei, S.A.; Zupkó, I.; et al. Pigments of the Moss Paraleucobryum longifolium: Isolation and Structure Elucidation of Prenyl-Substituted 8,8′-Linked 9,10-Phenanthrenequinone Dimers. J. Nat. Prod. 2020, 83, 268–276. [Google Scholar] [CrossRef]
- Ding, W.-Y.; Yan, Y.-M.; Meng, X.-H.; Nafie, L.A.; Xu, T.; Dukor, R.K.; Qin, H.-B.; Cheng, Y.-X. Isolation, Total Synthesis, and Absolute Configuration Determination of Renoprotective Dimeric N-Acetyldopamine–Adenine Hybrids from the Insect Aspongopus chinensis. Org. Lett. 2020, 22, 5726–5730. [Google Scholar] [CrossRef]
- Demarque, D.P.; Heinrich, S.; Schulz, F.; Merten, C. Sensitivity of VCD spectroscopy for small structural and stereochemical changes of macrolide antibiotics. Chem. Commun. 2020, 56, 10926–10929. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Nair, D.S.; Pillai, S.M.; Johnson, D.; Kallingathodi, Z.; Ibnusaud, I.; Polavarapu, P.L. Dissymmetry Factor Spectral Analysis Can Provide Useful Diastereomer Discrimination: Chiral Molecular Structure of an Analogue of (−)-Crispine A. ACS Omega 2019, 4, 6154–6164. [Google Scholar] [CrossRef] [PubMed]
- Felippe, L.G.; Batista Jr, J.M.; Baldoqui, D.C.; Nascimento, I.R.; Kato, M.J.; He, Y.; Nafie, L.A.; Furlan, M. VCD to determine absolute configuration of natural product molecules: Secolignans from Peperomia blanda. Org. Biomol. Chem. 2012, 10, 4208–4214. [Google Scholar] [CrossRef]
- Kerti, G.; Kurtán, T.; Illyés, T.-Z.; Kövér, K.E.; Sólyom, S.; Pescitelli, G.; Fujioka, N.; Berova, N.; Antus, S. Enantioselective Synthesis of 3-Methylisochromans and Determination of Their Absolute Configurations by Circular Dichroism. Eur. J. Org. Chem. 2007, 2007, 296–305. [Google Scholar] [CrossRef]
- Inoue, K.; Makino, Y.; Itoh, N. Production of (R)-chiral alcohols by a hydrogen-transfer bioreduction with NADH-dependent Leifsonia alcohol dehydrogenase (LSADH). Tetrahedron: Asymmetry 2005, 16, 2539–2549. [Google Scholar] [CrossRef]
- Ren, X.; She, X.; Peng, K.; Su, Y.; Xie, X.; Pan, X.; Zhang, H. First Enantioselective Synthesis of the Neolignans Rhaphidecursinol A and Virolongin B. J. Chin. Chem. Soc. 2004, 51, 969–974. [Google Scholar] [CrossRef]
- González-Liste, P.J.; León, F.; Arribas, I.; Rubio, M.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Highly Stereoselective Synthesis and Hydrogenation of (Z)-1-Alkyl-2-arylvinyl Acetates: A Wide Scope Procedure for the Preparation of Chiral Homobenzylic Esters. ACS Catal. 2016, 6, 3056–3060. [Google Scholar] [CrossRef]
- MacroModel; Schrödinger LLC. 2015. Available online: https://newsite.schrodinger.com/platform/products/macromodel (accessed on 12 July 2024).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, V.; Barone, G.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revisions C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef]
- Varetto, U. MOLEKEL 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009. [Google Scholar]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A Found. Adv. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Westrip, S. publCIF: Software for editing, validating and formatting crystallographic information files. J. Appl. Crystallogr. 2010, 43, 920–925. [Google Scholar] [CrossRef]
- Edgington, P.R.; McCabe, P.; Macrae, C.F.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar]
- Spek, A. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, V.; Barone, G.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revisions E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czenke, Z.; Mándi, A.; Király, S.B.; Kiss-Szikszai, A.; Kónya-Ábrahám, A.; Kurucz-Szabados, A.; Cserepes, K.; Bényei, A.; Zhang, C.; Kicsák, M.; et al. VCD Analysis of Axial Chirality in Synthetic Stereoisomeric Biaryl-Type bis-Isochroman Heterodimers with Isolated Blocks of Central and Axial Chirality. Int. J. Mol. Sci. 2024, 25, 9657. https://doi.org/10.3390/ijms25179657
Czenke Z, Mándi A, Király SB, Kiss-Szikszai A, Kónya-Ábrahám A, Kurucz-Szabados A, Cserepes K, Bényei A, Zhang C, Kicsák M, et al. VCD Analysis of Axial Chirality in Synthetic Stereoisomeric Biaryl-Type bis-Isochroman Heterodimers with Isolated Blocks of Central and Axial Chirality. International Journal of Molecular Sciences. 2024; 25(17):9657. https://doi.org/10.3390/ijms25179657
Chicago/Turabian StyleCzenke, Zoltán, Attila Mándi, Sándor Balázs Király, Attila Kiss-Szikszai, Anita Kónya-Ábrahám, Anna Kurucz-Szabados, Krisztián Cserepes, Attila Bényei, Changsheng Zhang, Máté Kicsák, and et al. 2024. "VCD Analysis of Axial Chirality in Synthetic Stereoisomeric Biaryl-Type bis-Isochroman Heterodimers with Isolated Blocks of Central and Axial Chirality" International Journal of Molecular Sciences 25, no. 17: 9657. https://doi.org/10.3390/ijms25179657